

Abstract
Campylobacter jejuni is the most frequent cause of human food-borne bacterial gastroenteritis but its physiology and biochemistry are poorly understood. Only a few amino-acids can be catabolised and these are known to be important for host colonization. Here we have established methods for rapid high throughput analyses of global metabolism in C. jejuni using direct injection mass spectrometry (DIMS) to compare metabolite fingerprints of wild-type and mutant strains. Principal component analyses show that the metabolic fingerprint of mutants that have a genomic deletion in genes for key amino-acid catabolic enzymes (either sdaA, serine dehydratase; aspA, aspartase or aspB, aspartate:glutamate transaminase) can easily be distinguished from the isogenic parental strain. Assignment of putative metabolites showed predictable changes directly associated with the particular metabolic lesion in these mutants as well as more extensive changes in the aspA mutant compared to the sdaA or aspB strains. Further analyses of a cj0150c mutant strain, which has no obvious phenotype, suggested a role for Cj0150 in the conversion of cystathionine to homocysteine. Our results show that DIMS is a useful technique for probing the metabolism of this important pathogen and may help in assigning function to genes encoding novel enzymes with currently unknown metabolic roles.
Electronic supplementary material
The online version of this article (doi:10.1007/s11306-014-0644-z) contains supplementary material, which is available to authorized users.
Keywords: Campylobacter jejuni, DIMS, Metabolism, aspA, aspB, sdaA
Introduction
Campylobacter jejuni is the leading cause of human bacterial gastroenteritis in the western world (Scallan et al. 2011) with over 400 million reported cases each year world wide (Ruiz-Palacios 2007), resulting in a large economic burden (Batz et al. 2012). The bacterium is commensal in the intestines of chickens and most infections arise from consumption of contaminated poultry meat. The physiology and metabolism of C. jejuni is less well understood compared to other food-borne pathogens. A limited ability to metabolise sugars along with the absence of phosphofructokinase (Parkhill et al. 2000) results in a major requirement for non-carbohydrate derived carbon sources in C. jejuni (Velayudhan and Kelly 2002). Previous studies have shown that only a limited range of amino acids and certain TCA cycle intermediates, are important carbon sources for C. jejuni (Velayudhan et al. 2004; Guccione et al. 2008; Hofreuter et al. 2008). The ability to use aspartate, glutamate (Leon-Kempis et al. 2006; Guccione et al. 2008), serine (Velayudhan et al. 2004; Hofreuter et al. 2012) and proline (Guccione et al. 2008; Hofreuter et al. 2008, 2012) is characteristic of most strains, with mutations in aspA (aspartase) aspB (aspartate:glutamate aminotransferase) and sdaA (serine dehydratase) genes found to result in an inability to catabolise these amino acids and defects in the colonisation of chicken and mouse animal models (Velayudhan et al. 2004; Guccione et al. 2008; Hofreuter et al. 2008, 2012).
Global metabolomic approaches have not previously been used to study the central carbon metabolism of C. jejuni. Although nuclear magnetic resonance methods have been used for this type of application (e.g. Raamsdonk et al. 2001), sensitivity of metabolite detection remains an issue. A potentially useful technique that has previously been used for high throughput fingerprinting is automated electrospray ionisation time of flight mass spectrometry (ESI-TOF MS) (Dunn et al. 2005). ESI-TOF MS is a method that enables rapid sample runtime and relatively simple mass spectra for complex samples, albeit at the expense of accurate mass identification and quantification (Dunn et al. 2005). Although originally utilised for high-throughput clinical screening there have also been several microbial applications of this technique, often in bacterial strain discrimination (Goodacre et al. 1998; Vaidyanathan et al. 2001), including MALDI-TOF mass spectrometry based identification of Campylobacter species (Bessède et al. 2011). The possibility of utilising Direct Injection ESI-TOF MS (DIMS) as a means of rapidly analysing gene knockout mutant libraries has also been suggested, with the major advantage of minimal sample preparation and processing. Work by Kaderbhai et al. (2003) found DIMS to be capable of discriminating between tryptophan metabolism mutants in Escherichia coli and ESI-TOF MS has been suggested as an advantageous methodology for assigning unknown gene function (Castrillo et al. 2003).
In this study, previously described C. jejuni null mutants in aspA, aspB (Guccione et al. 2008) and sdaA (Velayudhan et al. 2004) have been analysed by DIMS in order to assess whether the metabolic lesions in these strains can be confirmed by their metabolic fingerprints and whether other changes in the metabolome can be inferred from putative metabolite identifications. The relative abundances of key metabolites have been found to show the changes expected in these mutants and the techniques have also been applied to a mutant in cj0150c (Guccione et al. 2008), a gene of unknown function, to generate a hypothesis for the role of the encoded aminotransferase enzyme, Cj0150. Our results suggest that non-targeted, high sensitivity, low mass accuracy direct injection techniques can be useful for probing the metabolism of poorly understood pathogens like C. jejuni and for generating hypotheses for the function of genes with currently unknown roles.
Materials and methods
Bacterial strains, media and growth conditions
The strains used in this study were C. jejuni NCTC 11168 wild-type and a set of isogenic mutants carrying null mutations in the sdaA, aspA, aspB and cj0150c genes. These mutants have been fully described in previous publications (Velayudhan et al. 2004; Guccione et al. 2008) and were constructed by the insertion of antibiotic resistance cassettes into the appropriate genes. The mutants were obtained from the Kelly lab culture collection and verified by PCR analysis with gene specific primers. All bacterial strains were routinely grown on Columbia agar containing 5 % (v/v) lysed horse blood and 10 μg mL−1 amphotericin B and vancomycin at 37 °C under microaerobic conditions [10 % (v/v) O2, 5 % (v/v) CO2 and 85 % (v/v) N2] in a MACS growth cabinet (Don Whitley Scientific, Shipley, UK). For mutant strain analysis five biological replicate cultures were grown alongside five biological replicates of the isogenic parent strain in 25 mL volumes of brain heart infusion (BHI) (Oxoid) containing 5 % (v/v) foetal calf serum (FCS; GIBCO) under microaerobic conditions with continuous shaking at 180 rpm.
Extraction of C. jejuni metabolites
Cells taken for metabolite extraction were harvested by centrifugation (15,871 g, 1 min, 20 °C), the supernatants removed and the cell pellets flash frozen in liquid nitrogen. This decreased the possibility of cell lysis resulting in metabolites entering the supernatant and hence increased the number of measureable metabolites within the cells. Cell density was normalised by OD600 and centrifugation of a culture volume that would correspond to 1 mL of OD600 1.0 culture. Metabolite extraction was performed based on the method of Overy et al. (2005). Cell pellets were re-suspended in 1 mL methanol:chloroform 1:1 (−20 °C) and vortexed to disrupt the pellet. Samples were then stored at −80 °C for 1 h before further disruption through vigorous shaking with cold chloroform:methanol (1:1) washed stainless steel ball bearings (2 mm diameter). Samples were again stored at −80 °C for 1 h before the addition of 400 μL ice-cold ultra high pure (UHP) water. Tubes were mixed by vortexing before centrifugation (15, 871×g, 1 min, 4 °C) whereafter the top aqueous phase (methanol plus water) containing mainly polar metabolites was decanted into a new cooled 1.5 mL plastic tube. Then 400 μL ice-cold UHP water was added to the chloroform (organic) phase containing mainly non-polar metabolites and centrifuged (15, 871×g, 1 min, 4 °C). The top aqueous phase was again decanted into the 1.5 mL plastic tube containing the previously harvested aqueous phase. Both aqueous and organic phases were then stored at −80 °C until analysis. A negative control of the above full harvesting and extraction procedure was carried but without the addition of bacteria. The metabolite profiles of these negative controls were outliers in the subsequent PCA analysis (data not shown).
Electrospray ionisation time of flight mass spectrometry (ESI-TOF MS)
ESI-TOF MS was performed on a LCT spectrometer (Waters Ltd, Manchester, UK) based on the methods described in Davey et al. (2008) and Walker (2011). Data acquisition and processing was performed on MassLynx (version 4) to create centroid peak lists (m/z accurate to 4 decimal places vs. ion counts), which were then transferred to Microsoft Excel (Microsoft Corp, USA) as text files. The mass spectrometer was operated at a resolution of 4,000 (FWHM) at mass 200 m/z in positive and negative ion modes at a capillary voltage of 2,800 V (positive) and 2,500 V (negative), extraction cone at 3 V and sample cone at 20 V with a rangefinder lens voltage of 75 V chosen for detection of masses from 50 to 800 Da. Source temperature was 110 °C and desolvation temperature was 120 °C. Flow rates were 100 L h−1 for nebulisation and 400 L h−1 for desolvation. Spectra were collected in centroid mode at a rate of one spectrum s−1 (0.95 s scan time, 0.05 s interscan delay) with 180 summed over a 3-min period before being exported without background subtraction or smoothing.
Samples were either loaded using a syringe pump (Razel, Connecticut, USA) at a flow rate of 20 μL min−1 (organic phase) or loaded using an automated Waters 2695 Separations Module combining a HPLC pump and an autosampler (Waters, Hemel Hempstead, UK) (aqueous phase) with an inject volume of 100 μL at a flow rate of 50 μL min−1. A Lockspray™ interface was used to give an external standard and allow automated correction of mass measurements (5 ng μL−1 sulphadimethoxine giving a lockmass of 309.0653 or 311.0814 for negative and positive modes respectively). Samples were analysed in a randomised order to minimize effects of day-to-day machine variation.
Data processing
For each sample run, the summation of 180 centroid mode spectra were exported from MassLynx data systems as text file peak lists (Accurate mass to 4 decimal places vs. ion count). These were imported into Microsoft Excel (Microsoft Corp, USA) and an in-house macro programme (created by Prof. Mike Burrell, Sheffield University, UK) used to compare the accurate masses of three technical replicate analyses of each sample. The accepted range to give the maximum number of peaks with minimum false positives was proposed by Overy et al. (2005) to be defined best by plotting the acceptable mass variance as a linear function of the m/z values. The same equations were used: positive mode, y < 0.00003x + 0.0033; for negative mode y < 0.00003x + 0.044 where y is the standard deviation of the three masses and x is the mean of the three masses. Those masses found to have a y value within acceptable limits had their mean accurate mass and mean response (as % total ion count in order to minimize sample–sample variation and normalize data sets) exported to a separate table. This methodology negated the need for a noise threshold and enabled true low intensity metabolite peaks to be kept. principal component analysis (PCA) was carried out using Pareto scaled 0.2 Da binned data sets in Simca-P v12.0 software (Umetrics, Sweden). Significance values of the ion counts between the samples were determined using two-tailed t tests.
Putative metabolite identification
Preliminary identification of metabolites was performed through the comparison of monoisotopic masses likely to be present in extracts, including [M+H]+, [M−H]− and [M+Na]+ against the list of metabolites in the biocyc database (http://biocyc.org/) and to a subset of metabolites within biocyc predicted to be present in C. jejuni NCTC 11168 (http://biocyc.org/CJEJ192222/class-tree?object=Compounds) based on the genome annotation, to an accuracy of 0.2 Da (Davey 2011).
Results and discussion
Direct injection ESI-TOF MS methodologies can distinguish C. jejuni metabolomic profiles
Methodology for both the quenching of metabolism and extraction of metabolites is still a controversial area and can require significant optimisation for individual organisms to yield reproducible data (Meyer et al. 2013). Many studies have looked at the effectiveness of quenching and extraction techniques through statistical methods (Bolten et al. 2007; Faijes et al. 2007). However, very few have been focussed on the ability to see biologically relevant metabolite changes within a non-targeted metabolomics analysis as a measure of quenching and extraction effectiveness. A variety of quenching techniques have been adopted for bacterial metabolomics, with the most popular methods being quenching in cold organic solvents, followed by centrifugation and extraction (Bolten et al. 2007) or rapid filtration based methods, although metabolite leakage problems are still an issue. Agar plate membrane filter culture techniques followed by rapid solvent quenching is one solution for these problems (Bennett et al. 2008).
In this study, a complex liquid medium (BHI-FCS) was used for bacterial cultivation to ensure as similar growth as possible of wild-type and mutant strains as well as maintaining activity of amino-acid catabolic routes. The use of a simple centrifugation step to collect the cells meant a methodology could be employed that resulted in no metabolite loss, through the freezing of whole cell pellets in liquid nitrogen (Bolten et al. 2007) before solvent extraction. Although even brief centrifugation may cause some metabolic perturbation, the effects should be minimised when directly comparing wild-type and mutant cell metabolomes, as both sample groups are treated identically. Similarly, washing steps were avoided to prevent metabolite loss but contamination of cell pellets with residual medium will be the same for each sample compared and medium-only controls were also run. Although the efficiency of extraction techniques is organism specific (Faijes et al. 2007; Meyer et al. 2013), comparative studies have shown cold methanol based methodologies to be among the most efficient for extracting a wide range of metabolites, in a broad spectrum of bacteria (Maharjan and Ferenci 2003; Park et al. 2012). For this reason a cold methanol:chloroform based extraction protocol was adopted here (adapted from Overy et al. (2005)).
Multiple replicates of aspA, aspB and sdaA mutant extracts were analysed by ESI-TOF MS alongside extracts from the C. jejuni NCTC 11168 isogenic parent strain in both negative ionisation mode (to focus on detecting carboxylic acids and alcohols) and positive ionization mode (to focus on detecting amines). Background noise was removed by selecting peaks whereby mass variance between three technical replicates fell within an accepted range and data was normalized to the total ion count before being separated into 0.2 Da mass unit bins (Online resources 1 and 2). PCA can clearly be seen to separate mutant profiles from their isogenic parent strain (Fig. 1). Positive ionization datasets, containing a larger number of ionization species, could be separated in a similar fashion to negative ionization datasets.
Fig. 1.

Principal component analysis (PCA) of C. jejuni NCTC 11168 and amino acid catabolism mutant strains. Metabolite levels in multiple replicates of C. jejuni NCTC 11168 and aspB strains were analysed by ESI-TOF MS in positive and negative ionisation mode and processed so that centroid m/z peaks were converted to text file peak lists and placed into 0.2 Da bins. Principal component analysis was performed on these bin lists. PCA plot a and b show analysis of positive and negative ionization datasets respectively for C. jejuni NCTC 11168 (filled circles) and aspA (open circles). PCA plot (c) and (d) show analysis of positive and negative ionization datasets respectively for C. jejuni NCTC 11168 (filled circles) and aspB (open circles). PCA plot (e) and (f) show analysis of positive and negative ionization datasets respectively for C. jejuni NCTC 11168 (filled circles) and sdaA (open circles). In all instances mutant strains were observed to group separately from their isogenic parent strain
Allocation of putative metabolite identities to the C. jejuni metabolome
Dunn et al. (2005) have previously shown DIMS methods to be capable of identifying metabolites of the same nominal but different monoisotopic mass. In our work, protocols were adapted to minimize problems of fragmentation and ion suppression as previously highlighted (Dunn et al. 2005) and putative metabolite identities were preferentially assigned to negative ionization data due to the existence of fewer likely ionic species [M−H]−. A full list of m/z bin allocations and the associated metabolite identities can be seen in Online resources 1–3.
It has been stated that bacterial metabolomes contain low numbers of metabolites, i.e. ~600 predicted for E. coli and Saccharomyces cerevisiae (Wang et al. 2006) and 467 in a recent metabolic model of C. jejuni metabolism (Metris et al. 2011). Clearly a reliance on genomic analysis to make predictions (Oliver et al. 1998; Metris et al. 2011) will underestimate metabolome complexity, with many enzymes likely to work on broader substrate ranges within the cell than generally accepted, and many genes with unknown metabolic roles. In our work, the large metabolite list from the biocyc database has been used to maximise allocated peak identities. However, this obviously may result in metabolites being allocated that do not lie within predicted metabolic routes of C. jejuni. We therefore also present metabolite tables in Online resources 1–3 based only on a comparison with predicted metabolites in C. jejuni NCTC 11168 as derived from the automated genome annotation. It is essential that putative metabolite allocations relating to changes of interest are thoroughly reviewed and should be confirmed in future work by other targeted assays.
Putative metabolite identification in characterised metabolic mutants is consistent with known blocks in metabolism
Serine dehydratase catalyses the deamination of serine to form pyruvate and ammonium ions, in the first step of serine catabolism. In replicate extracts of the sdaA mutant compared to the wild-type, analyzed in negative ion ionisation mode, m/z 104.00, putatively corresponding to serine, was found to have undergone a 4.5 fold increase from 0.083 ± 0.016 to 0.37 ± 0.03 % of the total ion count with a p value of 8.2 × 10−7 (Fig. 2). Other key m/z values corresponding to metabolites of central metabolism within and connected with the citric-acid cycle were found to be highly similar with only m/z 117.00 (succinate) showing a statistically significant decrease in the mutant strain (Fig. 2). These results are consistent with the enzymatic block in the sdaA mutant resulting in a build-up of serine relative to wild-type. In total, however, we found only about 4.3 % of the total negative ion metabolite bins were significantly changed in the sdaA mutant (Table 1 and Online resource 1). The lack of other major central metabolite changes in this mutant is consistent with the only major effect on cell physiology being lack of growth on l-serine as C-source in minimal media (Velayudhan et al. 2004; Guccione et al. 2008). The fact that aspartate, proline and glutamate can still be utilised as carbon sources and that no growth defect is observed in complex media (Velayudhan et al. 2004) may explain why pyruvate levels are also maintained.
Fig. 2.

Key metabolite differences between C. jejuni NCTC 11168 and sdaA strains. Following ESI-TOF MS of five biological replicates in negative ionisation mode, m/z peaks were assigned putative metabolite identities to an accuracy of 0.2 Da for C. jejuni NCTC 11168 (black bars) and sdaA strains (open bars). Shown are those metabolites detected that are most closely related to amino-acid catabolism [Ser serine (104.00); Pyr pyruvate (87.00); Oxo 2-oxoglutarate (145.00); Suc Succinate (117.00); Fum Fumarate (115.00); Mal Malate (133.00); Oxa oxaloacetate (134.00); PEP phosphoenolpyruvate (167.00); Asp aspartate (132.00); Glu glutamate (146.00); Pro proline (115.00); Gln glutamine (145.00)] as a % of the total ion count. * p value <0.05 between C. jejuni NCTC 11168 and sdaA strain
Table 1.
Proportion of metabolite bins changed in each of the mutant strains analysed in this study, compared to the isogenic wild-type parent strain
| Strain | Ionisation mode | Total metabolite count | Significantly changed | % Metabolites changed |
|---|---|---|---|---|
| sdaA | − | 2,167 | 94 | 4.34 |
| aspB | − | 2,171 | 135 | 6.22 |
| aspA | − | 1,568 | 278 | 17.73 |
| cj0150c | − | 2,060 | 267 | 12.96 |
| sdaA | + | 1,474 | 72 | 4.88 |
| aspB | + | 1,532 | 135 | 8.81 |
| aspA | + | 1,568 | 223 | 14.22 |
The table shows how many bins have a value in at least one replicate and how many of those are significant at p < 0.05 when comparing each mutant with the wild-type strain
The metabolite profile of the aspA mutant was found to be distinctly different, with significant changes in the relative abundance of a much larger number of metabolites (Table 1 and Online resource 2), indicating a large-scale disruption to central as well as more peripheral metabolism. Aspartase catalyses the deamination of aspartate to fumarate plus ammonium ions and analysis of the metabolite levels show a relative accumulation of m/z 146.00 (glutamate) and m/z 132.00 (aspartate), and significantly reduced levels of m/z 115.00 (fumarate) and several putative TCA cycle metabolites, consistent with the metabolic lesion in this mutant (Fig. 3). An interesting exception is m/z 145.00 (2-oxoglutarate) (0.30 ± 0.02 and 0.30 ± 0.02 % total ion count in NCTC 11168 and aspA mutant respectively), but this cannot be distinguished from glutamine from our analysis. The many metabolite changes are consistent with the known phenotype of the aspA mutant, which cannot grow on aspartate, glutamate or proline and has a reduced growth rate compared to the wild-type, even in complex media (Guccione et al. 2008).
Fig. 3.
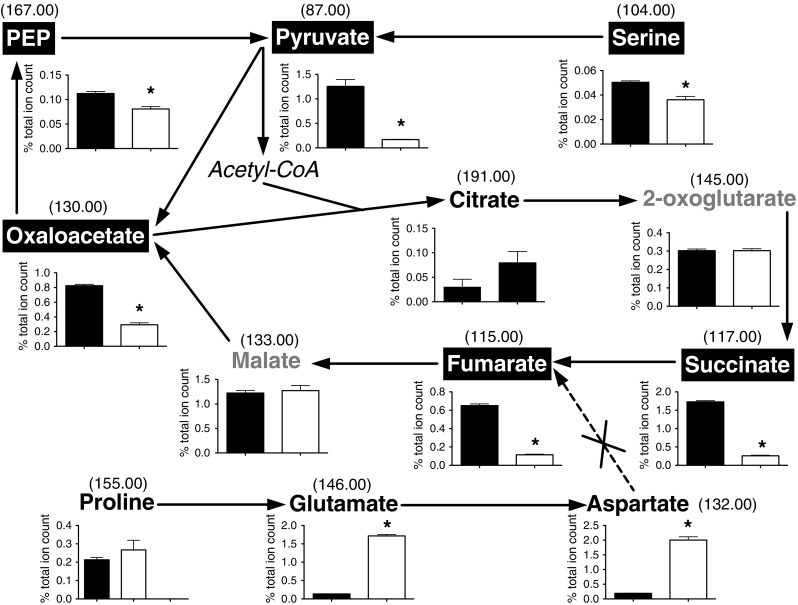
The effect of an aspA null mutation on central carbon metabolism. Metabolites in black typeface show an increase in relative abundance in the aspA mutant; white typeface a decrease in relative abundance; grey typeface no change and italicized typeface, not detected in either strain. *p value <0.05 between the C. jejuni NCTC 11168 (black bars) and aspA strain metabolite levels (open bars), displayed as a % of the total ion count with bin values depicted above. Aspartate and glutamate are 10.5 and 12.6 fold increased respectively in the aspA strain compared to the wild-type. Most central metabolites are lower in relative abundance in the aspA strain, exceptions being 2-oxoglutarate, a glutamate precursor, malate and citrate
AspB in C. jejuni is encoded by cj0762c and is essential for growth on glutamate, as it converts glutamate to aspartate using oxaloacetate as the amino-acceptor. In the aspB mutant a significant accumulation of m/z 146.00 (glutamate) is observed preceding the metabolic lesion as well as a significant increase in m/z 145.00 (2-oxoglutarate). Compared to the aspA mutant, fewer metabolites overall were significantly changed (Table 1 and Online resources 1 and 2). However, several glutamate precursors in various metabolic pathways have undergone significant changes (Fig. 4), which are not readily interpretable but might, for example, result from changes in enzyme allosteric control.
Fig. 4.
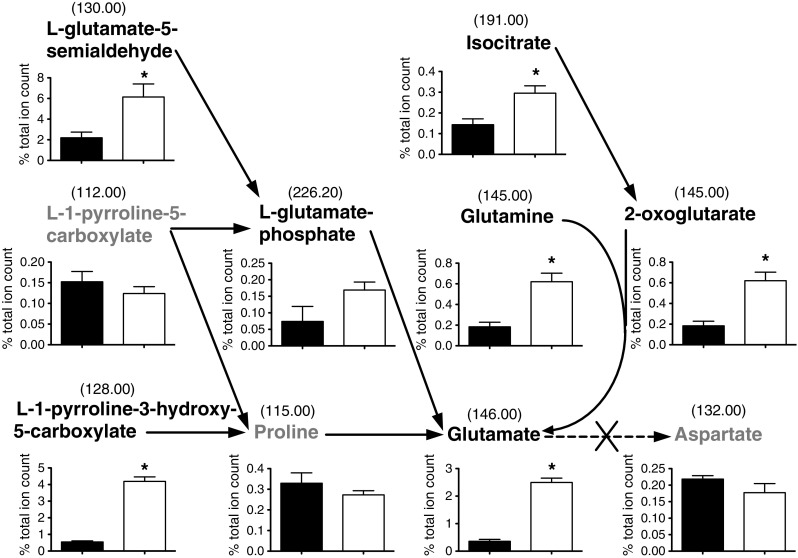
The effect of an aspB null mutation on glutamate metabolism. Having putatively identified metabolites from ESI-TOF MS negative ionisation mode the relative metabolite levels were analysed related to the lesion in metabolism caused by aspB mutation (black cross). Metabolites in black typeface show an increase in relative abundance in the aspB strain and in grey typeface a decrease. *p value < 0.05 between the C. jejuni NCTC 11168 (black bars) and aspA strain (white bars) metabolite levels, displayed as a % of the total ion count. Glutamate can be seen to have undergone a significant sevenfold increase and aspartate a slight decrease in the aspB mutant. Many metabolites linked to glutamate can be seen to have undergone an increase in the aspB strain
Analyses of these previously characterised metabolic mutants clearly shows that a simple ESI-TOF direct injection method is capable of detecting the corresponding metabolic lesion and provides unique information about how the mutation is affecting the wider metabolome (Online resources 1 and 2). Removal of AspA (by mutation) from the highly interconnected cellular enzyme network clearly has a “ripple effect” that spreads far beyond the mutant metabolic lesion. This has been observed in some other bacterial metabolic mutant studies [for a recent example see Lee et al. (2014)]. The observed metabolite changes will be the combined result of many processes e.g. altered allosteric control of enzyme activities by key metabolites and changed expression of enzyme encoding genes as a result of altered activity of regulatory proteins. It would therefore be highly informative to correlate mutant metabolomic data with transcriptomic and proteomic data to provide an integrated view that might give insights into the types of mechanisms involved.
Analysis of a cj0150c mutant reveals a possible role for Cj0150 as a cystathionine β lyase
In contrast to SdaA, AspA and AspB, Cj0150 is an enzyme of unknown function. Sequence analysis shows that it is a member of the amino-acid aminotransferase type I family (Guccione et al. 2008). These enzymes are known to be active with a wide range of substrates but it is not possible from sequence similarities alone to ascribe a physiological function to them. Purified recombinant Cj0150 has previously been shown to have aspartate:glutamate aminotransferase activity (Guccione et al. 2008), but this was not thought to be physiologically significant as a cj0150c mutant strain shows no growth defect on glutamate, whereas a cj0762c (aspB) mutant is completely unable to grow on or utilise glutamate or proline as a carbon source (Guccione et al. 2008). Here, we compared the metabolic fingerprints of the isogenic wild-type and cj0150c mutant by DIMS. PCA analysis of the data show ESI-TOF MS based metabolomics is capable of distinguishing the wild-type and mutant strain, even though the latter has no obvious growth or other known phenotype (Fig. 5). Analysis of the changes in the relative abundance of the putatively identified metabolites, suggests that although the cj0150c mutant is capable of growth on glutamate or proline, it is in fact providing some level of aspartate:glutamate aminotransferase activity in vivo as evidenced by a small but significant change in the ratio of glutamate and aspartate in the wild-type and mutant strains (Fig. 6). Most interestingly, we noted larger significant changes in two metabolites that we putatively identified as cystathionine and homocysteine. Changes in cystathionine and homocysteine levels would be consistent with Cj0150 being a cystathionine β lyase. Cystathionine β lyases are PLP dependent enzymes that function in the transulphuration pathway of methionine biosynthesis, converting cystathionine to homocysteine via an α/β elimination reaction (Auger et al. 2005). Significantly, Guccione et al. (2008) carried out a phylogenetic analysis of type I aminotransferases and found that Cj0150 clusters within the IΩ subgroup, along with enzymes experimentally found to possess cystathionine β lyase activity (Auger et al. 2005; Chu et al. 1995). In addition, the gene immediately downstream of cj0150c in strain NCTC 11168 encodes homoserine dehydrogenase; an enzyme that functions to produce homoserine from aspartate semialdehyde, an intermediate in the methionine biosynthetic pathway. It is important to note that C. jejuni does contain an annotated metC gene (cj1393), the major cystathionine β lyase known to perform this function (Lodha et al. 2010), and that methionine levels were maintained in the cj0150c strain. For this reason it is difficult to confidently predict whether the cystathionine β lyase activity of Cj0150 would be physiologically relevant for methionine biosynthesis, but determination of the kinetics of this activity for purified Cj0150 is clearly warranted. Although some changes in other putative metabolites related to aminotransferase activity were seen (tyrosine aminotransferase, phenylalanine aminotransferase; Fig. 6 and Online resource 3) the extent of the changes were less convincing, compared to those discussed above.
Fig. 5.

Principal component analysis of C. jejuni NCTC 11168 and cj0150c strains from negative ionisation mode metabolite profiles. Metabolite levels in multiple replicates of C. jejuni NCTC 11168 and cj0150c strains were analysed by ESI-TOF MS in negative ionisation mode and processed so that centroid m/z peaks were converted to text file peak lists and placed into 0.2 Da bins. Principal component analysis was performed on these bin lists. PCA plot (a) shows analysis of negative ionisation data with principal component 1 (PC1) against principal component 2 (PC2). C. jejuni NCTC 11168 and cj0150c are represented as closed circles and open circles respectively with the corresponding score loading plot shown in (b)
Fig. 6.

Significant metabolite changes for potential aminotransferase activities in the cj0150c strain. Significant changes were observed in metabolites related to: a cystathionine β lyase activity and metabolites related to general type I aminotransferase activity such as b tyrosine aminotransferase c phenylalanine aminotransferase and d aspartase activity. Wild type and cj0150c are shown as black and white bars respectively. *p value <0.05, an arrow represents an aminotransferase reaction and bin values for each metabolite seen above. Metabolites in black typeface show an increase in relative abundance in the cj0150 strain and in grey typeface, a decrease
Concluding remarks
We have shown that DIMS fingerprints can separate metabolomic profiles of metabolic mutants of C. jejuni and give insights into the degree to which the metabolome is changed by specific enzymatic blocks. Although the ESI-TOF MS based metabolomics method used here suffers from a lack of mass accuracy its high sensitivity and high throughput make it an ideal strategy for the initial analysis of differences in a large number of cellular metabolites in mutant screens. Information from the DIMS fingerprint of a mutant with no obvious phenotype has also been used here to suggest an in vivo activity for the Cj0150 enzyme.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online resource 1. Excel spread sheet showing 0.2 Da mass unit bin centres of peaks normalized to total ion count for C. jejuni NCTC 11168, sdaA and aspB strains run in positive and negative ionisation modes. Putative metabolite allocations based on the full and Campylobacter jejuni jejuni NCTC 11168 specific biocyc (www.biocyc.com) compounds lists and preliminary data analysis are also shown (XLS 8510 kb)
Online resource 2. Excel spread sheet showing 0.2 Da mass unit bin centres of peaks normalized to total ion count for C. jejuni NCTC 11168 and aspA strains run in positive and negative ionisation modes. Putative metabolite allocations based on the full and Campylobacter jejuni jejuni NCTC 11168 specific biocyc (www.biocyc.com) compounds lists and preliminary data analysis are also shown (XLS 6040 kb)
Online resource 3. Excel spread sheet showing 0.2 Da mass unit bin centres of peaks normalized to total ion count for C. jejuni NCTC 11168 and cj0150c strains run in negative ionisation mode. Putative metabolite allocations based on the full and Campylobacter jejuni jejuni NCTC 11168 specific biocyc (www.biocyc.com) compounds lists and preliminary data analysis are also shown (XLS 2486 kb)
Acknowledgments
This work was supported by a Biotechnology and Biological Sciences Research Council (BBSRC) Doctoral Training Award, to RMH. MPD acknowledges the support of a Wellcome Trust Value in People award—reference 083772/Z/07/Z. We thank Prof. Mike Burrell for help with data processing and Heather Walker for technical help.
References
- Auger S, Gomez MP, Danchin A, Martin-Verstraete I. The PatB protein of Bacillus subtilis is a C-S-lyase. Biochimie. 2005;87:231–238. doi: 10.1016/j.biochi.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Batz MB, Hoffmann S, Morris JG. Ranking the disease burden of 14 pathogens in food sources in the United States using attribution data from outbreak investigations and expert elicitation. Journal of Food Protection. 2012;75:1278–1291. doi: 10.4315/0362-028X.JFP-11-418. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Yuan J, Kimball EH, Rabinowitz JD. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nature Protocols. 2008;3:1299–1311. doi: 10.1038/nprot.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessède E, Solecki O, Sifré E, Labadi L, Mégraud F. Identification of Campylobacter species and related organisms by matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry. Clinical Microbiology and Infection. 2011;17:1735–1739. doi: 10.1111/j.1469-0691.2011.03468.x. [DOI] [PubMed] [Google Scholar]
- Bolten CJ, Kiefer P, Letisse F, Portais JC. Sampling for metabolome analysis of microorganisms. Analytical Chemistry. 2007;79:3843–3849. doi: 10.1021/ac0623888. [DOI] [PubMed] [Google Scholar]
- Castrillo JI, Hayes A, Mohammed S, Gaskell SJ, Oliver SG. An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry. 2003;62:929–937. doi: 10.1016/S0031-9422(02)00713-6. [DOI] [PubMed] [Google Scholar]
- Chu L, Burgum A, Kolodrubetz D, Holt SC. The 46-kilodalton-hemolysin gene from Treponema denticola encodes a novel hemolysin homologous to aminotransferases. Infection and Immunity. 1995;63:4448–4455. doi: 10.1128/iai.63.11.4448-4455.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey MP. Metabolite identification, pathways, and omic integration using online databases and tools. In: de Bruijn FJ, editor. Handbook of molecular microbial ecology: Metagenomics and complementary approaches. Hoboken: Wiley; 2011. [Google Scholar]
- Davey MP, Burrell MM, Woodward FI. Population-specific metabolic phenotypes of Arabidopsis lyrata ssp. petraea. New Phytologist. 2008;177:380–388. doi: 10.1111/j.1469-8137.2007.02282.x. [DOI] [PubMed] [Google Scholar]
- Dunn WB, Overy S, Quick WP. Evaluation of automated electrospray-TOF mass spectrometry for metabolic fingerprinting of the plant metabolome. Metabolomics. 2005;1:137–148. doi: 10.1007/s11306-005-4433-6. [DOI] [Google Scholar]
- Faijes M, Mars AE, Smid EJ. Comparison of quenching and extraction methodologies for metabolome analysis of Lactobacillus plantarum. Microbial Cell Factories. 2007;6:27. doi: 10.1186/1475-2859-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodacre R, Timmins EM, Burton R, Kaderbhai N, Woodward AM, et al. Rapid identification of urinary tract infection bacteria using hyperspectral whole-organism fingerprinting and artificial neural networks. Microbiology. 1998;144:1157–1170. doi: 10.1099/00221287-144-5-1157. [DOI] [PubMed] [Google Scholar]
- Guccione E, Leon-Kempis MR, Pearson BM, Hitchin E, Mullholland F, et al. Amino acid-dependent growth of Campylobacter jejuni: Key roles for aspartase (AspA) under microaerobic and oxygen-limited conditions and identification of AspB (Cj0762), essential for growth on glutamate. Molecular Microbiology. 2008;69:77–93. doi: 10.1111/j.1365-2958.2008.06263.x. [DOI] [PubMed] [Google Scholar]
- Hofreuter D, Mohr J, Wensel O, Rademacher S, Schreiber K, Schomburg D, Gao B, Galán JE. Contribution of amino acid catabolism to the tissue specific persistence of Campylobacter jejuni in a murine colonization model. PLoS One. 2012;7:e50699. doi: 10.1371/journal.pone.0050699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofreuter D, Novik V, Galán JE. Metabolic diversity in Campylobacter jejuni enhances specific tissue colonization. Cell Host & Microbe. 2008;4:425–433. doi: 10.1016/j.chom.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Kaderbhai NN, Broadhurst DI, Ellis DI, Goodacre R, Kell DB. Functional genomics via metabolic footprinting: Monitoring metabolite secretion by Escherichia coli tryptophan metabolism mutants using FT-IR and direct injection electrospray mass spectrometry. Comparative and Functional Genomics. 2003;4:376–391. doi: 10.1002/cfg.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Trostel A, Adhya S. Metabolite changes signal genetic regulatory mechanisms for robust cell behavior. MBio. 2014;5:e00972-13. doi: 10.1128/mBio.00972-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon-Kempis MR, Guccione E, Mulholland F, Williamson MP, Kelly DJ. The Campylobacter jejuni PEB1a adhesin is an aspartate/glutamate-binding protein of an ABC transporter essential for microaerobic growth on dicarboxylic amino acids. Molecular Microbiology. 2006;60:1262–1275. doi: 10.1111/j.1365-2958.2006.05168.x. [DOI] [PubMed] [Google Scholar]
- Lodha PH, Jaworski AF, Aitken SM. Characterization of site-directed mutants of residues R58, R59, D116, W340 and R372 in the active site of E. coli cystathionine β-lyase. Protein Science. 2010;3:383–391. doi: 10.1002/pro.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan RP, Ferenci T. Global metabolite analysis: the influence of extraction methodology on metabolome profiles of Escherichia coli. Analytical Biochemistry. 2003;313:145–154. doi: 10.1016/S0003-2697(02)00536-5. [DOI] [PubMed] [Google Scholar]
- Metris A, Reuter M, Gaskin DJH, Baranyi J, van Vliet AHM. In vivo and in silico determination of essential genes of Campylobacter jejuni. BMC Genomics. 2011;12:535. doi: 10.1186/1471-2164-12-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Weidman H, Laik M. Methodological approaches to help unravel the intracellular metabolome of Bacillus subtilis. Microbial Cell Factories. 2013;12:69. doi: 10.1186/1475-2859-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends in Biotechnology. 1998;16:373–378. doi: 10.1016/S0167-7799(98)01214-1. [DOI] [PubMed] [Google Scholar]
- Overy SA, Walker HJ, Malone S, Howard TP, Baxter CJ, et al. Application of metabolite profiling to the identification of traits in a population of tomato introgression lines. Journal of Experimental Botany. 2005;56:287–296. doi: 10.1093/jxb/eri070. [DOI] [PubMed] [Google Scholar]
- Park C, Yun S, Lee SY, Park K, Lee J. Metabolic profiling of Klebsiella oxytoca: Evaluation of methods for extraction of intracellular metabolites using UPLC/Q-TOF-MS. Applied Biochemistry and Biotechnology. 2012;167:425–438. doi: 10.1007/s12010-012-9685-9. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Mungall K, Ketley M, Churcher C, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- Raamsdonk LM, Teusink B, Broadhurst D, Zhang N, Hayes A, et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nature Biotechnology. 2001;19:45–50. doi: 10.1038/83496. [DOI] [PubMed] [Google Scholar]
- Ruiz-Palacios GM. The health burden of Campylobacter infection and the impact of antimicrobial resistance: Playing chicken. Clinical Infectious Diseases. 2007;44:701–703. doi: 10.1086/509936. [DOI] [PubMed] [Google Scholar]
- Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M, et al. Foodborne illness acquired in the United States—major pathogens. Emerging Infectious Diseases. 2011;17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan S, Rowland JJ, Kell DB. Discrimination of aerobic endospore-forming bacteria via electrospray-ionization mass spectrometry of whole cell suspensions. Analytical Chemistry. 2001;73:4134–4144. doi: 10.1021/ac0103524. [DOI] [PubMed] [Google Scholar]
- Velayudhan J, Jones MA, Barrow PA, Kelly DJ. l-serine catabolism via an oxygen-labile l-serine dehydratase is essential for colonization of the avian gut by Campylobacter jejuni. Infection and Immunity. 2004;72:260–268. doi: 10.1128/IAI.72.1.260-268.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayudhan J, Kelly DJ. Analysis of gluconeogenic and anaplerotic enzymes in Campylobacter jejuni: An essential role for phosphoenolpyruvate carboxykinase. Microbiology. 2002;148:685–694. doi: 10.1099/00221287-148-3-685. [DOI] [PubMed] [Google Scholar]
- Walker H. Metabolic profiling of plant tissues by electrospray mass spectrometry. In: de Bruijn FJ, editor. Handbook of molecular microbial ecology I—Metagenomics and complementary approaches. Hoboken: Wiley; 2011. [Google Scholar]
- Wang Q, Chen X, Yang Y, Zhao X. Genome-scale in silico aided metabolic analysis and flux comparisons of Escherichia coli to improve succinate production. Applied Microbiology and Biotechnology. 2006;73:887–894. doi: 10.1007/s00253-006-0535-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online resource 1. Excel spread sheet showing 0.2 Da mass unit bin centres of peaks normalized to total ion count for C. jejuni NCTC 11168, sdaA and aspB strains run in positive and negative ionisation modes. Putative metabolite allocations based on the full and Campylobacter jejuni jejuni NCTC 11168 specific biocyc (www.biocyc.com) compounds lists and preliminary data analysis are also shown (XLS 8510 kb)
Online resource 2. Excel spread sheet showing 0.2 Da mass unit bin centres of peaks normalized to total ion count for C. jejuni NCTC 11168 and aspA strains run in positive and negative ionisation modes. Putative metabolite allocations based on the full and Campylobacter jejuni jejuni NCTC 11168 specific biocyc (www.biocyc.com) compounds lists and preliminary data analysis are also shown (XLS 6040 kb)
Online resource 3. Excel spread sheet showing 0.2 Da mass unit bin centres of peaks normalized to total ion count for C. jejuni NCTC 11168 and cj0150c strains run in negative ionisation mode. Putative metabolite allocations based on the full and Campylobacter jejuni jejuni NCTC 11168 specific biocyc (www.biocyc.com) compounds lists and preliminary data analysis are also shown (XLS 2486 kb)