

Abstract
Zoonotic pathogens, including hantaviruses, are maintained in the environment by causing persistent infection in the absence of disease in their reservoir hosts. Spillover of hantaviruses to humans can cause severe disease that is mediated by excessive proinflammatory responses. The mechanisms mediating hantaviral persistence in rodent reservoirs remain largely unknown. Male Norway rats were inoculated with their species-specific hantavirus, Seoul virus (SEOV), and viral RNA, cytokine, and chemokine responses were evaluated in spleen and lung tissue. More viral RNA was detectable in the lungs than spleen, with copies of SEOV peaking 15-30 days post-inoculation (p.i.) and persisting for 60 days p.i. In the lungs, the expression and production of proinflammatory mediators (i.e. IL-1β, IL-6, TNF-α, IFN-γ, CCL5, CCL2, CX3CL1, CXCL10, VCAM, VEGF, and NOS2) remained at or below baseline throughout SEOV infection; whereas, regulatory factors, including TGF-β and FoxP3 were elevated. Conversely, in the spleen, proinflammatory responses were induced while regulatory responses remained unchanged during infection. To determine whether reduced proinflammatory responses mediate hantavirus persistence in the lungs, male rats were administered rIL-1β or vehicle for 30 days during SEOV infection. SEOV persistence and shedding were not affected by IL-1β treatment. Proinflammatory responses were elevated in rIL-1β-treated rats, but remained within physiological levels, suggesting that supra-physiological concentrations may be necessary for viral clearance at the cost of causing disease. Elevated regulatory responses may suppress excessively high proinflammatory responses at a site of elevated SEOV replication to contribute to viral persistence and prevent proinflammatory-mediated disease in reservoir hosts.
Keywords: HFRS, zoonotic, IL-1β, TGF-β, TNF, regulatory T cell
Introduction
Zoonotic pathogens are maintained in the environment in their natural reservoir hosts, often causing persistent infection in the absence of disease. In such highly coevolved systems, virulence and host antiviral responses have evolved to ensure survival of both the host and the pathogen [Hudson et al., 2002]. Several zoonotic viruses are persistently maintained in their natural reservoirs without causing disease, including Lassa virus in multimammate rats (Mastomys natalensis), SARS-CoV in horseshoe bat species (Rhinolophus spp.), Nipah virus in flying foxes (Pteropus spp.), Macupho virus in large vesper mice (Calomys callosus), Sin Nombre virus in deer mice (Peromyscus maniculatus), and Seoul virus (SEOV) in Norway rats (Rattus norvegicus) [Botten et al., 2003; Calisher et al., 2006; Childs et al., 1989; Johnson et al., 1965; Walker et al., 1975; Wang et al., 2006]. Spillover of these viruses to incidental human hosts often results in host-mediated disease, including Lassa fever, SARS, Nipah virus-associated disease, Bolivian hemorrhagic fever, hantavirus pulmonary syndrome (HPS), and hemorrhagic fever with renal syndrome (HFRS), respectively. Clinical symptoms in humans generally are caused by excessively elevated proinflammatory responses, often termed a “cytokine storm” [Huang et al., 2005; Khaiboullina and St Jeor, 2002; Mori et al., 1999; Villinger et al., 1999].
Whether virus persistence in the absence of clinical disease in natural reservoir hosts is caused by suppressed proinflammatory responses during viral infection has not been characterized because of a lack of available reagents for exotic host species. A notable exception is the persistence of an arenavirus, lymphocytic choriomeningitis virus (LCMV), in mice (Mus musculus), which is mediated by suppression of CD8+ T cell and proinflammatory responses [Hahm et al., 2005; Moskophidis et al., 1993]. Suppression of proinflammatory and cytotoxic T cell responses can be caused by the activity of regulatory T cells, including production of TGF-β, and recent studies demonstrate that these cells contribute to persistence of hantaviruses in their rodent hosts [Easterbrook et al., 2007; Schountz et al., 2007]. During SEOV infection, expression of Foxp3 and production of TGF-β is elevated during the persistent phase of infection at a site of elevated virus replication (i.e. lungs). Inactivation of regulatory T cells reduces persistence of SEOV in the lungs and the duration of shedding in rats [Easterbrook et al., 2007]. Whether host regulatory responses indirectly mediate hantavirus persistence through suppression of proinflammatory responses has not been examined and was one goal of the present study.
The kinetics of proinflammatory, anti-inflammatory, and regulatory responses during the acute and persistent phases of SEOV infection in Norway rats were evaluated. In the lungs (i.e. a site of elevated SEOV replication), proinflammatory responses generally were reduced, whereas regulatory responses were elevated throughout infection. In the spleen (i.e. a site of low SEOV replication and high immunological activity), proinflammatory responses were elevated and regulatory responses were not induced during infection. To establish whether reduced proinflammatory responses mediate SEOV persistence in rats, exogenous IL-1β was administered at physiological levels throughout infection. Moderate elevation of proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, did not reduce SEOV persistence, as initially hypothesized. Thus, these data illustrate that the balance between proinflammatory responses clearing virus and causing immunopathology may be tightly regulated in highly co-evolved host-pathogen systems.
Methods
Animals
Adult male (60-70 days of age) Long Evans rats (Rattus norvegicus) were purchased from Charles River Laboratories (Raleigh, NC) and housed individually in polypropylene cages covered with polyester filter bonnets. All infected males were housed in a pathogen-free Biosafety Level (BSL) 3 animal facility with food and water available ad libitum. Animals were maintained on a constant 14:10 light:dark cycle with lights on at 0600 hours Eastern Standard Time. The Johns Hopkins Animal Care and Use Committee (Protocol No. RA04H338) and the Johns Hopkins Office of Health, Safety and Environment (Registration No. P9902030107) approved all procedures described in this manuscript.
IL-1β treatment
One day prior to inoculation with SEOV, male rats were anaesthetized with isofluorane (Abbot Animal Health, North Chicago, IL) and CO2 and implanted s.c. with an osmotic minipump (Alzet, Cupertino, CA) that administered 1 μg of recombinant rat (r)IL-1β (Sigma, St. Louis, MO) in 0.9% saline (n = 10) or vehicle alone (n = 10) over a 24 h period for 30 days. Pumps were removed and new pumps containing either rIL-1β or saline were replaced s.c. after 15 days to ensure stability of the rIL-1β.
Infection
Male rats (n = 8-12/time-point/study) were inoculated i.p. with 104 plaque forming units (pfu) of purified SEOV (strain SR-11) suspended in 0.2 ml Minimum Essential Medium Eagle (MEM) (with Earle’s salts; Mediatech Cellgro, Herndon, VA) or vehicle alone, as described previously [Klein et al., 2001]. At Days 0, 3, 15, 30, and 60 post inoculation (p.i.), animals were anesthetized with isoflourane, blood was obtained from the retro-orbital sinus, and saliva was collected following an i.p. injection of 2.5 mg/kg of pilocarpine HCl (Sigma, St. Louis, MO). Rats were euthanized with CO2 and lung and spleen samples were collected and stored at -80°C until processed. Day 3 post-inoculation (p.i.) was selected to examine responses during the early acute phase of SEOV infection and samples collected 15 days p.i. were used to evaluate immune responses during the late acute phase of SEOV infection. Days 30 and 60 p.i. were selected to measure responses at different times during the persistent phase of infection. Rats assigned to ‘Day 0 p.i.’ were not infected, but were processed at different time-points during the study to control for potential effects of age.
Enzyme-linked immunosorbent assay (ELISA) for SEOV antibody
Microtiter plates were coated overnight at 4°C with SEOV infected or uninfected gamma-irradiated Vero E6 cells diluted 1:500 in carbonate buffer. Plasma samples and positive and negative control samples were diluted 1:100 in PBS-Tween 20 (PBS-T) with 2% fetal bovine serum (FBS), and added in duplicate to antigen-coated wells. The plates were washed and secondary antibody (alkaline-phosphatase conjugated goat anti-rat IgG (H + L), Kirkegaard and Perry Laboratories, Gaithersburg, MD), diluted 1:400 in PBS with 2% FBS, was added. Substrate buffer (0.5 mg/ml p-nitrophenyl phosphate (Sigma) diluted in diethanolamine buffer) was used to develop the reaction and the optical density (OD) was measured at 405 nm. To determine the average adjusted OD, the average OD for each set of uninfected Vero E6 duplicates was subtracted from the average OD for each set of infected Vero E6 duplicates. Samples were considered positive if the average adjusted OD was greater than 0.100 [Klein et al., 2001; Klein et al., 2004a]. To minimize intra- and inter-plate variability, the average adjusted OD was expressed as a percentage of the positive control OD run on each microtiter plate (anti-SEOV IgG, % of PC) for statistical analysis.
RNA Isolation
RNA was isolated from lung, spleen, and saliva using Trizol LS (Invitrogen, Carlsbad, CA) and the manufacturer’s protocol as described previously [Klein et al., 2001; Klein et al., 2004a]. Following isopropanol precipitation and washing in 75% ethanol, RNA pellets were briefly air dried and resuspended in DEPC-treated water.
Real-time quantitative RT – PCR for SEOV detection
First-strand synthesis of the S segment of SEOV cDNA was prepared as detailed elsewhere [Hannah et al., 2008] using a 0.1 μM gene-specific primer and the Invitrogen Superscript III First Strand Synthesis reagents. The positive control was SEOV RNA isolated from our virus stock (strain SR-11) and the negative control was DEPC-treated water included in the cDNA syntheses and amplification. Negative-strand genomic SEOV RNA was amplified in a reaction mixture containing Platinum Quantitative PCR SuperMix-UDG (Invitrogen), Rox reference dye, 4.5 mM MgCl2, 0.1 μM primers, and 0.2 μM probe (Applied Biosystems, Foster City, CA). All reactions were run in optical 96-well plates using the ABI 7300 Sequence Detection System (Applied Biosystems). A standard curve ranging from 106 to 10 copies of negative sense SR-11 S segment in plasmid pWRG7077 was run on each plate.
Real-time quantitative RT – PCR for host gene expression
First strand cDNA was prepared using 1 μg of RNA isolated from lung or spleen tissue, 2.5 μM oligo(dT)20 primer, and the manufacturer’s protocol for Superscript III First Strand Synthesis (Invitrogen). Custom primer and probe sets were generated for Gapdh, Il1β, Il6, Tnfα, Ifnγ, Nos2, Ccl2, Ccl5, Cx3cl1, Cxcl10, Vegf, Vcam, Tgfβ, Il10, Il1ra, and Foxp3 using Primer Express 2.0 software (Applied Biosystems). Complementary DNA (1 μl) was amplified in duplicate in a reaction mixture containing TaqMan Universal Master Mix, 900 nM of each primer, and 250 nM probe, using the manufacturer’s protocol (Applied Biosystems). All reactions were run in 96 well optical reaction plates using the ABI 7300 Sequence Detection System (Applied Biosystems). Serial dilutions of control cDNA from selected rats were used to generate a standard curve, ranging from 2.5 ng/μl to 25 pg/μl cDNA, which was run on each plate. Gene expression patterns from SEOV infected animals were normalized to Gapdh expression and are presented as relative to the expression levels from uninfected rats (i.e. Day 0 p.i.) and expressed as fold change.
Cytokine protein quantitation by ELISA
Lung and spleen tissues were homogenized in 1 ml lysis buffer, containing 50 mM Tris (pH 7.4), 0.5 mM EDTA, 1% Nonidet P40, 0.01 mM PMSF, and 0.1% protease inhibitor cocktail (Sigma), and centrifuged at 15000 g for 10 min. Cytokine protein in the supernatant was measured using ELISA kits for rat TNF-α, IL-1β, IL-10, and active mouse TGF-β1 (R&D Systems, Minneapolis, MN). Protein concentrations were normalized to mg total protein as determined using a BCA assay kit (Pierce, Rockford, IL) and are expressed as fold change relative to protein concentrations in uninfected rats (i.e. Day 0 p.i.).
Western blot for quantitation of CCL5 protein
Protein from lung and spleen tissue was extracted as described above. Because ELISA reagents are not available for rat CCL5, protein was measured by western blot. Protein (50 μg) bands were separated onto 4%-12% polyacrylamide-sodium dodecyl sulfate gels and transferred to nitrocellulose by standard procedures. Membranes were blocked for 2 h in TBS with 0.5% Tween 20 (TBS-T) and 5% nonfat dry milk. Monoclonal antibody against CCL5 (1:5000; AbCam, Cambridge, MA) was added and membranes were incubated with shaking at 4°C for 16 h. Membranes were washed in TBS-T and incubated for 1 h with HRP-conjugated goat anti-rabbit IgG (1:3000; AbCam). Membranes were washed in TBS-T and incubated for 2 h with mouse monoclonal β-actin conjugated to HRP (1:5000; Abcam) and detected with 3-aminophthathic hydrazide and 4-iodophenylboronic acid [Haan and Behrmann, 2007].
Immunohistochemistry
Uninflated lungs were fixed in 10% neutral, buffered formaldehyde, embedded in paraffin, cut into 5 μm sections, and mounted on glass slides. Formaldehyde-induced autofluorescence was photobleached with a 40 W cool white 800 fluorescent lamp at a distance of 6 in for 24 h [Neumann and Gabel, 2002]. Sections were deparaffinized with xylene, rehydrated in graded ethanol, and post-fixed with Streck’s Tissue Fixative (STF Streck, Omaha, NE). Antigen retrieval was maximized using Antigen Unmasking Solution (1:100; Vector Labs, Burlingame, CA) or proteinase K incubation (20 μg/ml; Sigma). Tissue sections were blocked with 10% normal horse serum, followed by blocking with avidin D and biotin to block all endogenous biotin, biotin receptors, and avidin binding sites. Slides were incubated in a humidified chamber with a mouse monoclonal antibody against the SEOV nucleocapsid protein (NP; 1:250; EC01-AB08, gift of Connie Schmaljohn, Fort Detrick, MD), mouse anti-rat CD68 mAb for macrophages (1:250, Serotec, Raleigh, NC), or rabbit anti-human von Willebrand factor mAb for endothelial cells (1:500; DakoCytomation, Carpinteria, CA). Sections were incubated with biotinylated, rat-adsorbed, anti-mouse IgG (5 μg/ml; Vector Labs) or rabbit multilinker (Biogenex, San Ramon, CA). The signal was revealed using streptavidin conjugated Fluorescein or Texas Red (15 μg/ml or 10 μg/ml, respectively; Vector Labs). Double staining was performed sequentially. Apoptosis was identified using a TUNEL staining kit (Roche, Nutley, NJ). Tissues were counterstained with 4’,6 diamidino-2-phenylindole (DAPI; Vector Labs) and stored in the dark at 4°C until viewed. Fluorescence emission images were acquired and merged using a SPOT charge-coupled device camera and software (Diagnostic Instruments, Sterling Heights, MI) at a magnification of 100x.
Pathological Analyses
Following 24 h fixation in 10% formaldehyde, inflated lung and spleen samples were embedded in paraffin, cut into 5 μm sections, and mounted on glass slides. The slides were stained with hematoxylin and eosin (H&E) to examine pathologic observations, including hemorrhage and edema, using light microscopy.
Statistical Analyses
Copies of SEOV RNA, antibody responses, gene expression, protein concentrations, pathological quantitation, and percentages of cells undergoing apoptosis were assessed by ANOVA with one between group variable (day post-inoculation). In cases where the data violated the assumptions of normality, the nonparametric Kruskal-Wallis test was used. Significant interactions were further analyzed using the Tukey or Dunn method for pairwise multiple comparisons. Mean differences were considered statistically significant if P < 0.05.
Results
SEOV RNA load is higher in the lungs than the spleen
Male rats have more copies of SEOV RNA in their lungs and shed more virus in saliva than females during infection [Hannah et al., 2008; Klein et al., 2001], suggesting that hantavirus persistence is elevated in males. The goal of the present study was to evaluate the mechanisms mediating viral persistence; therefore, only male rats were used. Anti-SEOV IgG responses increased significantly over the course of infection and were highest 60 days p.i. (Kruskal Wallis: H4 = 43.9, P < 0.001; Figure 1A). Copies of SEOV RNA were significantly higher in the lungs than spleen 15-60 days p.i. (Figure 1B; Kruskal-Wallis: H7 = 42.9, P < 0.001). In lung tissue, viral RNA was detected at all time-points post-inoculation, with viral copies being signficantly elevated 15 and 30 days p.i. as compared with values obtained 3 and 60 days p.i. (H3 = 12.7, P = 0.005; Figure 1B). SEOV RNA remained highly detectable through Day 60 p.i. (15,872 ± 9810 copies/g lung tissue) in the lungs of male rats, which is consistent with reports of lifelong viral persistence for other hantavirus infections in rodent reservoirs [Botten et al., 2003; Lee et al., 1981; Yanagihara et al., 1985].
Figure 1. SEOV RNA is higher in the lungs than spleen.
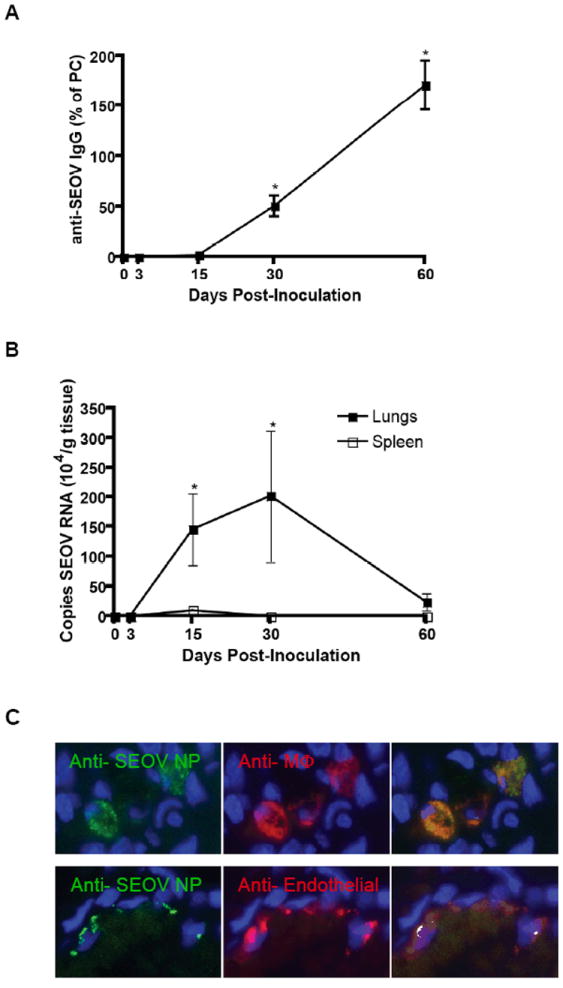
Male rats were inoculated with SEOV and virus-specific IgG was measured by ELISA and viral RNA was assessed by real-time RT-PCR. Anti-SEOV IgG was detectable by Day 15 p.i. and increased over time, as indicated (*), P<0.05 (A). SEOV RNA peaked 15 and 30 days p.i. in the lungs (*) and from Days 15 – 60 p.i., the amount of virus was higher in the lungs than spleen, P<0.05 (B). Using immunohistochemistry, SEOV N protein (green) was identified in macrophages with anti-ED1 mAb (C top; red) and endothelial cells with anti-von Willebrand’s factor mAb (C bottom; red).
SEOV antigen is localized in macrophages and endothelial cells
IHC was used to identify which cells in the lungs were infected with SEOV. Previous studies demonstrating hantavirus antigen in endothelial cells and macrophages have been solely based on morphological analysis or non-specific cell separation [Botten et al., 2003; Lee et al., 1986; Nagai et al., 1985]. In the present study, SEOV nucleocapsid (N) protein was definitively identified in alveolar macrophages, using CD68 (ED1) as a macrophage specific marker, and in endothelial cells, using von Willebrand factor as an endothelial cell marker, in the lungs of male rats during persistent infection (i.e. Day 30 p.i.; Figure 1C).
Proinflammatory responses are reduced in the lungs, but are elevated in the spleen, during SEOV infection
Proinflammatory cytokine gene expression was measured in the lungs, as a site of elevated SEOV replication, and in the spleen, as a representative secondary lymphoid organ that does not support substantial viral RNA load. In the lungs, the expression of proinflammatory cytokines, including Il1β, Tnfα, and Il6, generally was reduced during SEOV infection as compared with expression in lungs of uninfected males (Figure 2A; Il1β: Kruskal-Wallis, H4 = 8.87, P = 0.06; Tnfα: Kruskal Wallis: H4 = 22.1, P < 0.001; Il6: Kruskal Wallis: H4 = 22.1, P<0.001). In the spleen, however, expression of Il1β and Tnfα remained unchanged during infection and Il6 transcription was elevated Days 15 and 30 p.i. (Figure 2B; Il6: ANOVA: F4,58 = 6.15, P<0.001). SEOV infection had similar effects on proinflammatory cytokine protein production; the amount of IL-1β and TNF-α protein was reduced in the lungs (Figure 2C; IL-1β: ANOVA: F4,54 = 7.25, P<0.001; TNF-α: ANOVA: F4,54 = 4.20, P = 0.005) and remained unchanged in the spleen during the course of infection (Figure 2D).
Figure 2. Expression and production of proinflammatory mediators are reduced in the lungs of male rats throughout SEOV infection.
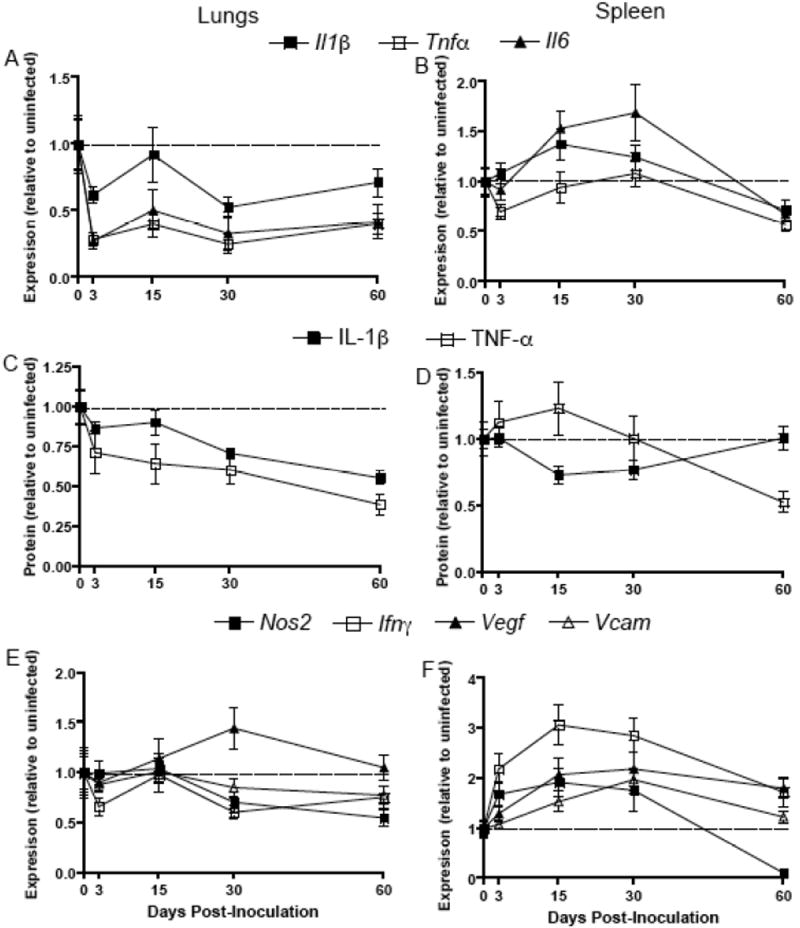
Expression of Il1β, Il6, and Tnfα in the lungs (A) and spleen (B) was measured at Days 0, 3, 15, 30, and 60 p.i. by real-time RT-PCR and is expressed as relative to tissue-specific concentrations in uninfected male rats (dotted line). For all dependent measures, samples were collected from uninfected rats at the same time as infected rats and are designated ‘Day 0 p.i.’. Expression of each cytokine was normalized to the expression of the endogenous control, Gapdh, for each sample. Protein concentrations of IL-1β and TNF-α were measured in the lungs (C) and spleen (D) by ELISA and are expressed as relative to protein concentrations in uninfected males. Transcription of proinflammatory mediators, including Nos2, Ifnγ, Vegf, and Vcam, also was measured in the lungs (E) and spleen (F).
The expression of proinflammatory mediators, including Nos2 (i.e. inducible nitric oxide synthase, iNOS), also was reduced at Days 30 and 60 p.i. (ANOVA: F4,58 = 2.67, P = 0.04) or unchanged (i.e. Ifnγ, Vegf, and Vcam) in the lungs of male rats during SEOV infection (Figure 2E). Conversely, in the spleen, the expression of Nos2, Ifnγ, Vegf, and Vcam was significantly elevated 15 and 30 days p.i. (Figure 2F; Nos2: Kruskal Wallis: H4 = 28.6, P<0.001; Ifnγ: ANOVA: F4,58 = 6.89, P<0.001; Vegf: Kruskal Wallis: H4 = 17.9, P = 0.001; Vcam: ANOVA: F4,58 = 6.28, P<0.001).
Chemokine gene expression is reduced in the lungs and elevated in the spleen during SEOV infection
Chemokines are critical mediators of inflammation as they recruit phagocytes and lymphocytes to sites of viral infection. In the lungs, the expression of Cxcl10 and Cx3cl1 was reduced (Cxcl10: Kruskal Wallis: H4 = 13.3, P = 0.01; Cx3cl1: ANOVA: F4,58 = 5.09, P = 0.001) and the expression of Ccl2 was not affected by SEOV infection (Figure 3A). Conversely, in the spleen, the expression of chemokines, including Ccl2 and Cxcl10, was elevated Days 15 and 30 p.i. (Ccl2: ANOVA: F4,58 = 6.20, P<0.001; Cxcl10: ANOVA: F4,58 = 3.09, P = 0.023) and the expression of Cx3cl1 remained unchanged during infection (Figure 3B). The expression of Ccl5 was elevated in both the lungs (Figure 3A; ANOVA: F4,58 = 5.09, P = 0.002) and spleen (Figure 3B; ANOVA: F4,58 = 6.78, P<0.001), but protein concentrations in the lungs remained unchanged during infection and were below limits of detection in the spleen (Figure 3C and data not shown). Whether discordance of CCL5 gene expression and protein production is indicative of specific post-transcriptional regulation remains to be determined.
Figure 3. Expression of chemokines is reduced in the lungs and elevated in the spleen.
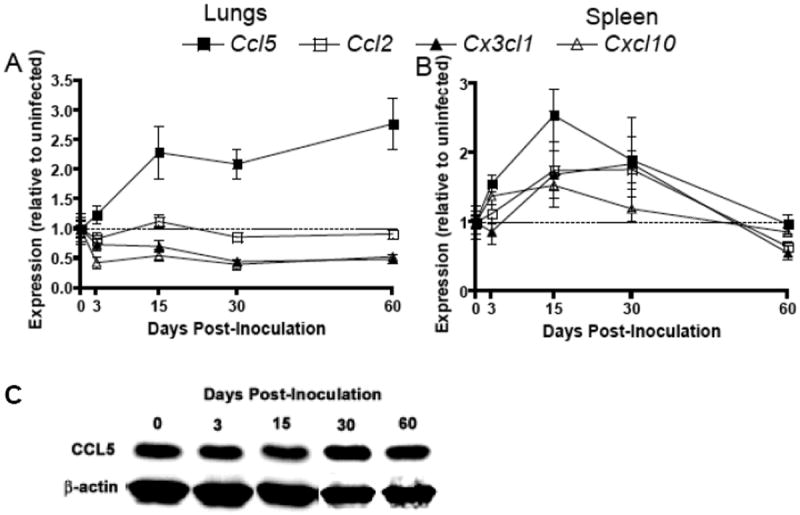
The expression of chemokines, including Ccl5, Ccl2, Cx3cl1, and Cxcl10, was measured in the lungs (A) and spleen (B) at Days 0, 3, 15, 30, and 60 p.i. by real-time RT-PCR and expressed as relative to expression in uninfected males. Gene expression was normalized to expression of Gapdh for each sample. Lung and spleen samples were collected from uninfected and infected rats at the same time-points and are labeled ‘Day 0 p.i.’. Protein concentrations of CCL5 in the lungs during acute and persistent infection were measured by western blot and representative samples at each time-point post-inoculation are pictured (C).
Regulatory responses are elevated in the lungs but not in the spleen during persistent SEOV infection
Regulatory T cells can suppress proinflammatory responses and contribute to SEOV pesistence [Belkaid and Rouse, 2005; Easterbrook et al., 2007]. Both TGF-β gene expression and protein concentrations were elevated in the lungs during the persistent phase of SEOV infection (i.e. Days 30 and 60 p.i.) when compared to baseline concentrations in uninfected males (Figure 4A and C; mRNA: Kruskal Wallis: H4 = 17.0, P = 0.002; protein: ANOVA: F4,49 = 3.95, P = 0.009). The expression of the regulatory T cell transcription factor, Foxp3, also was elevated in the lungs of male rats during persistent infection (Figure 4A; ANOVA: F4,49 =5.03, P = 0.002). In the spleen, regulatory responses, including expression and production of TGF-β and Foxp3, were not altered by SEOV infection (Figure 4B and D).
Figure 4. Regulatory responses are elevated in the lungs, but not in the spleen during persistent SEOV infection.
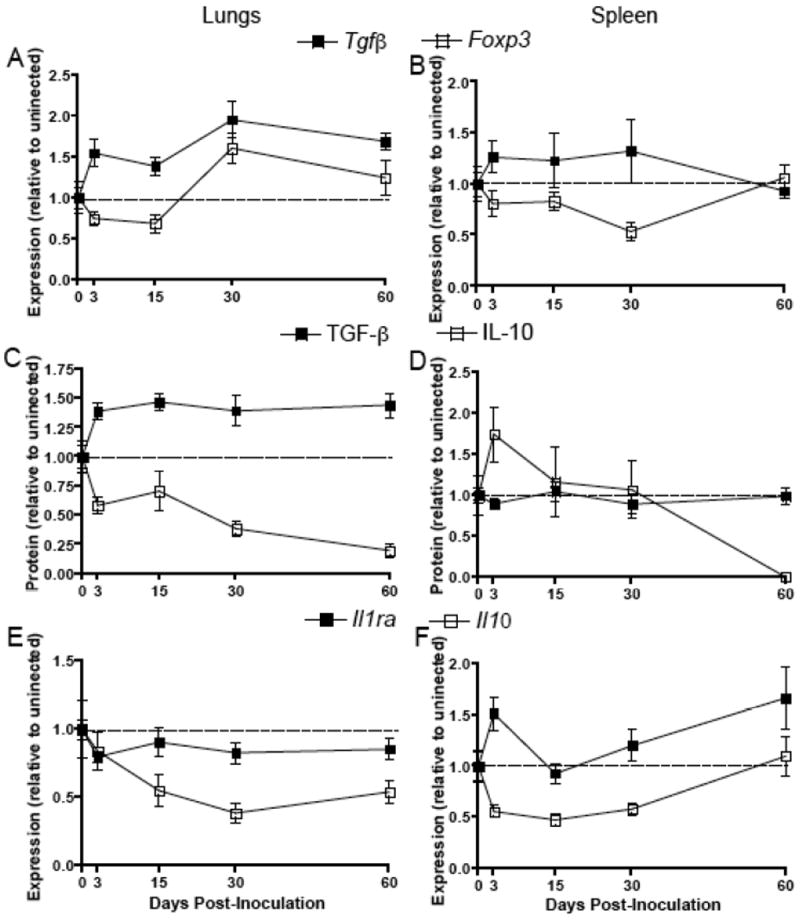
Expression of Foxp3 and Tgfβ in the lungs (A) and spleen (B) at Days 0, 3, 15, 30, and 60 p.i. was measured by real-time RT-PCR, expressed as relative to expression in uninfected males (dotted line), and normalized to the expression Gapdh for each sample. Tissue samples were collected from uninfected rats at the same time as infected rats and are designated ‘Day 0 p.i.’. Protein concentrations of active TGF-β1 and IL-10 were measured in the lungs (C) and spleen (D) by ELISA and are expressed as relative to protein production in uninfected males. The expression of Il10 and Il1ra was measured in the lungs (E) and spleen (F) of male rats during the acute and persistent phases of SEOV infection.
Expression of anti-inflammatory cytokines is not induced by SEOV infection in either the lungs or the spleen
In addition to TGF-β, transcription of proinflammatory factors can be regulated by other anti-inflammatory cytokines, including IL-10 and IL-1ra. Interleukin-10 is produced by a range of cell types, including lymphocytes and macrophages, and serves as a general anti-inflammatory cytokine; whereas, IL-1ra, also produced by several cell types, specifically inhibits IL-1β activity by binding to the IL-1 receptor [Janeway C.A., 2001]. Interleukin-10 gene expression and protein production were reduced in the lungs throughout SEOV infection (Figures 4C and E; mRNA: Kruskal Wallis: H4 = 12.6, P = 0.014; protein: ANOVA: F4,58 = 8.09, P<0.001). Similarly, in the spleen, the expression of Il10 was reduced from Days 3-30 p.i. and protein concentrations were reduced at Day 60 p.i. (Figures 4D and F; mRNA: ANOVA: F4,50 = 7.01, P<0.001; protein: F4,49 = 2.82, P = 0.039). Expression of Il-1ra mRNA, however, did not increase above expression levels in uninfected males in either the lungs or spleen of male rats throughout SEOV infection (Figure 4E and F).
Exogenous administration of IL-1β does not affect SEOV persistence
During the classical acute phase response, IL-1β production is elevated and causes downstream induction of other proinflammatory responses, including TNF-α and IL-6 [Granowitz et al., 1992; Ikejima et al., 1990]. In humans, concentrations of proinflammatory cytokines are elevated during acute hantavirus infection and if survival is the final outcome, then virus is cleared [Krakauer et al., 1995; Mori et al., 1999]. Proinflammatory responses are consistently reduced or remain unchanged in male rats at sites of elevated virus replication, throughout SEOV infection. Whether the lack of induction of proinflammatory responses directly influences the ability of SEOV to persist in its host in the absence of clinical pathology was evaluated by administering rIL-1β or vehicle during the course of infection at a dose (1 μg/day, s.c.) reported to elevate circulating IL-1β without causing manifestations of sickness [Busbridge et al., 1993]. Because we were interested in the role of proinflammatory cytokines in mediating SEOV persistence, the effects of exogenous IL-1β were measured at a time-point during persistent infection when viral replication was high (i.e. Day 30 p.i.). Administration of rIL-1β significantly elevated circulating concentrations of IL-1β, as well as expression of Il6 and Tnfα in the lungs, as compared with uninfected rats that were administered vehicle alone (Figure 5; IL-1β: T = 17.5, P = 0.032; Il6: t = -2.13, df = 7, P = 0.071; Tnfα: t = -2.52, df = 7, P = 0.04). Administration of exogenous IL-1β did not, however, affect the amount of SEOV RNA in the lungs and saliva nor did it affect the proportion of rats shedding virus in saliva (Figure 6A and B).
Figure 5. Elevated concentrations of circulating IL-1β increases expression of Il6 and Tnfα in the lungs.
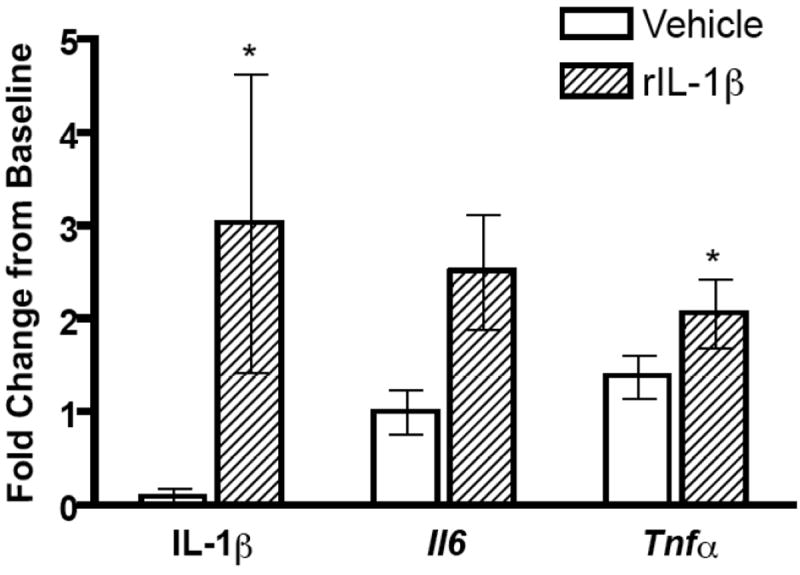
Following administration of exogenous rat IL-1β (1 μg/day) or vehicle for 30 days in SEOV-infected and uninfected male rats, IL-1β protein was measured in serum by ELISA and Il6 and Tnfα expression in the lungs was measured by real-time RT-PCR. Protein concentrations and gene expression (normalized to Gapdh expression) in infected rats are represented as fold change from baseline (i.e. concentrations in uninfected vehicle-treated male rats).
Figure 6. Administration of exogenous IL-1β does not affect SEOV persistence.
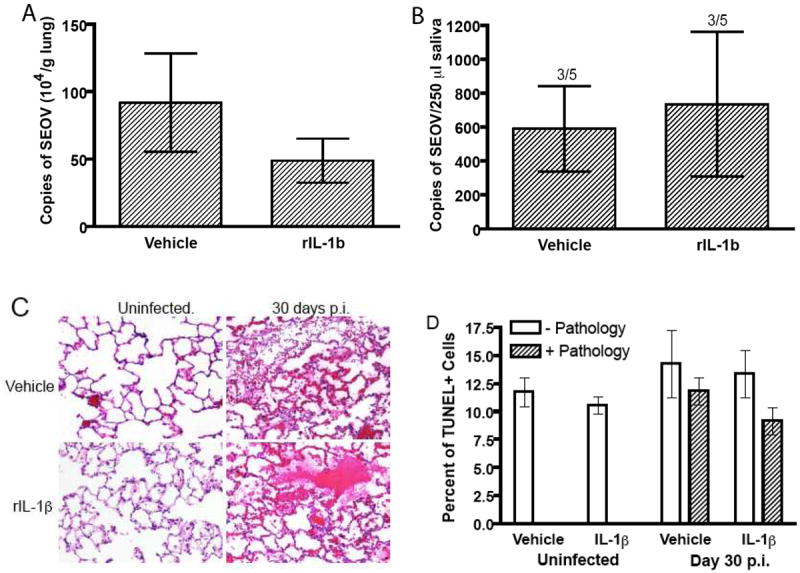
Exogenous rat IL-1β (1 μg/day) or vehicle was administered to male rats and SEOV RNA was measured in the lungs (A) and saliva (B) 30 days after inoculation. The proportion of rats shedding SEOV in saliva is represented above the appropriate bar. Lung tissues from male rats that received rIL-1β or vehicle for 30 days also were prepared for pathologic examination using H&E staining (C). Lungs were collected from uninfected rats at the same time as infected rats. The top panels are representative lung sections from vehicle-treated rats and the bottom panels are lungs sections from rats treated with exogenous rIL-1β for 30 days, but that either remained uninfected or were infected with SEOV (10X magnification). Representative foci of hemorrhage and edema in the lungs of male rats that were infected with SEOV are illustrated in both of the right panels. Apoptosis was identified in lung tissue from uninfected male rats and from males infected with SEOV for 30 days using an TUNEL staining kit for IHC (D). Percentages of cells in the lungs of infected and uninfected rats that received rIL-1β or vehicle for 30 days and that were positive for apoptosis (TUNEL+) were measured in regions with (Pathology+) and without (Pathology-) subclinical hemorrhage and edema.
SEOV infection in rats causes subclinical, acute, and focal hemorrhage and edema in the lungs [Easterbrook et al., 2007]. Similar foci of hemorrhage and edema were observed in the lungs of male rats 30 days after inoculation with SEOV and treatment with rIL-1β had no effect on the amount of pathological observations (Figure 6C). Although proinflammatory cytokines, including IL-1β, TNF-α, and IL-6, may contribute to clinical disease during human hantavirus infection, proinflammatory infiltration was not observed in the lungs of infected rats, whether treated with exogenous rIL-1β or vehicle. Pathology was not observed in the lungs of uninfected male rats, regardless of rIL-1β treatment (Figure 6C). Additionally, pathological lesions were not observed in the spleen (data not shown).
The mechansims mediating hemorrhage and edema are not known. Hantaviruses generally do not cause apoptosis in human cell lines (i.e. HUVEC) or cells in tissues from patients who died from hantavirus infection [Khaiboullina et al., 2004; Mori et al., 1999]. Hantaviruses do not appear to cause cytopathic effects in their rodent hosts, but whether apoptosis occurs has not been determined [Botten et al., 2000; Yanagihara et al., 1985]. Similar percentages of apoptotic cells were observed in the lungs of uninfected male rats and males infected with SEOV for 30 days; exogenous administration of rIL-1β had no effect on the number of cells undergoing apoptosis (Figure 6D). Taken together, these data suggest that hemorrhage and edema in the lungs that is caused by SEOV infection is not affected by the modest elevation of concentrations of proinflammatory cytokines achieved by administration of rIL-1β or apoptosis.
Discussion
SEOV disseminates to several tissues in rats, but the lungs are a primary site of elevated virus replication [Hinson et al., 2004; Klein et al., 2004b]. Consistent with previously reported tissue tropism, copies of SEOV RNA were higher in the lungs than spleen and virus persisted for at least 60 days p.i. Hantaviruses maintain lifelong infections in their rodent reservoirs; thus, SEOV RNA likely remains detectable well after 60 days p.i. [Botten et al., 2003; Lee et al., 1981; Yanagihara et al., 1985]. Elevated viral load in the lungs was correlated with reduced proinflammatory and elevated regulatory responses during persistent infection. Conversely, in the spleen, where SEOV RNA was low, proinflammatory responses were elevated and regulatory responses were not induced during either acute or persistent phases of SEOV infection.
Humans are incidental hosts that develop acute disease during infection and do not maintain persistent infection. Excessive proinflammatory responses in humans are correlated with the development of clinical symptoms and death during hantavirus infection. Proinflammatory cytokines, including IL-1β, TNF-α, and IL-6, are elevated in circulation and in tissues during symptomatic or fatal hantavirus infection in humans [Mori et al., 1999; Temonen et al., 1996]. Production of chemokines, including CCL5 and CXCL10, is induced following exposure of human endothelial cells to hantaviruses and likely contributes to the infiltration of lymphocytes to the site of infection [Sundstrom et al., 2001]. When symptoms of hantavirus infection are most severe in humans, nitric oxide responses are elevated, which is indicative of an activated macrophage response [Davis et al., 2002; Groeneveld et al., 1995]. Following the innate response to hantavirus infection, hantavirus-specific CD4+ and CD8+ T cells are recruited to infected tissues and overall activity of CD8+ T cells (e.g. production of IFN-γ and TNF-α) is elevated during symptomatic hantavirus infection in humans and contributes to the manifestations of disease and mortality [Ennis et al., 1997; Kilpatrick et al., 2004; Klingstrom et al., 2006; Terajima et al., 2007].
In contrast to humans, the expression and production of proinflammatory cytokines, including IL-1β, TNF-α, and IL-6, was reduced in the lungs of male rats infected with SEOV. The absence or suppression of chemokine stimulation in the lungs of male rats may contribute to reduced lymphocyte infiltration to sites of viral replication, which may prevent proinflammatory-mediated pathology and contribute to viral persistence. Reduced expression of Nos2 in the lungs of infected male rats suggests that macrophage activity may be reduced during SEOV infection [Lyons et al., 1992]. By contrast, proinflammatory responses, including production of proinflammatory cytokines and chemokines, were elevated in the spleen. Male rats that are infected with SEOV do not appear to be globally immunosuppressed, but rather have a site-specific reduction of proinflammatory responses. There is no evidence that male rats that are naturally infected with SEOV are more likely to acquire additional pathogens, further demonstrating that infected rats are not immunocompromised during hantavirus infection [Easterbrook et al., 2007]. Responses mounted in the spleen may represent an attempt by the rodent host to control viral replication, but do not appear to eliminate viral RNA in other organs.
Mechanisms mediating localized suppression of proinflammatory responses in the lungs of male rats during SEOV infection are not known, but may be mediated by host regulatory responses. Regulatory T cells (CD4+CD25+FoxP3+) contribute to the persistence of several pathogens, including hantaviruses, by suppressing host proinflammatory and effector T cell responses locally at the site of infection [Belkaid and Rouse, 2005; Easterbrook et al., 2007; Schountz et al., 2007]. Consistent with previous data, the expression of Foxp3 and the production of TGF-β were elevated in the lungs, but not in the spleen, during persistent SEOV infection in male rats [Easterbrook et al., 2007]. The effects of regulatory T cells on virus persistence likely are mediated by TGF-β, and not IL-10, as IL-10 transcription and synthesis consistently was suppressed in both the lungs and spleen throughout infection. Functional inactivation of regulatory T cells eliminates the suppression of proinflammatory cytokines in the lungs during the persistent phase of SEOV infection in male rats [Easterbrook et al., 2007], but whether regulatory T cells suppress proinflammatory or CD8+ T cell responses to mediate hantavirus persistence in rodents has not been determined.
To test the hypothesis that suppression of proinflammatory responses during infection may be a critical component of hantavirus persistence in rodents, IL-1β was manipulated in vivo. Male rats were administered doses of rIL-1β that elevated circulating IL-1β and expression of IL-6 and TNF-α in their lungs, but that did not cause pathology, as is observed in humans during acute hantavirus infection [Khaiboullina and St Jeor, 2002; Krakauer et al., 1995; Mori et al., 1999]. Treatment of male rats with physiological doses of rIL-1β had no effect on the amount of SEOV RNA present in the lungs or shed in saliva. From these data, we conclude that suppression of proinflammatory cytokines below baseline concentrations may not be the critical factor in mediating SEOV persistence, as elevation of proinflammatory cytokines within physiological ranges is not sufficient to affect persistence of SEOV in rats. Supra-physiological concentrations of proinflammatory cytokines may be necessary for viral clearance, but at the cost of causing immunopathology and disease. An alternative hypothesis is that enhanced regulatory T cell activity in the lungs may reduce local CD8+ T cell responses to cause SEOV persistence. Several viruses, including LCMV and HCV suppress CD8+ T cell activity to establish and maintain persistent infection [Moskophidis et al., 1993; Neumann-Haefelin et al., 2007] and although a non-natural host, in newborn mice, Hantaan virus persistence is correlated with reduced IFNγ-producing CD8+ T cells [Araki et al., 2004]. Whether SEOV-specific CD8+ T cell activity (e.g. production of IFN-γ and TNF-α) is altered by infection requires further investigation.
Hantaviral persistence may be mediated not only by alterations in rodent host immune responses, but also directly by the virus. Segment reassortment has been observed in several natural rodent hosts that are infected with multiple strains of a single hantavirus species [Feuer et al., 1999]. Development of quasispecies during the course of infection with a single strain of hantavirus also is suggestive of potential virus-mediated mechanisms of immune evasion [Chu et al., 2006]. Additionally, cyclical accumulation of deletions and insertions in areas of the SEOV genome necessary for the initiation of transcription has been observed in infected VeroE6 cells and presents another alternative mechanism of virus-mediated persistence [Meyer and Schmaljohn, 2000]. Hantavirus persistence in rodents is likely mediated by both the virus and host response to infection.
Zoonotic viruses, including hantaviruses, have cospeciated with their natural reservoir hosts and have calibrated their virulence to support virus persistence without causing notable disease. Strategies of hantaviral persistence in their rodent reservoirs may include not only alteration of host behavior to increase behavioral transmission (i.e. increased aggression), but also modulation of host immunity to establish and maintain persistent infection [Hannah et al., 2008; Klein et al., 2004a; Meyer and Schmaljohn, 2000]. Future studies must continue to determine whether hantaviral persistence, mediated by regulatory T cells in the lungs, is due to suppression of excessively high proinflammatory or CD8+ T cell responses at a site of elevated virus replication that may otherwise be necessary for viral clearance. Immune responses against zoonotic pathogens in other mammalian reservoirs should be evaluated to better understand the evolution of pathogens and their reservoir hosts as well as the mechanisms contributing to maintenance of these pathogens in the environment.
Acknowledgments
We thank Connie Schmaljohn, Cindy Rossi, and Kristen Spik, at the U.S. Army Medical Research Institute of Infectious Diseases, for providing hantavirus reagents. We also would like to thank Michele Hannah for assistance with IHC and M. Christine Zink for providing the anti-von Willebrand factor antibody and assistance with evaluation of pathological observations. Financial support was provided by NIH R01 AI 054995 (SLK).
References
- Araki K, Yoshimatsu K, Lee BH, Kariwa H, Takashima I, Arikawa J. A new model of Hantaan virus persistence in mice: the balance between HTNV infection and CD8(+) T-cell responses. Virology. 2004;322(2):318–327. doi: 10.1016/j.virol.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6(4):353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- Botten J, Mirowsky K, Kusewitt D, Bharadwaj M, Yee J, Ricci R, Feddersen RM, Hjelle B. Experimental infection model for Sin Nombre hantavirus in the deer mouse (Peromyscus maniculatus) Proc Natl Acad Sci. 2000;97(19):10578–10583. doi: 10.1073/pnas.180197197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botten J, Mirowsky K, Kusewitt D, Ye CY, Gottlieb K, Prescott J, Hjelle B. Persistent Sin Nombre virus infection in the deer mouse (Peromyscus maniculatus) model: Sites of replication and strand-specific expression. J Virol. 2003;77(2):1540–1550. doi: 10.1128/JVI.77.2.1540-1550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busbridge NJ, Dascombe MJ, Rothwell NJ. Chronic effects of Interleukin-1beta on fever, oxygen consumption and food intake in the rat. Hormone Metab Res. 1993;25:222–227. doi: 10.1055/s-2007-1002081. [DOI] [PubMed] [Google Scholar]
- Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19(3):531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs JE, Glass GE, Korch GW, Leduc JW. Effects of hantaviral infection on survival, growth and fertility in wild rat (Rattus Norvegicus) populations of Baltimore, Maryland. J Wild Dis. 1989;25(4):469–476. doi: 10.7589/0090-3558-25.4.469. [DOI] [PubMed] [Google Scholar]
- Chu YK, Milligan B, Owen RD, Goodin DG, Jonsson CB. Phylogenetic and geographical relationships of hantavirus strains in eastern and western Paraguay. Am J Trop Med Hyg. 2006;75(6):1127–1134. [PMC free article] [PubMed] [Google Scholar]
- Davis IC, Zajac AJ, Nolte KB, Botten J, Hjelle B, Matalon S. Elevated generation of reactive oxygen/nitrogen species in hantavirus cardiopulmonary syndrome. J Virol. 2002;76(16):8347–8359. doi: 10.1128/JVI.76.16.8347-8359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easterbrook JD, Kaplan JB, Vanasco NB, Reeves WK, Purcell RH, Kosoy MY, Glass GE, Watson J, Klein SL. A survey of zoonotic pathogens carried by Norway rats in Baltimore, Maryland, USA. Epidemiol Infect. 2007;135(7):1192–1199. doi: 10.1017/S0950268806007746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easterbrook JD, Zink MC, Klein SL. Regulatory T cells enhance persistence of the zoonotic pathogen Seoul virus in its reservoir host. Proc Natl Acad Sci. 2007;104(39):15502–15507. doi: 10.1073/pnas.0707453104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis FA, Cruz J, Spiropoulou CF, Waite D, Peters CJ, Nichol ST, Kariwa H, Koster FT. Hantavirus pulmonary syndrome: CD8+ and CD4+ cytotoxic T lymphocytes to epitopes on Sin Nombre virus nucleocapsid protein isolated during acute illness. Virology. 1997;238(2):380–390. doi: 10.1006/viro.1997.8827. [DOI] [PubMed] [Google Scholar]
- Feuer R, Boone JD, Netski D, Morzunov SP, St Jeor SC. Temporal and spatial analysis of Sin Nombre virus quasispecies in naturally infected rodents. J Virol. 1999;73(11):9544–9554. doi: 10.1128/jvi.73.11.9544-9554.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granowitz EV, Clark BD, Vannier E, Callahan MV, Dinarello CA. Effect of interleukin-1 (IL-1) blockade on cytokine synthesis: I. IL-1 receptor antagonist inhibits IL-1-induced cytokine synthesis and blocks the binding of IL-1 to its type II receptor on human monocytes. Blood. 1992;79(9):2356–2363. [PubMed] [Google Scholar]
- Groeneveld PH, Colson P, Kwappenberg KM, Clement J. Increased production of nitric oxide in patients infected with the European variant of hantavirus. Scand J Infect Dis. 1995;27(5):453–456. doi: 10.3109/00365549509047045. [DOI] [PubMed] [Google Scholar]
- Haan C, Behrmann I. A cost effective non-commercial ECL-solution for Western blot detections yielding strong signals and low background. J Immunol Methods. 2007;318(1-2):11–19. doi: 10.1016/j.jim.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Hahm B, Trifilo MJ, Zuniga EI, Oldstone MBA. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22(2):247–257. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Hannah MF, Bajic VB, Klein SL. Sex differences in the recognition of and innate antiviral responses to Seoul virus in Norway. Brain Behav Immunity. 2008 doi: 10.1016/j.bbi.2007.10.005. in press, [EPub Nov 27, 2007] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson ER, Shone SM, Zink MC, Glass GE, Klein SL. Wounding: the primary mode of Seoul virus transmission among male Norway rats. Am J Trop Med Hyg. 2004;70(3):310–317. [PubMed] [Google Scholar]
- Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, Lei HY. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol. 2005;75(2):185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson PJ, Rizzoli A, Grenfell BT, Heesterbeek H, Dobson AP. The ecology of wildlife diseases. Oxford (UK): Oxford University Press; 2002. [Google Scholar]
- Ikejima T, Okusawa S, Ghezzi P, van der Meer JW, Dinarello CA. Interleukin-1 induces tumor necrosis factor (TNF) in human peripheral blood mononuclear cells in vitro and a circulating TNF-like activity in rabbits. J Infect Dis. 1990;162(1):215–223. doi: 10.1093/infdis/162.1.215. [DOI] [PubMed] [Google Scholar]
- Janeway CA, P T, Walport M, Schlomick M. Immunobiology. New York: Garland Publishing; 2001. [Google Scholar]
- Johnson KM, Mackenzie RB, Webb PA, Kuns ML. Chronic infection of rodents by Machupo virus. Science. 1965;150(703):1618–1619. doi: 10.1126/science.150.3703.1618. [DOI] [PubMed] [Google Scholar]
- Khaiboullina SF, Rizvanov AA, Otteson E, Miyazato A, Maciejewski J, St Jeor S. Regulation of cellular gene expression in endothelial cells by Sin Nombre and Prospect Hill viruses. Viral Immunol. 2004;17(2):234–251. doi: 10.1089/0882824041310504. [DOI] [PubMed] [Google Scholar]
- Khaiboullina SF, St Jeor SC. Hantavirus immunology. Viral Immunol. 2002;15(4):609–625. doi: 10.1089/088282402320914548. [DOI] [PubMed] [Google Scholar]
- Kilpatrick ED, Terajima M, Koster FT, Catalina MD, Cruz J, Ennis FA. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J Immunol. 2004;172(5):3297–3304. doi: 10.4049/jimmunol.172.5.3297. [DOI] [PubMed] [Google Scholar]
- Klein SL, Bird BH, Glass GE. Sex differences in immune responses and viral shedding following Seoul virus infection in Norway rats. Am J Trop Med Hyg. 2001;65(1):57–63. doi: 10.4269/ajtmh.2001.65.57. [DOI] [PubMed] [Google Scholar]
- Klein SL, Cernetich A, Hilmer S, Hoffman EP, Scott AL, Glass GE. Differential expression of immunoregulatory genes in male and female Norway rats following infection with Seoul virus. J Med Virol. 2004a;74:180–190. doi: 10.1002/jmv.20163. [DOI] [PubMed] [Google Scholar]
- Klein SL, Zink MC, Glass GE. Seoul virus infection increases aggressive behaviour in male Norway rats. Animal Behaviour. 2004b;67:421–429. [Google Scholar]
- Klingstrom J, Hardestam J, Stoltz M, Zuber B, Lundkvist A, Linder S, Ahlm C. Loss of cell membrane integrity in Puumala hantavirus-infected patients correlates with levels of epithelial cell apoptosis and perforin. J Virol. 2006;80(16):8279–8282. doi: 10.1128/JVI.00742-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer T, Leduc JW, Krakauer H. Serum levels of tumor necrosis factor-alpha, interleukin-1, and interleukin-6 in hemorrhagic fever with renal syndrome. Viral Immunol. 1995;8(2):75–79. doi: 10.1089/vim.1995.8.75. [DOI] [PubMed] [Google Scholar]
- Lee HW, Lee PW, Baek LJ, Song CK, Seong IW. Intraspecific transmission of Hantaan virus, etiologic agent of Korean Hemorrhagic fever, in the rodent. Am J Trop Med Hyg. 1981;30(5):1106–1112. doi: 10.4269/ajtmh.1981.30.1106. [DOI] [PubMed] [Google Scholar]
- Lee PW, Yanagihara R, Gibbs CJ, Jr, Gajdusek DC. Pathogenesis of experimental Hantaan virus infection in laboratory rats. Arch Virol. 1986;88(1-2):57–66. doi: 10.1007/BF01310890. [DOI] [PubMed] [Google Scholar]
- Lyons CR, Orloff GJ, Cunningham JM. Molecular cloning and functional expression of an inducible nitric oxide synthase from a murine macrophage cell line. J Biol Chem. 1992;267(9):6370–6374. [PubMed] [Google Scholar]
- Meyer BJ, Schmaljohn C. Accumulation of terminally deleted RNAs may play a role in Seoul virus persistence. J Virol. 2000;74(3):1321–1331. doi: 10.1128/jvi.74.3.1321-1331.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BJ, Schmaljohn CS. Persistent hantavirus infections: characteristics and mechanisms. Trends Microbiol. 2000;8(2):61–67. doi: 10.1016/s0966-842x(99)01658-3. [DOI] [PubMed] [Google Scholar]
- Mori M, Rothman AL, Kurane I, Montoya JM, Nolte KB, Norman JE, Waite DC, Koster FT, Ennis FA. High levels of cytokine-producing cells in the lung tissues of patients with fatal hantavirus pulmonary syndrome. J Infect Dis. 1999;179(2):295–302. doi: 10.1086/314597. [DOI] [PubMed] [Google Scholar]
- Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362(6422):758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- Nagai T, Tanishita O, Takahashi Y, Yamanouchi T, Domae K, Kondo K, Dantas JR, Takahashi M, Yamanishi K. Isolation of hemorrhagic fever with renal syndrome virus from leukocytes of rats and virus- replication in cultures of rat and human macrophages. J Gen Virol. 1985;66:1271–1278. doi: 10.1099/0022-1317-66-6-1271. [DOI] [PubMed] [Google Scholar]
- Neumann-Haefelin C, Spangenberg HC, Blum HE, Thimme R. Host and viral factors contributing to CD8+ T cell failure in hepatitis C virus infection. World J Gastroenterol. 2007;13(36):4839–4847. doi: 10.3748/wjg.v13.i36.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Gabel D. Simple method for reduction of autofluorescence in fluorescence microscopy. J Histochem Cytochem. 2002;50(3):437–439. doi: 10.1177/002215540205000315. [DOI] [PubMed] [Google Scholar]
- Schountz T, Prescott J, Cogswell AC, Oko L, Mirowsky-Garcia K, Galvez AP, Hjelle B. Regulatory T cell-like responses in deer mice persistently infected with Sin Nombre virus. Proc Natl Acad Sci. 2007;104(39):15496–15501. doi: 10.1073/pnas.0707454104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundstrom JB, McMullan LK, Spiropoulou CF, Hooper WC, Ansari AA, Peters CJ, Rollin PE. Hantavirus infection induces the expression of RANTES and IP-10 without causing increased permeability in human lung microvascular endothelial cells. J Virol. 2001;75(13):6070–6085. doi: 10.1128/JVI.75.13.6070-6085.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temonen M, Mustonen J, Helin H, Pasternack A, Vaheri A, Holthofer H. Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: An immunohistochemical study. Clin Immunol Immunopathol. 1996;78(1):47–55. doi: 10.1006/clin.1996.0007. [DOI] [PubMed] [Google Scholar]
- Terajima M, Hayasaka D, Maeda K, Ennis FA. Immunopathogenesis of hantavirus pulmonary syndrome and hemorrhagic fever with renal syndrome: Do CD8(+) T cells trigger capillary leakage in viral hemorrhagic fevers? Immunol Lett. 2007;113(2):117–120. doi: 10.1016/j.imlet.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, Zaki SR, Swanepoel R, Ansari AA, Peters CJ. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J Infect Dis. 1999;179:S188–S191. doi: 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- Walker DH, Wulff H, Lange JV, Murphy FA. Comparative pathology of Lassa virus infection in monkeys, guinea-pigs, and Mastomys natalensis. Bull World Health Organ. 1975;52(4-6):523–534. [PMC free article] [PubMed] [Google Scholar]
- Wang LF, Shi Z, Zhang S, Field H, Daszak P, Eaton BT. Review of bats and SARS. Emerg Infect Dis. 2006;12(12):1834–1840. doi: 10.3201/eid1212.060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara R, Amyx HL, Gajdusek DC. Experimental infection with Puumala virus, the etiologic agent of nephropathia epidemica, in bank voles (Clethrionomys glareolus) J Virol. 1985;55(1):34–38. doi: 10.1128/jvi.55.1.34-38.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]