

Abstract
The bone microenvironment (BME) is the main hub of all skeletal related pathological events in osteosarcoma leading to tumor induced bone destruction, and decreasing overall bone quality and bone strength. The role of extra-cellular membrane vesicles (EMVs) as mediators of intercellular communication in modulating osteosarcoma-BME is unknown, and needs to be investigated. It is our hypothesis that osteosarcoma-EMVs contain pro-osteoclastogenic cargo which increases osteoclastic activity, and dysregulated bone remodeling in the osteosarcoma-BME. In this study, EMVs were isolated from the conditioned media of 143B and HOS human osteosarcoma cell cultures using differential ultracentrifugation. Nano-particle tracking analysis determined EMVs in the size range of 50-200 nm in diameter. The EMV yield from 143B cells was relatively higher compared to HOS cells. Transmission electron microscopy confirmed the ultrastructure of 143B-EMVs and detected multivesicular bodies. Biochemical characterization of 143B-EMVs detected the expression of bioactive pro-osteoclastic cargo including matrix metalloproteinases-1 and -13 (MMP-1, -13), transforming growth factor-β (TGF-β), CD-9, and receptor activator of nuclear factor kappa-β ligand (RANKL). Detection of a protein signature that is uniquely pro-osteoclastic in 143B-EMVs is a novel finding, and is significant as EMVs represent an interesting mechanism for potentially mediating bone destruction in the osteosarcoma-BME. This study further demonstrates that 143B cells actively mobilize calcium in the presence of ionomycin, and forskolin, and induce cytoskeleton rearrangements leading to vesicular biogenesis. In conclusion, this study demonstrates that 143B osteosarcoma cells generate EMVs mainly by mechanisms involving increased intracellular calcium or cAMP levels, and contain pro-osteoclastic cargo.
Introduction
Osteosarcoma is an aggressive malignancy of bone, mainly affecting adolescents and young adults. Interactions between osteosarcoma and bone microenvironment (BME) promote tumor growth and osteoclastic bone destruction. The main goal of this study is to understand the role of extracellular membrane vesicles (EMVs) as potential modulators of osteosarcoma BME and to identify the key biochemical components of EMVs mediating cellular dynamics and dysregulated pathologic remodeling of the matrix and bone. EMVs are membrane-invested structures that are derived from a number of cells including osteosarcoma cells [1], [2]. In recent years, EMVs have received much attention for their role in various diseases and as biomarkers of therapy and disease burden [3]. Recent studies report that tumor cell–derived EMVs support cancer cell growth, survival, metastasis, and angiogenesis, evade host immune surveillance, modulate tumor microenvironment (TMN), and initiate the formation of premetastatic sites [4], [5], [6], [7], [8], [9], [10], [11], [12]. Tumor-derived EMVs, in general, originate through the fusion of multivesicular bodies (MVBs) with the plasma membrane (exosomes) or by budding (shed vesicles or microvesicles), followed by exocytotic release [13], [14], [15], [16]. Detection of EMVs and osteoblastic and osteoclastic lesions in the bioluminescent osteosarcoma orthotopic mouse (BOOM) model provides a strong rationale to investigate the role of EMVs in modulating osteosarcoma BME [2]. Biochemical analyses of EMV cargo will be informative as it will identify the key EMV mediators underlying osteosarcoma pathobiology.
Biomechanical stress in the bone TMN leads to increased intracellular calcium levels that, in turn, may promote EMV biogenesis, increase the expression of extracellular remodeling enzymes such as matrix metalloproteinases (MMPs), and stimulate exocytotic delivery of bioactive cargo. These biochemical events may result through the activation of G protein–coupled receptors (GPCRs) or calcium-dependent signaling pathways. A study by Ancha et al. showed the role of H2 receptor, a GPCR, in the regulation of MMP-1 expression and secretion in cultured gastric cells [17]. Savina et al. demonstrated that increased intracellular calcium concentrations in K562 leukemia cells trigger Rab11-mediated fusion of MVBs with the plasma membrane and release exosomes [18]. Another study suggested the role of cAMP/protein kinase A pathway in the release of tumor necrosis factor receptor 1–associated exosomes [19]. In the osteosarcoma BME, neither the role of cAMP/protein kinase A pathway nor of calcium-dependent pathway and their downstream effects on cytoskeleton rearrangements leading to vesicle biogenesis are known and are subjects of the current study.
Functional implications of EMVs depend on the cargo composition that, in turn, is governed by the metabolic status of the donor cell from which they originate. For instance, EMVs containing MMPs and proteases such as plasminogen activator promote tumor invasion and metastases, whereas those enriched in cytokines such as transforming growth factor β (TGF-β) evade host immune response. Little is known about the mechanisms underlying EMV-mediated intercellular dynamics in the TMN. Peinado et al. reported a role for melanoma exosomes in establishing premetastatic niches by reprogramming bone marrow–derived cells [20]. Exosomes derived from prostate, breast, and lung cancer cells activate fibroblasts or mesenchymal stem cells by increasing their motility and rendering them resistant to apoptosis [21], [22] or by stimulating myofibroblastic differentiation [23], [24].
Extracellular matrix remodeling is an important process mainly mediated by metalloproteinases, such as MMPs in the tumor BME, which enable the tumor cells to grow, invade, and metastasize. Another important role of MMPs besides extra cellular matrix (ECM) degradation is in the activation of membrane-associated proteins and regulation of cell signaling pathways. Increased expression of MMP-1, MMP-2, and MMP-9 and down-regulation of micro RNA (miRNA) 143, which targets MMP-13, correlates to poor prognostic outcomes in patients with osteosarcoma [25], [26], [27], [28]. A recent study by Husmann et al. clearly outlines the importance of MMP-1 in osteosarcoma pathobiology where in short hairpin RNA (shRNA)-mediated down regulation of MMP-1 expression in 143B cells generated smaller primary tumors and fewer micrometastases and macrometastases in the lungs, and overexpression of MMP-1 in nonmetastatic HOS cells resulted in osteolytic primary tumors and lung metastasis [29].
It is our hypothesis that osteosarcoma EMVs contain pro-osteoclastogenic cargo that increases osteoclastic activity and dysregulated bone remodeling in the osteosarcoma BME. In this study, we demonstrate that 143B osteosarcoma cells generate EMVs by mechanisms involving cAMP/calcium-dependent signaling pathways and contain pro-osteoclastic cargo.
Materials and Methods
Detection of Tumor-Induced Histopathologic Changes in the Osteosarcoma BME
For evaluating tumor-induced histopathologic changes in the osteosarcoma BME, excised tibias (tumor bearing) from the BOOM model [2] were fixed in 4% paraformaldehyde (pH 7.4). The in vivo studies using the BOOM model were done with the assistance of the Proof of Concept Laboratory at The University of Kansas Cancer Center, with the approval of the University of Kansas Institutional Animal Care and Use Committee. For histopathologic evaluation, tibias were decalcified in 10% EDTA (pH 7.5) for 2 weeks before sectioning and paraffin embedding. The sections were processed for hematoxylin and eosin staining and immunohistochemistry (IHC). To detect osteoblastic-mediated mineralization in the tumor tissue, von Kossa staining was done using non-decalcified tumor tissue sections. To detect the immunoexpression of MMPs in the tumor tissue of the BOOM model, MMP-1 and MMP-13 IHC was done using primary antibodies (MMP-1, RB-1536; MMP-13, MS-825) purchased from Lab Vision Thermo Scientific (Kalamazoo, MI), followed by detection. The detection reagents were purchased from Biocare Medical (Concord, CA) and Dako (Carpinteria, CA). For negative control, primary antibody was excluded, and human placenta tissue sections were used as positive control in MMP IHC.
Preparation of Conditioned Media from 143B Cell Cultures
Human osteosarcoma cell lines 143B (highly aggressive and metastatic; k-ras activated) and HOS (nonaggressive and nonmetastatic; k-ras wild type) were purchased from American Type Culture Collection (Manassas, VA). The 143B cells were genetically engineered to express luciferase gene (FUW-Luc-mCherry-puro), and cultured in Dulbecco's modified Eagle's medium according to the previously described method [2]. The 143B-luc-mCherry cell line was authenticated for its ability to grow in the presence of puromycin in vitro and to proliferate in the tibia of Nu/Nu mice and metastasize to the lungs, as described in the BOOM model [2]. At subconfluence, conditioned media (CM) were prepared by culturing 143B or HOS cells in serum-free media for 24 hours and subjected to differential ultracentrifugation for isolation of EMVs.
Isolation, Quantitation, and Characterization of EMVs Derived from the CM of 143B and HOS Cell Cultures
Differential ultracentrifugation
We used differential ultracentrifugation (low speed followed by ultracentrifugation at 110,000g for 2 hours) to isolate EMVs from the CM prepared from osteosarcoma cells according to the scheme shown in Figure 1.
Figure 1.
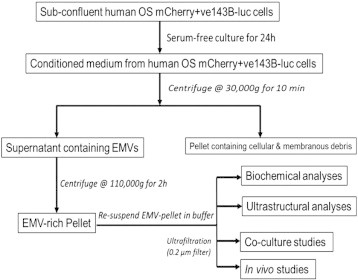
Schematic shows the isolation of EMVs from CM of serum-starved human OS 143B and HOS cells.
Nanoparticle tracking analysis
To determine the EMV concentration and size distribution profile of EMVs isolated from CM of osteosarcoma cell cultures, vesicles were analyzed using the NanoSight (Amesbury, UK) NTA 2.3: Nanoparticle Tracking and Analysis instrument and software (release version build 11 RC1, 2012, hardware: LM14). The samples were injected in the sample chamber according to the manufacturer's recommendations. EMVs were analyzed in phosphate-buffered saline solution under Brownian motion at 22°C to 24°C with laser wavelength at 638 nm. Multiple video frames were captured for 60 seconds per reading. Screen gain remained at 1.0, and detection threshold ranged from 13 to 14. The number of readings for EMVs, at dilutions 1:5000, 1:2000, 1:1000, and 1:100, ranged from 5 to 20 measurements. Analysis displayed plots describing particle diameter (size) distribution cumulative of each reading, 2-dimensional (2-D) and 3-dimensional (3-D) particle light intensity against particle concentration per milliliter, as well as a video snapshot.
Alkaline phosphatase assay
The alkaline phosphatase (ALP) activity of EMVs was assayed using ALP colorimetric kit (AnaSpec, Fremont, CA). Briefly, 50-μg vesicles were incubated with a colorimetric substrate, para-nitrophenyl phosphate, and the conversion of para-nitrophenyl phosphate to p-nitrophenol on release of phosphate ions was monitored at 405 nm. The protein concentration of the EMV samples was measured by Bradford assay (Bio-Rad Laboratories, Hercules, CA).
Flow cytometry and fluorescence microscopy
For detection of mCherry fluorescence on EMVs derived from mCherry-labeled, 143B luciferase–expressing, puromycin-resistant cells, EMV suspensions were examined microscopically using the Olympus (Center Valley, PA) IX71 inverted fluorescent microscope equipped with a xenon arc lamp and monochromatic complementary metal oxide semiconductor camera. In addition, flow cytometric data were acquired on EMV suspensions using the BD LSR II flow cytometer integrated with FACSDiva software (BD Biosciences, San Jose, CA, USA).
Transmission electron microscopy (TEM) analyses
For TEM, 143B EMV pellets were fixed in 2.5% glutaraldehyde, postfixed in 1% osmium tetraoxide (OsO4), dehydrated, embedded in epon resin, and cut into ultrathin sections. The sections were stained with uranyl acetate and lead acetate before mounting on EM grids. The sections were examined and photographed using a JEM 1400 electron microscope (JEOL USA, Inc., Peabody, MA, USA) (80 kV).
Western blot analyses
To determine the biochemical composition of the 143B EMV cargo, Western blot analyses were performed according to the previously described method [30]. 143B EMVs were homogenized in Tris lysis buffer (20 mM Tris, 137 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM PMSF, and 1 mM DTT). Crude lysates of 143B cells (12.5-25 μg) and EMVs (25-40 μg) were denatured in sodium dodecyl sulfate sample buffer, electrophoresed on 12% denaturing polyacrylamide gels, and visualized by Ponceau stain. For immunoblot analysis, the proteins from the gel were transferred on to a polyvinylidene fluoride (PVDF) membrane and incubated with the following primary antibodies: anti–MMP-1 and anti–MMP-13 (Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); 200 μg/ml each) at 1:200, anti-CD-9 (SBI: System Biosciences (Mountain View, CA, USA); 0.25 mg/ml) at 1:1000, and anti-RANKL and anti–TGF-β (GeneTex (Irvine, CA, USA); 1 mg/ml) at 1:1000 dilution. Detection of the immunostained bands was done by ECL chemiluminescence detection system (Thermo Scientific, Rockford, IL). Image acquisition was done using LabWorks Image Acquisition and Analysis Software 4.6.00.0 (UVP Bioimaging Systems, Upland, CA) and Image Lab software for the ChemiDoc MP system (Bio-Rad Laboratories) at incremental exposure time frame of 15, 30, 60, 180, and 300 seconds.
Calcium sensitization and 143B EMV biogenesis
To measure the effect of intracellular rise in calcium concentrations on EMV biogenesis, mCherry-labeled 143B cells were cultured on 35-mm dishes to subconfluence and prepared for calcium imaging. Briefly, 143B cells were loaded with 1 μM Fura-2 AM, a fluorescent dye which binds to free intracellular calcium by incubating at 37οC for 30 minutes according to previously described methods [31]. The ratio of Fura-2 excitations at 340 to 350 nm and 375 to 380 nm of light corresponds to the intracellular calcium concentration [Ca++]. Specifically, we evaluated the effects of two agents that modulate intracellular [Ca++]: ionomycin, an ionophore, which increases intracellular calcium levels through store-dependent mechanisms and forskolin, an activator of cAMP generating adenylate cyclase on EMV biogenesis. Osteosarcoma cells were either stimulated with ionomycin (alone) at three different concentrations, i.e., 1, 3, and 10 μM, or pretreated with forksolin at 10 μM before the addition of ionomycin. Measurements of increase in calcium concentrations in 143B osteosarcoma cells were recorded using a Photon Technology International (PTI Technologies Inc, Birmingham, NJ) automated spectroflurometer connected to an inverted microscope (Leica DMI-4000B; Leica Microsystems, Wetzlar, Germany) equipped with a 14-bit CoolSNAP charge-coupled device camera (Photometrics, Tucson, AZ). Data acquisition, calibration, and analysis were done using the EasyPro (PTI) software. Changes in the cellular morphology and induced EMV biogenesis on forskolin and/or ionomycin stimulation were observed in high power (× 40) by fluorescence microscopy. Forskolin pretreatment was done using 10 μM concentrations at 37οC for 5 minutes. Ionomycin stimulation was done at 1, 3, and 10 μM. Intracellular calcium concentration was estimated from the Fura ratio by using Grynkiewicz equation [32].
Statistical analysis
Data presented represent means (+ SD) from three or more independent experiments. Statistical analysis was performed using Prism 5 (GraphPad Software, La Jolla, CA). All experimental data are presented as means ± SD. Student's t test and one-way analysis of variance were used for determining statistical significance between resting cells (before stimulation) versus ionomycin or forskolin + ionomycin–treated cells. A P value of < 0.05 was considered statistically significant.
Results
Rapidly Growing Tumor Induces Histopathologic Changes in the Osteosarcoma BME
Histopathologic studies on the tumor tissue obtained from the BOOM model detected remarkable tumor-induced morphologic changes as evidenced by varying cortical bone thickness and destruction of tibia of tumor-bearing mice (Figure 2, A and B). Detection of resorptive pits and multinucleate osteoclasts in the tibial sections of the BOOM model demonstrates high osteoclastic activity (Figure 2C). Intense von Kossa staining of tumor-bearing bones suggests tumor-induced prolific osteoblastic activity (Figure 2D). Light microscopy revealed the presence of numerous osteocytes in the tumor-bearing bone (Figure 2, A and B). Whether those osteocytes are transformed by 143B EMVs is unknown at present and is a subject for future investigation. Furthermore, detection of numerous vesicles in the vicinity of the resorption pit suggests an active procatabolic role for osteoclasts in osteosarcoma pathobiology (Figure 2E). Ultrastructural examination of the extracellular matrix of the tumor tissue from the BOOM model revealed the presence of EMVs interspersed among collagen fibrils (Figure 2F). Immunohistochemical studies detected the expression of MMP-1 and MMP-13 in the tumor and nontumor cells such as osteocytes, osteoclasts, and osteoblasts of the osteosarcoma BME (Figure 3).
Figure 2.
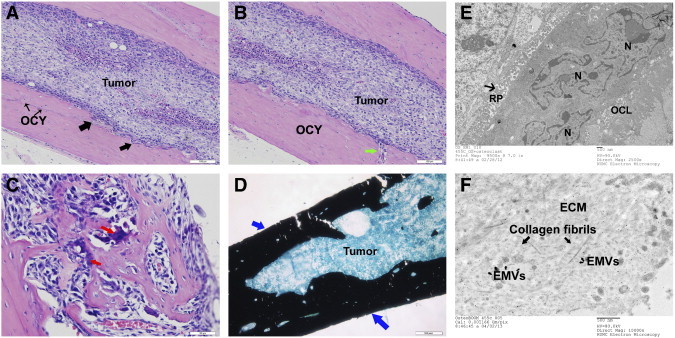
Osteosarcoma-induced osteolytic and osteoblastic changes in the BME of the BOOM model. A and B are hematoxylin and eosin–stained tumor-bearing tibial sections showing the presence of rapidly expanding tumor within the marrow cavity. The cortical bone is abnormal, thickened, and has uneven or ruffled borders due to active osteoclastic bone resorption. The black arrows indicate resorption pits, and the green arrow indicates a capillary infiltrating into the cortical bone. The tumor-bearing bone also shows several osteocytes (OCY). C shows multinucleate osteoclasts actively remodeling the bone in the osteosarcoma BME. D shows mineralizing osteoblasts as detected by von Kossa staining. E and F show electron micrographs of an active osteoclast in the vicinity of a resorption pit filled with hydrolytic acidic vesicles (E) and extracellular matrix containing numerous EMVs interspersed among collagen fibrils (F).
Figure 3.
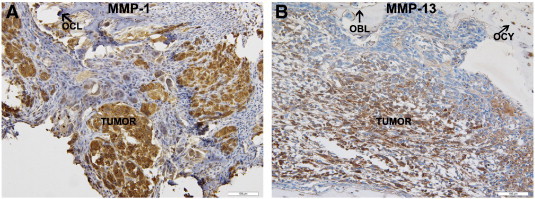
Immunodetection and localization of MMP-1 and MMP-13 in the tibial sections of the BOOM model. Brown peroxidase staining indicates MMP expression in the tumor and nontumor cells (osteoblasts (OBL); osteoclasts (OCL); and osteocytes (OCY). Primary antibodies (MMP-1, RB-1536; MMP-13, MS-825) were used at 1:100 dilutions.
Isolation and Characterization of 143B EMVs Includes a Multimodality Approach Using Differential Centrifugation, Nanoparticle Tracking Analysis, and TEM
Osteosarcoma EMVs were isolated from the CM of mCherry + ve, 143B-luc, and HOS cells by differential ultracentrifugation (Figure 1). The size distribution profile of isolated EMVs as determined by nanoparticle tracking analysis (NTA) was in the range of 50 to 200 nm (Figures 4, A and B, and W1). The EMV yield generated from 143B cells was higher as reflected by their mean EMV number per milliliter (711 × 108 bEMVs per milliliter) and protein concentration (1.2 mg/ml) compared to HOS cells (mean EMV number per milliliter = 7.3 × 108 hEMVs per milliliter) and protein concentration (0.33 mg/ml) (Figure W2). Because 143B EMV output was greater (100 ×) than HOS EMVs, and for the sake of focus of the current study, further characterization was done on 143B EMVs. Ultrastructural characterization of EMVs derived from 143B cells revealed the presence of numerous vesicles in the size range of 50 to 200 nm (Figure 4, C and D). TEM revealed the presence of MVBs and perivesicular mineral clusters in the osteosarcoma BME (Figure 4, C and D). Presence of ALP enzyme activity in 143B-derived EMVs confirmed their mineralization competence as observed by TEM (Figure 5A). Flow cytometry and fluorescence microscopy detected the retention of mCherry fluorescence in EMVs derived from mCherry + ve, 143B luciferase–expressing cells (Figure 5, B and C).
Figure 4.
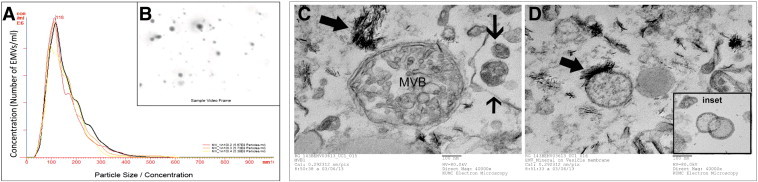
NTA and TEM of 143B EMVs. A shows NTA measurements of size and concentration of EMVs in three different samples. B shows a screenshot of video from NanoSight LM14 showing light scatter caused by 143 EMVs. C and D show electron micrographs of 143B EMV pellet. C shows the presence of an MVB containing intraluminal vesicles and several EMVs in the size range of 50 to 200 nm. D shows several mineralizing EMVs and the presence of polarized membrane-associated deposition of hydroxyapatitelike crystals.
Figure W1.
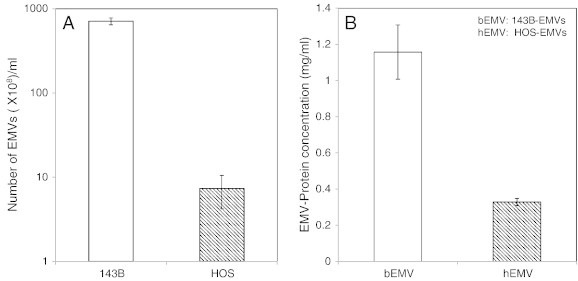
Comparison of EMV output between 143B, an aggressive osteosarcoma cell line versus HOS, a nonaggressive nonmetastatic cell line. A shows the number of EMVs (× 108/ml) as determined by NTA, and B compares the protein concentration (mg/ml) of EMVs derived from 143B (bEMVs) versus HOS (hEMVs) osteosarcoma cells.
Figure W2.
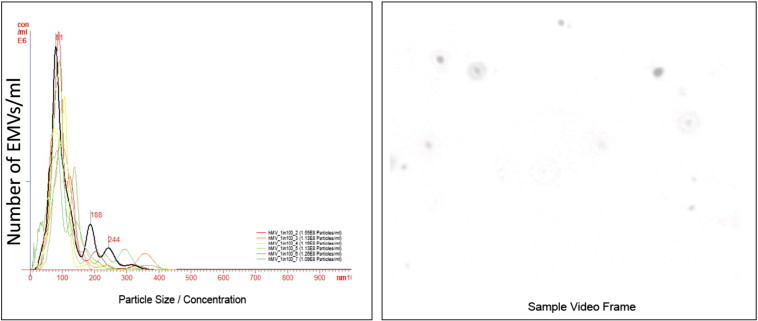
NTA of HOS EMVs. A shows NTA measurements of size and concentration of EMVs in six different samples. B shows a screenshot of video from NanoSight LM14 showing light scatter caused by HOS EMVs.
Figure 5.
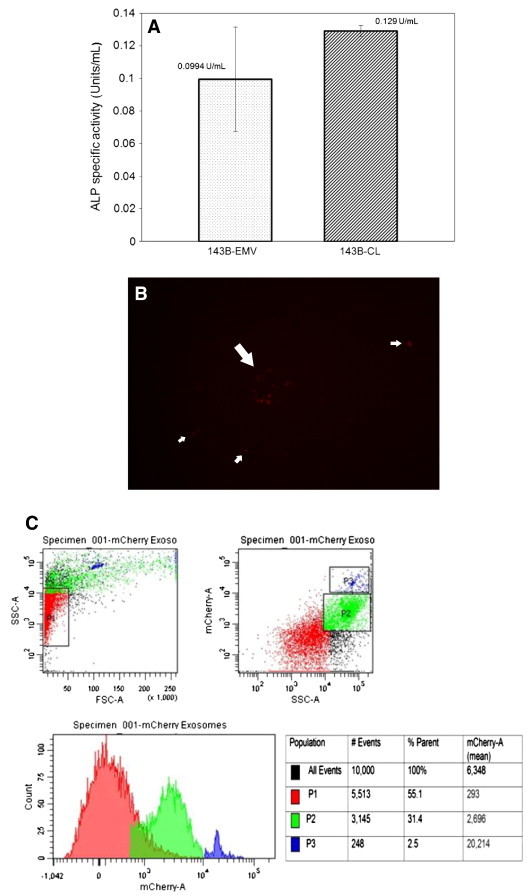
Determination of ALP activity and detection of mCherry fluorescence in 143B EMVs. A shows bar graphs comparing ALP activity (U/ml) in143B EMVs versus 143B cell lysates (n ≥ 3). B shows detection of mCherry fluorescence in EMVs derived from 143B-luc-mCherry OS cells (× 20). C shows mCherry-positive 143B EMV forward scatter (FSC) and side scatter (SSC) by flow cytometry.
143B EMVs Contain Pro-Osteoclastogenic Cargo
Biochemical characterization of cargo proteins of 143B-derived EMVs by Western blot analysis demonstrates the expression of a pro-osteoclastogenic cargo, which includes MMPs (MMP-1 and MMP-13), CD-9, RANKL, and TGF-β (Figure 6). Detection of a clear band at 52 kDa in 143B EMV lysates corresponds to the predicted band size for MMP-1 as previously reported by Husmann et al., in the 143B cell lysates [29] (Figure 6A). This band is likely to be a proenzyme as reported previously [33]. Immunodetection for MMP-13 expression revealed the presence of a major band at 68 to 70 kDa that was selectively enriched in 143B EMVs (Figure 6A). This band is very likely to be the proenzyme form of MMP-13 as previous studies report the detection of the proenzyme or the latent form at 60 to 65 kDa, whereas the active form is detected at 30 to 48 kDa [34], [35].
Figure 6.
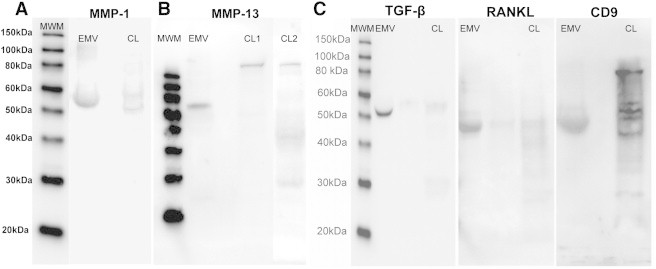
Detection of pro-osteoclastogenic cargo in 143B EMVs by Western blot analysis. Crude lysates of 143B cells (12.5-25 μg) and EMVs (25-40 μg) were analyzed for MMP-1, MMP-13 (A and B), TGF-β, RANKL, and CD-9 (C) expression by Western blot analysis. All samples were analyzed in triplicate.
Further characterization revealed that 143B EMVs contain pro-osteoclastogenic cargo, i.e., CD-9, RANKL, and TGF-β (Figure 6C). All the three proteins are potent stimulators of osteoclastogenesis [36], [37], [38], [39], [40], but their presence in EMVs derived from osteosarcoma has never been previously reported. Immunoblot analysis of 143B EMVs with CD-9 antibody detected a band at 48 to 50 kDa, which is very likely the trimeric form. Recent studies have reported the presence of multimeric forms of CD-9 detected at 24 kDa (monomeric), 38 kDa (homodimer), 52 to 54 kDa (trimer), and 70 to 72 kDa (tetramer), which most likely form due to spontaneous intermolecular disulfide bonding of membrane-proximal cysteine residues [41], [42]. Immunoblot analysis of 143B EMVs with anti-RANKL antibody revealed the presence of multimeric form of RANKL at 48 kDa. Previous studies report the existence of the following three different RANKL isoforms: RANKL1, which is similar to the original RANKL, contains both the intracellular and transmembrane spanning domain; RANKL2, which has a shorter intracellular domain than RANKL; and RANKL3, which lacks the transmembrane domain, constitutes the soluble form of RANKL and inhibits osteoclastogenesis [43]. Immunoblot analysis of 143B EMVs with anti–TGF-β antibody revealed the presence of latent form of TGF-β at 52 kDa, which was also detected in exosomes derived from brain tumors [44].
Ionomycin and Forskolin Stimulate Calcium Mobilization within 143B Cells Leading to Vesicle Biogenesis
Calcium imaging studies revealed that 143B cells actively mobilize calcium in the presence of ionomycin, a calcium ionophore, and cause cytoskeleton rearrangements leading to vesiculation. Confocal microscopy showed that ionomycin induced morphologic changes within 143B cells such as loss of cell-cell contact, distortion of cellular margins, changes in the cytoskeleton architecture, formation of membrane blebs, and accumulation of intracellular, perinuclear vesicles (Figure 7, A1, and B1). Addition of 1, 3, and 10 μM ionomycin to 143B cells induced a significant increase (P < 0.0001) in intracellular [Ca++] within 300,000 milliseconds (Figures 7C1, and W3). Pretreatment with 10 μM forskolin, an adenylate cyclase activator, increased calcium mobilization in both naïve and ionomycin-sensitized 143B OS cells and resulted in increased intracellular [Ca++] within 100,000 milliseconds (Figures 7D2, and W3). The above events stimulated cytoskeleton rearrangements within 143B cells leading to vesicular biogenesis (Figure 7, A2, B2, and C2).
Figure 7.
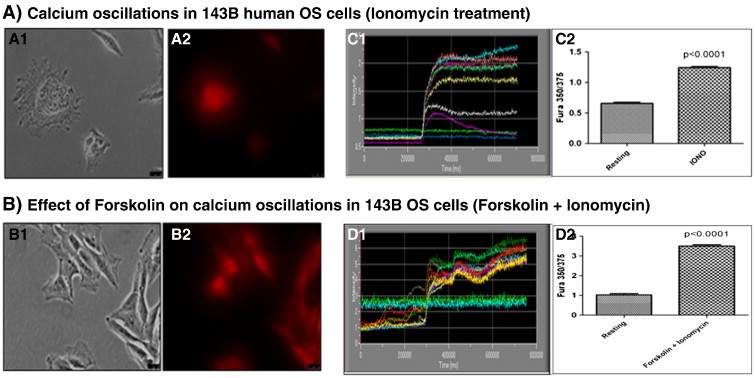
Calcium mobilization and induction of EMV biogenesis in 143B OS cells in the presence of forskolin and ionomycin. A1 and A2, show morphologic changes within ionomycin-sensitized 143B cells leading to the accumulation of intracellular, vesicles, whereas B1 and B2, show intracellular vesiculation in forskolin-pretreated and ionomycin-sensitized 143B cells as observed by confocal microscopy. C1 and D1, show kinetic changes in the Fura-2 ratio in ionomycin (C1) and in forskolin-pretreated, ionomycin-sensitized (D1) 143B cells. C2 and D2, compare Fura 350/375 between resting versus ionomycin (alone)–treated 143B osteosarcoma cells (C2) and resting versus forskolin-pretreated and ionomycin-sensitized (forskolin + ionomycin combination) 143B osteosarcoma cells (D2).
Figure W3.
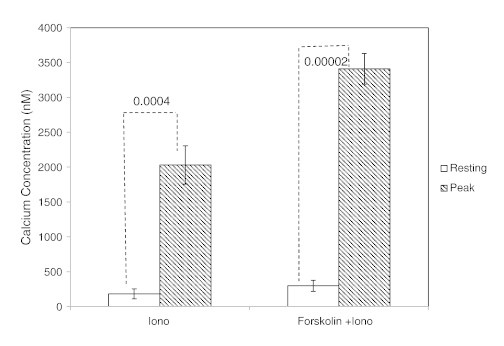
Estimated values of cytosolic calcium concentrations in 143B osteosarcoma cells in the presence of forskolin and ionomycin, as determined by Grynkiewicz equation. The estimated values of cytosolic calcium concentrations for resting and ionomycin-sensitized 143B cells are 182 and 2030 nM, respectively, and for resting versus forskolin-pretreated and ionomycin-sensitized cells are 298 and 3410 nM, respectively.
Discussion
Emerging evidence suggests the role of EMVs in supporting tumor microenvironment niches and as potential mediators of intercellular communication mainly through horizontal transfer of oncogenic cargo [45], [46]. Although EMVs were previously detected in the BOOM model [2], their role as potential drivers of cancer-induced bone destruction and as key mediators of osteolytic activity in the osteosarcoma BME needs further investigation. This study for the first time reports isolation and characterization of EMVs derived from 143B human osteosarcoma cells and its potential implications on the TMN. It clearly demonstrates that majority of the EMVs derived from 143B cells are in the size range of 50 to 200 nm in diameter. The use of NTA allows quantitative and rapid determination of EMV sample size, size range, and concentration. It is highly reliable for accurately determining the size distribution of cell-derived EMVs as it is based on Brownian motion, does not consider the refractive index of the nanoparticle, and is free from sample shrinkage artifacts commonly encountered during fixation for microscopy [47]. Vesicles obtained from 143B CM were devoid of contaminating vesicles from FBS [48]. Detection of MVBs by TEM in 143B EMV samples suggests that the mode of biogenesis and release of EMVs is most likely through endocytic invagination followed by the formation of early endosomes that mature to form MVBs. Size range of 143B EMVs as determined by NTA (50-200 nm), evidence of MVBs by TEM, and the presence of CD-9, an exosome-specific biomarker as listed in ExoCarta database (Bundoora, Victoria, Australia), suggest that 143B EMVs contain exosomes.
To our best knowledge, this is the first study to report the presence of a pro-osteoclastogenic cargo in EMVs isolated from 143B cells. Detection of MMPs (MMP-1 and MMP-13) in 143B EMVs is an important and novel finding because MMP-1– and MMP-13 (MMP)–expressing EMVs could be used as disease biomarkers for evaluating osteosarcoma prognosis. Detection of RANKL in osteosarcoma EMVs is novel and significant as it plays an important role in the activation of MMPs and for stimulating osteoclastogenesis. Targeting MMP-1 expression and activity through RANKL inhibition is promising as recent studies by Casimiro et al. demonstrates a role of RANKL in the activation of MMP-1 expression and activity in breast cancer metastasis [49]. Whether selective inhibition of EMV-derived RANKL and/or MMP-1 and MMP-13 inhibits osteosarcoma pathobiology remains to be investigated. Targeting RANK/RANKL/osteoprotegrin (OPG) signaling in osteosarcoma is currently under intense investigation, and studies with OPG and RANK-Fc demonstrate inhibition of osteolytic lesions in mouse models and improved survival rates [50], [51].
Detection of TGF-β in 143B EMVs is an important finding especially in the context of regulating the bone TMN. In the BME, TGF-β is generated mainly from the mineralized bone matrix by osteoclastic resorption and further stimulates the production of osteolytic and proneoplastic factors [52], [53]. It can stimulate migration of osteoblast progenitors and osteosarcoma cells either directly [54] or indirectly through osteoclast-mediated chemokine (C-X-C motif) ligand 16 (CXCL16) chemokine secretion [55]. It plays an important role in the osteoclastogenic differentiation of uncommitted monocytes by stimulating RANKL and/or tumor necrosis factor α (TNF-α)-induced nuclear factor of activated T-cells cytoplasmic, calcineurin dependent 1 (NFATc1) expression [38]. Tumor exosomes and microvesicles secrete TGF-β that blocks the differentiation of monocytes and increase the accumulation of immature myeloid cells including myeloid-derived suppressor cells (MDSCs) [56] Recently, the role of MDSCs in the osteolytic bone tumor microenvironment in promoting osteoclastic bone resorption was demonstrated [57], [58]. Whether EMV-derived TGF-β increases MDSC-mediated osteoclastic resorption in the OS BME is currently unknown and is the subject of our future studies. Blocking exosome-derived TGF-β is an attractive therapeutic strategy to reduce osteoclastic activity from MDSCs in the tumor microenvironment and increase the efficacy of antitumor immune therapies.
Detection of CD-9, a tetraspanin protein in the EMVs derived from 143B cells, is a novel finding. To our best knowledge, the role of this protein in osteosarcoma pathobiology has never been investigated. Besides being a designated exosome-specific marker, CD-9 is also a pro-osteoclastogenic fusogenic protein as it regulates osteoclast differentiation and the formation of mature polykaryons [36], [59]. It is overexpressed in osteotropic cancers and not only promotes the homing of cancer cells in the bone marrow but also induces osteoclastic bone resorption [37]. Studies report that inhibition of CD-9 by KMC8, a widely used antibody against CD-9, suppresses osteoclastogenesis [60], whereas RANKL-stimulated expression of CD-9 and other fusogenic genes such as CD-47 in osteoclast precursors promotes mature polykaryotic, tartarate-resistant acid phosphatase and osteoclast-specific transmembrane protein expressing osteoclast phenotype [61]. A recent study demonstrated the role of CD-9 in mediating MMP-9–induced migration and invasion in fibrosarcoma cells [62].
Elevation of intracellular calcium concentration on forskolin pretreatment and ionomycin sensitization of 143B cells leads to changes in the cytoskeleton architecture and vesicle biogenesis. This finding is important especially in the context of osteosarcoma BME where actively metabolizing cancer cells maintain energy homeostasis by regulating cytosolic calcium through induction of oscillatory events that eventually trigger cytoskeleton rearrangements and vesicle biogenesis. Previous studies have reported that elevated intracellular calcium concentration [Ca++]i, cAMP levels, and P2X7 receptor (purinergic receptor ion channels mediating calcium and influx across the plasma membrane) activation modulate the pool of EMV output and sorting of cargo by regulating docking, priming, and exocytosis of vesicles [19], [63], [64]. Identification of targets associated with EMV biogenesis in response to elevated calcium or adenylate cyclase remains to be elucidated. Therapies targeting the osteosarcoma BME could be designed to either inhibit EMV biogenesis directly or inactivate their bone-destructive, proneoplastic cargo.
In conclusion, this study suggests a novel role of EMVs in driving osteoclastic bone resorption by virtue of their pro-osteoclastogenic cargo and disrupting bone remodeling homeostasis in the osteosarcoma BME.
Appendix A. Supplementary data
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.tranon.2014.04.011.
Acknowledgments
We thank Clarke Anderson for his discussions and expert advice on EMVs. We thank Shrikant Anant for access to ultracentrifuge for isolation of EMVs and helpful suggestions, Lane Christenson and Peggy Petroff for allowing us to use the NanoSight equipment for NTA, Jeremy Chien for access to the ChemiDoc MP system, Marsha Danley for helping with IHC, Barbara Fegley for assistance with the electron microscopy at the Electron Microscopy Research Laboratory, which is supported, in part, by funds from NIH Centers of Biomedical Research Excellence (COBRE) grant 9P20GM104936 and NIH grant S10RR027564. We thank Lynda Bonewald and Sarah Dallas (University of Missouri-Kansas City) for helpful discussions. We thank Van Veldhuizen, Sullivan, and Perez for their support.
Footnotes
Disclosures: None.
Grant support: Funding sources for this study include Division of Hematology and Oncology, Department of Internal Medicine, School of Health Professions, The University of Kansas Cancer Center research support to R.G., and National Institutes of Health (NIH)-National Institutes of Aging Program Project grant P01 AG039355-01-A1 and the Thompson Endowment Fund to M.B.
Authors' contributions: Study conception and design: R.G. Development of methodology: R.G., L.W., and M.B. Study conduct: R.G., L.W., J.I., and M.B. Data collection: R.G., L.W., J.I., J.V., M.S., and M.B. Data analyses: R.G., L.W., J.I., and M.B. Data interpretation: R.G., M.B., O.T., P.R., and R.P. Writing and drafting the manuscript: R.G. Revising and approving manuscript content: R.G., L.W., J.I., J.V., M.S., M.B., O.T., P.R., and R.P. Study supervision: R.G.
This article refers to supplementary materials, which are designated by Figures W1 to W3 and are available online at www.transonc.com.
References
- 1.Thouverey C, Malinowska A, Balcerzak M, Strzelecka-Kiliszek A, Buchet R, Dadlez M, Pikula S. Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J Proteomics. 2011;74:1123–1134. doi: 10.1016/j.jprot.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Garimella R, Eskew J, Bhamidi P, Vielhauer G, Hong Y, Clarke Anderson H, Tawfik O, Rowe P. Biological characterization of preclinical bioluminescent osteosarcoma orthotopic mouse (BOOM) model: a multi-modality approach. J Bone Oncol. 2013;2:11–21. doi: 10.1016/j.jbo.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson HC, Mulhall D, Garimella R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab Invest. 2010;90:1549–1557. doi: 10.1038/labinvest.2010.152. [DOI] [PubMed] [Google Scholar]
- 4.Dolo V, Ginestra A, Cassará D, Ghersi G, Nagase H, Vittorelli ML. Shed membrane vesicles and selective localization of gelatinases and MMP-9/TIMP-1 complexes. Ann N Y Acad Sci. 1999;878:497–499. doi: 10.1111/j.1749-6632.1999.tb07707.x. [DOI] [PubMed] [Google Scholar]
- 5.Ginestra A, La Placa MD, Saladino F, Cassarà D, Nagase H, Vittorelli ML. The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res. 1998;18:3433–3437. [PubMed] [Google Scholar]
- 6.Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr., Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y, Kappes JC, Barnes S, Kimberly RP, Grizzle WE. Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J Immunol. 2006;176:1375–1385. doi: 10.4049/jimmunol.176.3.1375. [DOI] [PubMed] [Google Scholar]
- 8.Clayton A, Turkes A, Dewitt S, Steadman R, Mason MD, Hallett MB. Adhesion and signaling by B cell-derived exosomes: the role of integrins. FASEB J. 2004;18:977–979. doi: 10.1096/fj.03-1094fje. [DOI] [PubMed] [Google Scholar]
- 9.Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011;71:3792–3801. doi: 10.1158/0008-5472.CAN-10-4455. [DOI] [PubMed] [Google Scholar]
- 10.Taraboletti G, D'Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia. 2006;8:96–103. doi: 10.1593/neo.05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim CW, Lee HM, Lee TH, Kang C, Kleinman HK, Gho YS. Extracellular membrane vesicles from tumor cells promote angiogenesis via sphingomyelin. Cancer Res. 2002;62:6312–6317. [PubMed] [Google Scholar]
- 12.Graves LE, Ariztia EV, Navari JR, Matzel HJ, Stack MS, Fishman DA. Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res. 2004;64:7045–7049. doi: 10.1158/0008-5472.CAN-04-1800. [DOI] [PubMed] [Google Scholar]
- 13.Bobrie A, Théry C. Unraveling the physiological functions of exosome secretion by tumors. Oncoimmunology. 2013;2:e22565. doi: 10.4161/onci.22565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Théry C. Exosomes: secreted vesicles and intercellular communications. F1000 Biol Rep. 2011;3:15. doi: 10.3410/B3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muralidharan-Chari V, Clancy JW, Sedgwick A, D'Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123:1603–1611. doi: 10.1242/jcs.064386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee TH, D'Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as mediators of intercellular communication in cancer—the emerging science of cellular 'debris'. Semin Immunopathol. 2011;33:455–467. doi: 10.1007/s00281-011-0250-3. [DOI] [PubMed] [Google Scholar]
- 17.Ancha HR, Kurella RR, Stewart CA, Damera G, Ceresa BP, Harty RF. Histamine stimulation of MMP-1(collagenase-1) secretion and gene expression in gastric epithelial cells: role of EGFR transactivation and the MAP kinase pathway. Int J Biochem Cell Biol. 2007;39:2143–2152. doi: 10.1016/j.biocel.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Savina A, Fader CM, Damiani MT, Colombo MI. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic. 2005;6:131–143. doi: 10.1111/j.1600-0854.2004.00257.x. [DOI] [PubMed] [Google Scholar]
- 19.Islam A, Jones H, Hiroi T, Lam J, Zhang J, Moss J, Vaughan M, Levine SJ. cAMP-dependent protein kinase A (PKA) signaling induces TNFR1 exosome-like vesicle release via anchoring of PKA regulatory subunit RIIβ to BIG2. J Biol Chem. 2008;283:25364–25371. doi: 10.1074/jbc.M804966200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar C. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castellana D, Zobairi F, Martinez MC, Panaro MA, Mitolo V, Freyssinet JM, Kunzelmann C. Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: a role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Res. 2009;69:785–793. doi: 10.1158/0008-5472.CAN-08-1946. [DOI] [PubMed] [Google Scholar]
- 22.Wysoczynski M, Ratajczak MZ. Lung cancer secreted microvesicles: underappreciated modulators of microenvironment in expanding tumors. Int J Cancer. 2009;125:1595–1603. doi: 10.1002/ijc.24479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010;70:9621–9630. doi: 10.1158/0008-5472.CAN-10-1722. [DOI] [PubMed] [Google Scholar]
- 24.Cho JA, Park H, Lim EH, Lee KW. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol. 2012;40:130–138. doi: 10.3892/ijo.2011.1193. [DOI] [PubMed] [Google Scholar]
- 25.Ferrari C, Benassi S, Ponticelli F, Gamberi G, Ragazzini P, Pazzaglia L, Balladelli A, Bertoni F, Picci P. Role of MMP-9 and its tissue inhibitor TIMP-1 in human osteosarcoma: findings in 42 patients followed for 1-16 years. Acta Orthop Scand. 2004;75:487–491. doi: 10.1080/00016470410001295-1. [DOI] [PubMed] [Google Scholar]
- 26.Bjørnland K, Flatmark K, Pettersen S, Aaasen AO, Fodstad O, Maelandsmo GM. Matrix metalloproteinases participate in osteosarcoma invasion. J Surg Res. 2005;127:151–156. doi: 10.1016/j.jss.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 27.Uchibori M, Nishida Y, Nagasaka T, Yamada Y, Nakanishi K, Ishiguro N. Increased expression of membrane-type matrix metalloproteinase-1 is correlated with poor prognosis in patients with osteosarcoma. Int J Oncol. 2006;28:33–42. [PubMed] [Google Scholar]
- 28.Osaki M, Takeshita F, Sugimoto Y, Kosaka N, Yamamoto Y, Yoshioka Y, Kobayashi E, Yamada T, Kawai A, Inoue T. MicroRNA-143 regulates human osteosarcoma metastasis by regulating matrix metalloprotease-13 expression. Mol Ther. 2011;19:1123–1130. doi: 10.1038/mt.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Husmann K, Arlt MJ, Muff R, Langsam B, Bertz J, Born W, Fuchs B. Matrix Metalloproteinase 1 promotes tumor formation and lung metastasis in an intratibial injection osteosarcoma mouse model. Biochim Biophys Acta. 2013;1832:347–354. doi: 10.1016/j.bbadis.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Nahar NN, Missana LR, Garimella R, Tague SE, Anderson HC. Matrix vesicles are carriers of bone morphogenetic proteins (BMPs), vascular endothelial growth factor (VEGF), and noncollagenous matrix proteins. J Bone Miner Metab. 2008;26:514–519. doi: 10.1007/s00774-008-0859-z. [DOI] [PubMed] [Google Scholar]
- 31.Pan Z, Zhao X, Brotto M. Fluorescence-based measurement of store-operated calcium entry in live cells: from cultured cancer cell to skeletal muscle fiber. J Vis Exp. 2012;60:3415. doi: 10.3791/3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2 + indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 33.Zong W, Stein SE, Starcher B, Meyn LA, Moalli PA. Alteration of vaginal elastin metabolism in women with pelvic organ prolapse. Obstet Gynecol. 2010;115:953–961. doi: 10.1097/AOG.0b013e3181da7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez M, Valenzuela MA, Lopez-Otin C, Alvarez J, Lopez JM, Vernal R, Gamonal J. Matrix metalloproteinase-13 is highly expressed in destructive periodontal disease activity. J Periodontol. 2006;77:1863–1870. doi: 10.1902/jop.2006.050461. [DOI] [PubMed] [Google Scholar]
- 35.Wigner NA, Kulkarni N, Yakavonis M, Young M, Tinsley B, Meeks B, Einhorn TA, Gerstenfeld LC. Urine matrix metalloproteinases (MMPs) as biomarkers for the progression of fracture healing. Injury. 2012;43:274–278. doi: 10.1016/j.injury.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanio Y, Yamazaki H, Kunisada T, Miyake K, Hayashi SI. CD9 molecule expressed on stromal cells is involved in osteoclastogenesis. Exp Hematol. 1999;27:853–859. doi: 10.1016/s0301-472x(99)00011-9. [DOI] [PubMed] [Google Scholar]
- 37.Kischel P, Bellahcene A, Deux B, Lamour V, Dobson R, DEP E, Clezardin P, Castronovo V. Overexpression of CD9 in human breast cancer cells promotes the development of bone metastases. Anticancer Res. 2012;32:5211–5220. [PubMed] [Google Scholar]
- 38.Fox SW, Evans KE, Lovibond AC. Transforming growth factor-β enables NFATc1 expression during osteoclastogenesis. Biochem Biophys Res Commun. 2008;366:123–128. doi: 10.1016/j.bbrc.2007.11.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 40.Dougall WC. RANKL signaling in bone physiology and cancer. Curr Opin Support Palliat Care. 2007;1:317–322. doi: 10.1097/SPC.0b013e3282f335be. [DOI] [PubMed] [Google Scholar]
- 41.Kovalenko OV, Yang X, Kolesnikova TV, Hemler ME. Evidence for specific tetraspanin homodimers: inhibition of palmitoylation makes cysteine residues available for cross-linking. Biochem J. 2004;377:407–417. doi: 10.1042/BJ20031037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akuthota P, Melo RC, Spencer LA, Weller PF. MHC Class II and CD9 in human eosinophils localize to detergent-resistant membrane microdomains. Am J Respir Cell Mol Biol. 2012;46:188–195. doi: 10.1165/rcmb.2010-0335OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ikeda T, Kasai M, Suzuki J, Kuroyama H, Seki S, Utsuyama M, Hirokawa K. Multimerization of the receptor activator of nuclear factor-κB ligand (RANKL) isoforms and regulation of osteoclastogenesis. J Biol Chem. 2003;278:47217–47222. doi: 10.1074/jbc.M304636200. [DOI] [PubMed] [Google Scholar]
- 44.Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, Mitchell DA, Bigner DD. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009;23:1541–1557. doi: 10.1096/fj.08-122184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006;20:847–856. doi: 10.1038/sj.leu.2404132. [DOI] [PubMed] [Google Scholar]
- 46.Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Urbanowicz B, Brański P, Ratajczak MZ, Zembala M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol Immunother. 2006;55:808–818. doi: 10.1007/s00262-005-0075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJ, Hole P, Carr B, Redman CW, Harris AL, Dobson PJ. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine. 2011;7:780–788. doi: 10.1016/j.nano.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Webber J, Clayton A. How pure are your vesicles? J Extracell Vesicles. 2013;2 doi: 10.3402/jev.v2i0.19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casimiro S, Mohammad KS, Pires R, Tato-Costa J, Alho I, Teixeira R, Carvalho A, Ribeiro S, Lipton A, Guise TA. RANKL/RANK/MMP-1 molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells in vitro. PLoS One. 2013;8:e63153. doi: 10.1371/journal.pone.0063153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamoureux F, Picarda G, Rousseau J, Gourden C, Battaglia S, Charrier C, Pitard B, Heymann D, Rédini F. Therapeutic efficacy of soluble receptor activator of nuclear factor-κB-Fc delivered by nonviral gene transfer in a mouse model of osteolytic osteosarcoma. Mol Cancer Ther. 2008;7:3389–3398. doi: 10.1158/1535-7163.MCT-08-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lamoureux F, Richard P, Wittrant Y, Battaglia S, Pilet P, Trichet V, Blanchard F, Gouin F, Pitard B, Heymann D. Therapeutic relevance of osteoprotegerin gene therapy in osteosarcoma: blockade of the vicious cycle between tumor cell proliferation and bone resorption. Cancer Res. 2007;67:7308–7318. doi: 10.1158/0008-5472.CAN-06-4130. [DOI] [PubMed] [Google Scholar]
- 52.Uy HL, Guise TA, De La Mata J, Taylor SD, Story BM, Dallas MR, Boyce BF, Mundy GR, Roodman GD. Effects of parathyroid hormone (PTH)-related protein and PTH on osteoclasts and osteoclast precursors in vivo. Endocrinology. 1995;136:3207–3212. doi: 10.1210/endo.136.8.7628353. [DOI] [PubMed] [Google Scholar]
- 53.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massagué J, Mundy GR, Guise TA. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Celotti F, Colciago A, Negri-Cesi P, Pravettoni A, Zaninetti R, Sacchi MC. Effect of platelet-rich plasma on migration and proliferation of SaOS-2 osteoblasts: role of platelet-derived growth factor and transforming growth factor-β. Wound Repair Regen. 2006;14:195–202. doi: 10.1111/j.1743-6109.2006.00110.x. [DOI] [PubMed] [Google Scholar]
- 55.Ota K, Quint P, Weivoda MM, Ruan M, Pederson L, Westendorf JJ, Khosla S, Oursler MJ. Transforming growth factor beta 1 induces CXCL16 and leukemia inhibitory factor expression in osteoclasts to modulate migration of osteoblast progenitors. Bone. 2013;57:68–75. doi: 10.1016/j.bone.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer. 2009;124:2621–2633. doi: 10.1002/ijc.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sawant A, Deshane J, Jules J, Lee CM, Harris BA, Feng X, Ponnazhagan S. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res. 2013;73:672–682. doi: 10.1158/0008-5472.CAN-12-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Danilin S, Merkel AR, Johnson JR, Johnson RW, Edwards JR, Sterling JA. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology. 2012;1:1484–1494. doi: 10.4161/onci.21990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ishii M, Iwai K, Koike M, Ohshima S, Kudo-Tanaka E, Ishii T, Mima T, Katada Y, Miyatake K, Uchiyama Y. RANKL-induced expression of tetraspanin CD9 in lipid raft membrane microdomain is essential for cell fusion during osteoclastogenesis. J Bone Miner Res. 2006;21:965–976. doi: 10.1359/jbmr.060308. [DOI] [PubMed] [Google Scholar]
- 60.Yi T, Kim HJ, Cho JY, Woo KM, Ryoo HM, Kim GS, Baek JH. Tetraspanin CD9 regulates osteoclastogenesis via regulation of p44/42 MAPK activity. Biochem Biophys Res Commun. 2006;347:178–184. doi: 10.1016/j.bbrc.2006.06.061. [DOI] [PubMed] [Google Scholar]
- 61.Mensah KA, Ritchlin CT, Schwarz EM. RANKL induces heterogeneous DC-STAMPlo and DC-STAMPhi osteoclast precursors of which the DC-STAMPlo precursors are the master fusogens. J Cell Physiol. 2010;223:76–83. doi: 10.1002/jcp.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herr MJ, Kotha J, Hagedorn N, Smith B, Jennings LK. Tetraspanin CD9 promotes the invasive phenotype of human fibrosarcoma cells via upregulation of matrix metalloproteinase-9. PLoS One. 2013;8:e67766. doi: 10.1371/journal.pone.0067766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gutiérrez-Martín Y, Bustillo D, Gómez-Villafuertes R, Sánchez-Nogueiro J, Torregrosa-Hetland C, Binz T, Gutiérrez LM, Miras-Portugal MT, Artalejo AR. P2X7 receptors trigger ATP exocytosis and modify secretory vesicle dynamics in neuroblastoma cells. J Biol Chem. 2011;286:11370–11381. doi: 10.1074/jbc.M110.139410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takai E, Tsukimoto M, Harada H, Sawada K, Moriyama Y, Kojima S. Autocrine regulation of TGF-β1-induced cell migration by exocytosis of ATP and activation of P2 receptors in human lung cancer cells. J Cell Sci. 2012;125:5051–5060. doi: 10.1242/jcs.104976. [DOI] [PubMed] [Google Scholar]