

Abstract
More than 130 different mutations in the Cu/Zn superoxide dismutase (SOD1) gene have been associated with Amyotrophic lateral sclerosis (ALS) but the mechanism of this toxicity remains controversial. To gain insight into the importance of the zinc site in the pathogenesis of SOD1 in vivo, we generated a Drosophila model with transgenic expression of a zinc-deficient human SOD1. Expression of zinc-deficient SOD1 in Drosophila resulted in a progressive movement defect with associated mitochondrial cristae vacuolization and reductions in ATP levels. Furthermore, these flies are sensitized to mitochondrial toxins, paraquat and zinc. Importantly, we show that the zinc-deficient SOD1-induced motor defect can be ameliorated by supplementing the endogenous fly respiratory chain machinery with the single-subunit NADH–ubiquinone oxidoreductase (Ndi1) from yeast. These results demonstrate that zinc-deficient SOD1 is neurotoxic in vivo and suggest that mitochondrial dysfunction plays a critical role in this toxicity. The robust behavioral, pathological and biochemical phenotypes conferred by zinc-deficient SOD1 in Drosophila have general implications for the role of the zinc ion in both familial and sporadic ALS.
Keywords: Amyotrophic Lateral Sclerosis (ALS), fly, Ndi1, paraquat, mitochondria, respiration, superoxide dismutase (SOD1)
Introduction
Amyotrophic lateral sclerosis (ALS) patients suffer from progressive paralysis and muscle weakness that will eventually lead to respiratory failure and death. About 90% of ALS patients are sporadic cases whereas the other 10% are familial cases, 20% of which inherit mutations in the copper, zinc-superoxide dismutase (Cu, Zn SOD or SOD1) gene (Rosen et al., 1993; Vucic and Kiernan, 2009). The pathology and symptoms of SOD1-patients closely resemble those of sporadic ALS cases. Therefore, transgenic animal models expressing SOD1 mutations may provide not only insight into the pathogenic mechanisms of both familial and sporadic ALS, but also valuable tools to develop and test therapeutic approaches. Although it is known that mutations result in a toxic gain of function of the SOD1 enzyme, which normally functions as a free radical scavenger, the mechanisms underlying disease progression have not been clearly elucidated (Beckman et al., 2001; Bruijn et al., 2004).
In the last decade, perturbations in mitochondrial energy metabolism have emerged as a focal point in studies to elucidate the molecular mechanisms of aging and age-related diseases (Lin and Beal, 2006; Wallace, 2005). Mitochondria dysfunction has also been implicated in the pathophysiology of ALS (Hervias et al., 2006). Biochemical and morphological mitochondrial abnormalities have been demonstrated in postmortem spinal cords of ALS patients (Borthwick et al., 1999; Wiedemann et al., 2002; Wiedemann et al., 1998) and in rodent models of SOD1-linked ALS (Kong and Xu, 1998; Wong et al., 1995). Although it is not fully understood how mutant SOD1 could damage mitochondria, a number of recent studies have shown that ALS-linked SOD1 mutations are selectively recruited to mitochondria and induce respiratory defects (Ferri et al., 2006; Liu et al., 2004; Pasinelli et al., 2004; Vande Velde et al., 2008).
The SOD1 mutations associated with ALS occur in all of the functional elements of the protein structure, and many SOD ALS mutants have superoxide-scavenging activity comparable to that of wild-type SOD (Vucic and Kiernan, 2009; Bruijn et al., 2004). However, ALS mutations make the SOD1 mutants more susceptible to the loss of their metal cofactors (Crow et al., 1997; Lyons et al., 1996; Trumbull and Beckman, 2009). SOD has approximately a 7000-fold lower affinity for zinc than it does for copper (Crow et al., 1997) and, therefore, zinc is more likely to disassociate than copper. Zinc deficiency dramatically increases the reduction of the active-site copper by low molecular weight reductants such as ascorbate (Estévez et al., 1999). Delivery of zinc-deficient SOD into cultured motor neurons is sufficient to induce apoptosis, associated with increased redox activity of the remaining copper (Estévez et al., 1999). Furthermore, zinc-deficient wild-type SOD was shown to be just as toxic to cultured motor neurons as zinc-deficient mutant SOD. This observation leads to the idea that loss of zinc from wild-type SOD could be involved in the 98% of ALS patients without SOD mutations (Crow et al., 1997; Trumbull and Beckman, 2009). In the zinc-deficient SOD hypothesis, the mutations to SOD do not directly cause the toxic gain-of-function but rather increase the propensity of SOD to become zinc deficient (Crow et al., 1997).
A significant limitation of the ‘zinc deficient’ hypothesis has been a lack of in vivo data. Moreover, although mitochondrial dysfunction has been widely reported as an early event in ALS pathogenesis (Hervias et al., 2006), it is not yet known whether strategies to enhance respiratory chain function can improve the behavioral defects associated with ALS. In this study, we set out to examine the physiological and phenotypic consequences of expression of zinc-deficient SOD1 in the fruit fly Drosophila. A previous attempt to construct a Drosophila model of ALS demonstrated that expression of G41S mutant allele of human SOD in Drosophila motorneurons had no adverse effects on life-span but, somewhat surprisingly, enhanced survival both under normal and under oxidative stress conditions (Elia et al., 1999). Later studies also confirmed that expression of mutant SOD alleles in Drosophila does not result in a dominant gain-of-function as observed in humans and mice (Mockett et al., 2003). Recently, a Drosophila model of ALS was constructed by transgenic expression of mutant SOD1 (A4V and G85R) in motorneurons using D42-GAL4 driver; the resulting strains exhibit a progressive loss of locomotor function along with an impaired neural circuit electrophysiology (Watson et al., 2008). While the latter models mimic certain phenotypes of ALS, these SOD1 alleles are not ideal for the purpose of our study in that the relatively short lifespan of the fruit flies may either be insufficient for these mutant alleles of SOD1 to lose their zinc-cofactor or it may simply be too late in the life cycle to induce any visible mitochondrial phenotypes. Consequently, we have constructed transgenic flies expressing zinc-deficient SOD throughout their entire lifespan. We report that zinc-deficient SOD1 expression confers an age-related movement disorder, a hallmark of ALS in humans. Moreover, zinc-deficient SOD1 expression produces alterations in mitochondrial structure as well as ATP production and sensitizes flies to mitochondrial toxins such as paraquat and zinc. Finally, we demonstrate that transgenic enhancement of respiratory chain function ameliorates the motor defect associated with zinc-deficient SOD1, suggesting that mitochondrial dysfunction may be an important intermediate in development of ALS associated phenotypes.
Results
Transgenic expression of D83S SOD (zinc-deficient SOD1 or CuSOD) in Drosophila
D83S is a zinc-deficient mutant SOD in which the zinc-binding residue Asp83 was mutated to Ser using PCR mutagenesis (Roberts et al., 2007). In this mutant protein, the zinc-binding site has no electron density for zinc; the copper-binding site, however, maintains its electron density for copper and the coordination of copper remains similar to that of wild-type SOD. Here, we expressed D83S SOD in flies using the GAL4/UAS system. We transformed flies with UAS-constructs containing the D83S SOD gene and performed ten rounds of backcrossing into a w1118 background. In all subsequent experiments, w1118 was used as a control strain. Except for the insertion of the UAS-D83S SOD transgene, these control flies were genetically very similar to the experimental flies. Activation of UAS-D83S transgene under the control of the ubiquitous da-GAL4 driver strongly (more than 5-fold) induces expression of SOD protein in transgenic flies, as assayed by Western blot analysis (Figure 1A). Next, we examined SOD activity in UAS-D83S transgenic flies to determine whether or not D83S mutant SOD maintains its catalytic activity. As Figure 1B illustrates, expression of D83S SOD under the control of da-GAL4 confers additional SOD activities in adult flies, as evaluated by an in-gel NBT SOD assay. These activities are attributed to production of Cu, Zn SOD-CuSOD heterodimers and CuSOD-CuSOD dimers upon expression of D83S SOD1 transgene.
Fig. 1.
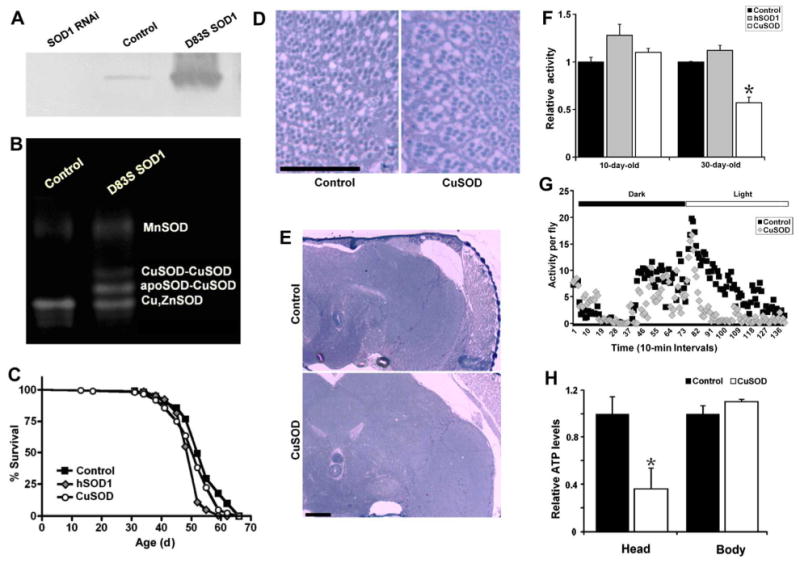
Ubiquitous expression of zinc-deficient superoxide dismutase (SOD1, CuSOD, or D83S SOD1) in Drosophila produces locomotor defects without neurodegeneration or shortened life span. (A) Western blot analysis of SOD protein level in adult fly extracts confirmed daGAL4-mediated expression of the CuSOD transgene (third lane). The first lane indicates the knockdown of SOD protein on daGAL4-mediated activation of a UAS-SOD1-RNAi transgene. (B) Expression of CuSOD leads to formation of heterodimers between wild type SOD and CuSOD. SOD activity was measured using the in-gel nitroblue tetrazolium assay. (C) Expression of wild type human SOD1 (hSOD1) or CuSOD has a minor negative effect on the adult life span. (D) and (E) Expression of CuSOD does not promote neurodegeneration in Drosophila eye or brain (60-day-old flies; scale bar, 50 μm). (F) Expression of CuSOD significantly compromises locomotor activity (p=0.002) in 30-day-old flies. Ubiquitous expression of wild type hSOD1, however, had no adverse effect on physical activity in young (p=0.067) or old (p = 0.093) flies. (G) Representative activity pattern for 30-day-old flies during a 24-hour period. (H) Expression of CuSOD lowers ATP levels in heads (p=0.029) but not the rest of the body (p=0.21) of 10-day-old flies. Genotypes were as follows: control (da-Gal4/+), SOD1 RNAi (da-Gal4/UAS-SOD1-RNAi); and D83S SOD1 or CuSOD (da-Gal4/UAS-D83S), hSOD1 (da-Gal4/UAS-hSOD1). For (F) and (H), the significance of the difference (Note that RNAi is an abbrevation for double stranded interference RNA.) between means was analyzed using 1-way analysis of variance. Abbreviations: apoSOD, endogenous Drosophila SOD1 monomer; MnSOD, manganese superoxide dismutase (SOD2).
Effects of zinc-deficient SOD1 on fly lifespan, neurodegeneration and motor activity
Next, we tested the effects of zinc-deficient SOD1 on longevity, neurodegeneration and motor performance of adult flies. As Figure 1C and Table S1 illustrate, ubiquitous expression of wild-type human SOD1 under the control of the da-GAL4 driver slightly shortens lifespan of adult flies (5%-7%). Importantly, expression of zinc-deficient SOD1 with the same GAL4 drives had no impact, negative or positive, on adult longevity (Figure 1C).
Next, we investigated whether expression of zinc-deficient SOD1 resulted in gross neurodegeneration in the fly brain or retina. As figures 1D and 1E illustrate, no gross anatomical degeneration of the brain or retina was observed in response to zinc-deficient SOD1 expression even at 60 days of age. Finally, we assessed motor activity in zinc-deficient SOD1 transgenic flies by measuring and comparing their locomotor activity to control lines; physical activity was quantified using a Drosophila Activity Monitor (DAM) system, in which two rings of infrared beams record fly movement in a small chamber. As Figure 1F and 1G illustrates, expression of zinc-deficient SOD1 produces an age-related movement disorder. At 10 days of age, zinc-deficient SOD1 expressing flies did not display reduced movement compared to isogenic controls. At 30 days of age, however, ubiquitous expression of zinc-deficient SOD1 reduces physical activity by 43% compared to isogenic controls; ubiquitous expression of wild-type human SOD1 had no adverse effect on physical activity. Superficially at least, this phenotype of our fly model is similar to that of ALS patients who suffer from a progressive paralysis.
Finally, to determine whether expression of zinc-deficient SOD resulted in a respiratory deficit, we measured ATP production in these flies. In doing so, we observed that ATP levels were decreased by >60% in the heads of flies expressing zinc-deficient SOD1 (Figure 1H).
Expression of zinc-deficient SOD leads to alterations in mitochondrial structure
A growing body of evidence suggests that mitochondrial dysfunction may play an important role in the pathophysiology of ALS (Hervias et al., 2006). Here, we examined the impact of zinc-deficient SOD1 expression on mitochondrial structure and function in Drosophila. Using electron microscopy, we identified a striking mitochondrial pathology within the flight muscle in response to zinc-deficient SOD1 expression. Insect flight muscle is well-suited for studies of mitochondrial ultrastructure. The mitochondria typically lie in columns, aligned longitudinally between the myofibrils (Figure 2). The cristae within these mitochondria are packed very densely so that any changes in their configuration are easily seen. Zinc-deficient SOD1 expression did not affect the structure of the myofibrils. Expression of wild-type human SOD1 (hSOD1) did not affect mitochondrial ultrastructure (Figure 2B, 2E, 2H, 2K). However, we observed a localized rearrangement of the cristae within individual mitochondria of zinc-deficient SOD1 flies (Figure 2C, 2F, 2I, 2L). These observations were consistent for both 10-day-old and 30-day-old flies, with cristae derangement in zinc-deficient SOD1 flies being more severe on day 30 compared to day 10 (Figure 2).
Fig. 2.
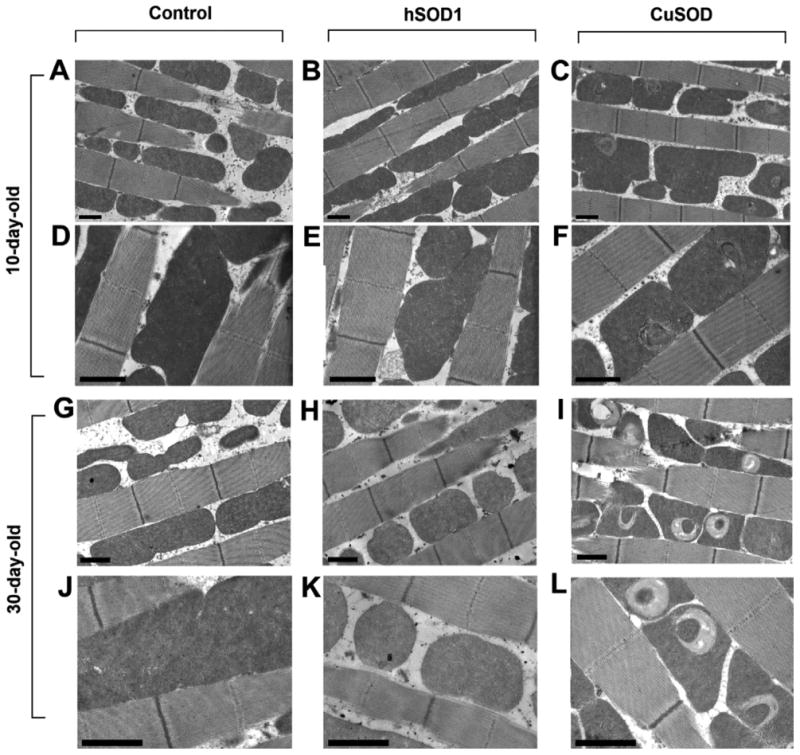
Ubiquitous expression of zinc-deficient SOD (or CuSOD) in Drosophila deteriorates mitochondrial structure. Electron micrographs of flight muscle of 10-day-old flies (A–F) and 30-day-old flies (G–L). (A), (D), (G), and (J) control flies show normal mitochondrial ultrastructure whereas expression of CuSOD (C), (F), (I), (L) but not wild-type human superoxide dismutase (hSOD1) (B), (E), (H), (K) produces a localized rearrangement of the cristae within individual mitochondria (scale bar, 1 μm). CuSOD phenotypes were more severe on day 30 compared to day 10. Genotypes were as follows: Control (da-Gal4/+), hSOD1 (da-Gal4/UAS-hSOD1), and CuSOD (da-Gal4/UAS-D83S).
Expression of zinc-deficient SOD1 in Drosophila neurons and glial cells produces locomotor defects
To test the tissue-specific effects of zinc-deficient SOD1 in Drosophila, we analyzed locomotor function and lifespan of flies expressing zinc-deficient SOD1 in neurons, glial cells, and muscle cells respectively (Figure 3A-H). As with ubiquitous expression, expression of zinc-deficient SOD1 mediated by the pan-neuronal GAL4 driver Elav-GAL4 had no major impact on longevity (Figure 3A; Table S1). Using the D42-GAL4 driver, we observed that expression of wild-type human SOD in motor neurons produces a moderate decrease in lifespan but we observed no further deficit in zinc-deficient SOD1-expressing flies (Figure 3B; Table S1). Expression of zinc-deficient SOD1 mediated by the glial driver Repo-GAL4 (Figure 3C; Table S1) or the muscle driver MHC-GAL4 (Figure 3D; Table S1) did not produce major effects on longevity.
Fig. 3.
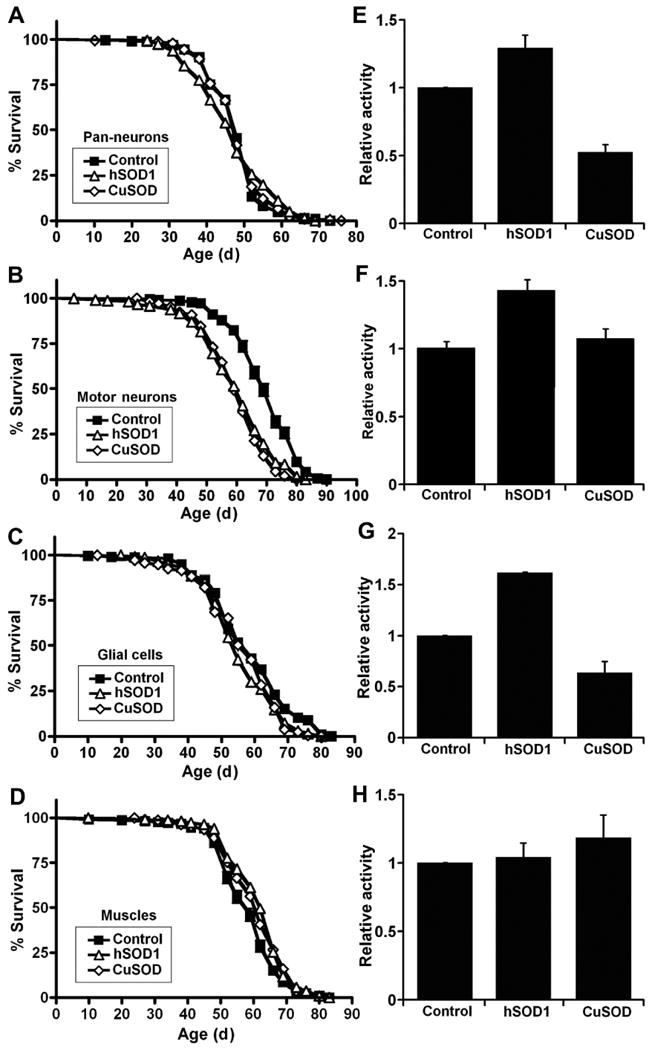
Expression of zinc-deficient superoxide dismutase (SOD1, CuSOD, or D83S) in Drosophila neurons and glial cells produces locomotor defects without major changes in life span. (A–D) Expression of zinc-deficient SOD (CuSOD) or human SOD1 (hSOD1) in Drosophila neurons, glial cells, or muscles has a minor effect on adult life span. Expression of CuSOD in (E) neurons (p=0.0003), (F) motor neurons (p=0.014) and (G) glial cells (p=0.0001) but not (H) muscles (p=0.27) produces locomotor defects in 30-day-old flies as compared to wild-type hSOD1 flies. Genotypes were as follows: (A) and (E) Control: Elav-GAL4/+; hSOD1: Elav-GAL4/UAS-hSOD1; CuSOD: Elav-GAL4/UAS-D83S. (B) and (F) Control: D42-GAL4/+; hSOD1: D42-GAL4/UAS-hSOD1; CuSOD: D42-GAL4/UAS-D83S. (C) and (G) Control: Repo-GAL4/+; hSOD1: Repo-GAL4/UAS-hSOD1; CuSOD: Repo-GAL4/UAS-D83S. (D) and (H) Control: MHC-GAL4/+; hSOD1: MHC-GAL4/UAS-hSOD1; CuSOD: MHC-GAL4/UAS-D83S. For (E), (F), (G), and (H), the significance of the difference between means was analyzed using one-way analysis of variance.
Next, using the Drosophila Activity Monitor, we examined the tissue-specific requirements for the zinc-deficient SOD1-induced movement defect. We assayed physical activity in 30-day-old flies and observed that expression of zinc-deficient SOD1 in neurons and glial cells, but not muscles, reduces physical activity by about 40% to 50% compared to wild-type hSOD1 (Figure 3E-3H). Expression of zinc-deficient SOD1 mediated by the pan-neuronal Elav-GAL4 (Figure 3E) and the glial driver Repo-GAL4 (Figure 3G) produced a motor defect compared to isogenic controls. As expression of zinc-deficient SOD1 mediated by the pan-neuronal Elav-GAL4 produced the most robust movement defect, we focused on characterizing this model. In comparison to aged (30-day-old) flies where physical activity was reduced by almost 48% (Figures 3E and S1B), pan-neuronal expression of zinc-deficient SOD1 in young (10-day-old) flies reduced physical activity by only 13% (Figure S1A), indicating that neuronal expression of zinc-deficient SOD1 causes an age-related decline in motor activity.
Neuronal expression of zinc-deficient SOD1 reduces ATP levels and sensitizes flies to paraquat and zinc toxicity
To investigate the mechanism by which neuronal zinc-deficient SOD1 expression results in a locomotor defect, we focused on the role of mitochondrial energy metabolism. Firstly, to determine whether expression of zinc-deficient SOD1 affects overall energy production in neuronal tissue, we examined ATP levels in heads of zinc-deficient SOD1-expressing flies and controls. In doing so, we observed a significant decrease in ATP levels in the heads of flies expressing zinc-deficient SOD1 (Figure 4A). Next, we examined whether neuronal expression of zinc-deficient SOD1 affected tolerance to dietary paraquat (PQ), which is widely used as a redox cycler to stimulate superoxide production in organisms, cells, and mitochondria. We observed that neuronal expression of zinc-deficient SOD1 greatly sensitized flies to PQ toxicity (Figure 4B). In contrast, neuronal expression of wild-type hSOD1 increased flies' resistance to PQ (Figure 4C).
Fig. 4.
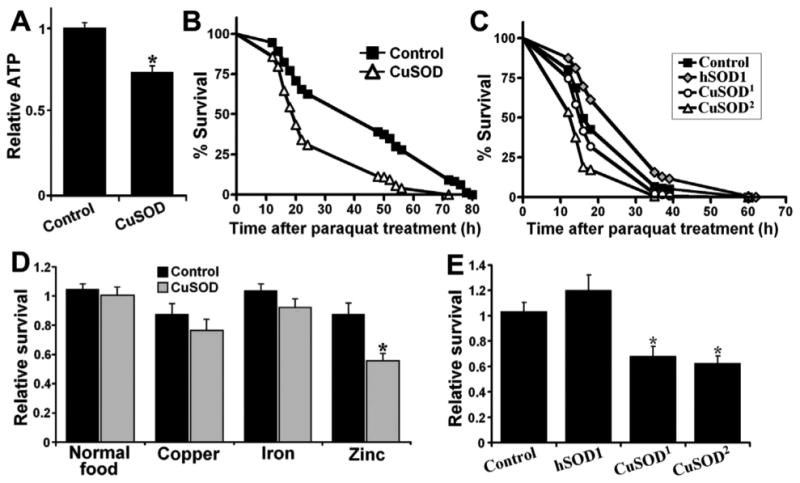
Neuronal expression of zinc-deficient SOD (CuSOD) sensitizes flies to zinc and paraquat toxicity. (A) Neuronal expression of zinc-deficient superoxide dismutase (SOD1 or CuSOD) leads to a decrease in ATP levels of fly heads (p=0.005). (B) and (C) Neuronal expression of CuSOD sensitizes flies to paraquat toxicity, whereas expression of wild-type human SOD1 (hSOD1) enhances resistance to paraquat (p<0.0001). (D) Neuronal expression of CuSOD sensitizes larvae to zinc (p=0.003) but not iron or copper toxicity (p>0.05). Relative survival represents eclosion ratio of females (Elav-GAL4/+ for control, Elav-GAL4/UAS-D83S for CuSOD, Elav-GAL4/UAS-hSOD1 for hSOD1) to male files (+/+ for control, +/UAS-D83S for CuSOD, +/UAS-hSOD1 for hSOD1) on different media. (E) Expression of hSOD1 does not sensitize flies to zinc toxicity. Genotypes were as follows: control (Elav-GAL4/+), hSOD1 (Elav-GAL4/UAS-hSOD1), CuSOD1 (Elav-GAL4/UAS-D83S1), CuSOD or CuSOD2 (Elav-GAL4/UAS-D83S2). For panels (A), (D), and (E), the significance of the difference between means was analyzed using 1-way analysis of variance. For (B) and (C), the significance of the difference between survival curves was analyzed using log-rank test.
Next, we assayed whether zinc-deficient SOD1 affects tolerance to the metals copper, iron and zinc. Neuronal expression of zinc-deficient SOD1 results in decreased tolerance to zinc but not to copper or iron (Figure 4D). In contrast, neuronal expression of wild-type hSOD1 did not alter sensitivity to zinc (Figure 4E).
Neuronal expression of Ndi1 restores ATP levels and motor function of zinc-deficient SOD1 flies
To further investigate the relationship between mitochondrial dysfunction and the locomotor phenotype associated with zinc-deficient SOD1, we set out to enhance respiratory chain activity in zinc-deficient SOD1 flies. Recently, we reported the generation of a novel transgenic approach to supplement the endogenous respiratory chain machinery of the fly with the alternative single-subunit NADH–ubiquinone oxidoreductase (Ndi1) of the baker's yeast Saccharomyces cerevisiae (Bahadorani et al., 2010). Expression of Ndi1 in wild-type flies leads to an increase in NADH–ubiquinone oxidoreductase activity, oxygen consumption and ATP levels (Bahadorani et al., 2010). Here, we have extended this study to zinc-deficient SOD1 flies. To eliminate the possible contribution of induction of endogenous genes at the site of insertion of the Ndi1 transgene, we tested two independent insertions of the UAS-Ndi1 transgene. We observed that neuronal expression of Ndi1 can increase ATP levels in heads of zinc-deficient SOD1 flies (Figure 5B). Next, we set out to determine whether Ndi1 expression could improve motor function in zinc-deficient SOD1 flies. Using the Drosophila Activity Monitor, we assayed physical activity in zinc-deficient SOD1 flies with and without Ndi1 gene expression. In doing so, we observed that Ndi1 gene expression in neurons is sufficient to ameliorate the movement disorder associated with zinc-deficient SOD1 expression (Figure 5C), suggesting that the zinc-deficient SOD1 locomotor defects are linked to respiratory chain defects.
Fig. 5.
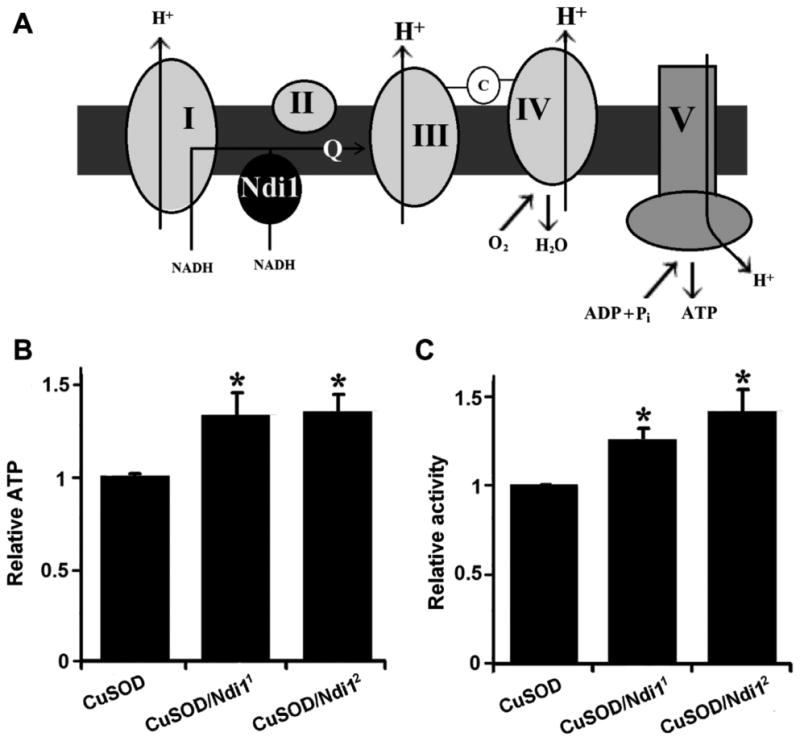
Neuronal expression of NADH–ubiquinone oxidoreductase increases ATP production and rescues locomotor defects of zinc-deficient superoxide dismutase (SOD1, CuSOD, or D83S) flies. (A) Schematic drawing of the mechanism of NADH-ubiquinone oxidoreductase (Ndi1) enhanced energy production in the electron transport chain. Coexpression of the single-subunit yeast Ndi1 with CuSOD (B) increases ATP levels in heads and (C) boosts physical activity in 30-day-old flies. The significance of the difference between means was analyzed using 1-way analysis of variance (p < 0.05). Genotypes were as follows: CuSOD (Elav-GAL4/Y; +/UAS-GFP; +/UAS-D83S), CuSOD/Ndi11 (Elav-GAL4/Y; +/UAS-Ndi1; +/UAS-D83S), CuSOD/Ndi12 (Elav-GAL4/Y; +; UAS-Ndi1/UAS-D83S). Abbreviations: ADP, adenosine diphosphate; C, cytochrome c; Pi, inorganic phosphate; Q, coenzyme Q; +, a wild-type chromosome containing no mutations or transgenes.
Discussion
To better understand the mechanism by which mutations in SOD1 leads to the neurodegenerative manifestations of ALS, we have developed a Drosophila model that expresses a zinc-deficient from of human SOD1. To date, most studies have failed to demonstrate any significant phenotype arising from the expression of human mutant ALS-associated human ALS SOD1 in Drosophila. Gradual loss of the Zn cofactor has been identified as a critical pathogenic event in the neurodegenerative phenotypes associated with ALS. It has been argued that a possible shortcoming of the Drosophila model might be that the short adult lifespan is insufficient for the gradual loss of the Zn cofactor and the ensuing pathology to manifest phenotypically. To address this possibility, we have used a mutant form of human SOD1 (D83S SOD) that is structurally incapable of binding Zn but retains the catalytically-important Cu cofactor along with robust enzymatic activity. Expression of zinc-deficient SOD1 (D84S SOD) in drosophila confers a robust, age-dependent movement disorder reminiscent of ALS in humans. Expression of zinc-deficient SOD1 in neurons or glial cells is sufficient to impair motor function in aging flies. The zinc-deficient SOD1-associated movement defect is accompanied by mitochondrial structural damage, presumably due to zinc-deficient SOD1 penetration into mitochondria owing to the absence of the zinc cofactor (Field et al., 2003). Hypothetically, it could be possible that the consequent penetration and accumulation of zinc-deficient SOD1 in mitochondria may be the cause of cristae disruption and mitochondrial vacuolization in these flies.
We provide genetic evidence that such respiratory defect underlies the movement disorder, and that expression of the alternative single-subunit yeast NADH–ubiquinone oxidoreductase (Ndi1) is sufficient to enhance ATP levels and motor activity of zinc-deficient SOD1 flies. These findings support the link between zinc-deficient SOD1 and mitochondrial respiratory chain dysfunction.
Expression of zinc-deficient SOD1 produces an age-related movement defect without shortening lifespan
We did not detect any major effect on longevity or neuronal integrity in response to zinc-deficient SOD1 expression in Drosophila. Similar negative findings have been reported with expression of ALS-associated SOD1 mutations in Drosophila (Elia et al., 1999; Mockett et al., 2003; Watson et al., 2008; Parkes et al., 1998). Although the reasons for this are unclear, it is possible that the short adult lifespan of Drosophila relative to mammals may preclude the manifestation of all but the most catastrophic neurodegenerative phenotypes.
It is worth noting that expression of wild-type human SOD1 (hSOD1) in neurons and glial cells significantly increases physical activity (Figure 3A and 3C). Similar findings were recently reported and attributed to increased production of hydrogen peroxide and the consequent alterations in Drosophila activity (Grover et al., 2009). Therefore, it is particularly striking that zinc-deficient SOD1 expression leads to a decrease in physical activity.
Mitochondrial respiratory chain dysfunction underlies the zinc-deficient SOD1-induced movement defect: a novel therapeutic strategy to treat ALS
A number of previous studies have reported that ALS-linked mutant forms of SOD1 can produce mitochondrial respiratory defects (Ferri et al., 2006; Liu et al., 2004; Pasinelli et al., 2004; Vande Velde et al., 2008), presumably through mechanisms involving nitric oxide-mediated toxicity (Cassina et al., 2008; Chen et al., 2010). Nitric oxide is synthesized by the enzyme nitric oxide synthase and associates with mitochondrial dysfunction (Park et al., 2011). Previous studies demonstrated that supplementation with nitric oxide synthase inhibitors prevented neuronal loss and ameliorated phenotypes of G93A-mSOD1 ALS mice (Cassina et al., 2008; Chen et al., 2010). These observations suggest that mitochondrial dysfunction may be a central mechanism of mutant SOD toxicity. Considering that zinc-deficient SOD neurotoxicity is also dependent on endogenous production of nitric oxide (Estévez et al., 1999), we hypothesized that mitochondrial dysfunction may be responsible for locomotor defects in our ALS flies. Our results demonstrate that a mutation affecting the zinc-binding site of SOD1 is sufficient to induce mitochondrial vacuolization and to reduce ATP levels. Furthermore, zinc-deficient SOD1-expressing flies are severely sensitized to mitochondrial toxins paraquat and zinc: paraquat lowers mitochondrial complex I activity while zinc ion interacts with nitric oxide to induce mitochondrial dysfunction and neuronal cell death (Bossy-Wetzel et al., 2004; Tawara et al., 1996).
Similar to our findings, one of the major pathological changes in transgenic mouse models of ALS includes mitochondrial vacuolization resulting from expansion of mitochondrial intermembrane space and extension of the outer membrane (Xu et al., 2004). Our interpretation of these findings is that it is the loss of zinc from the ALS-linked mutant SODs that leads to mitochondrial structural damage. While the etiology of this toxicity is unknown, the fact that loss of zinc cofactor enhances SOD1 penetration into mitochondria proposes a potential mechanism for zinc-deficient SOD1-mediated mitochondrial vacuolization (Field et al., 2003). It is also worth noting that when we assayed ATP levels, zinc-deficient SOD1 flies displayed a defect in heads but not in the rest of the body. Thus, it is possible that zinc-deficient SOD1-induced mitochondrial damage is more pronounced in neuronal tissue.
In rodent models of SOD1-ALS, mitochondrial dysfunction precedes behavioral manifestations of the disorder (Hervias et al., 2006). In a similar fashion, while robust behavioral defects were observed in 30-day-old zinc-deficient SOD1 flies, we detected alterations in mitochondrial structure and function in 10-day old flies. An important advance reported in the present study is the finding that the locomotor defect conferred by zinc-deficient SOD1 can be treated by expression of the Ndi1 enzyme. In the first place, the observation that genetic enhancement of the respiratory chain can suppress zinc-deficient SOD1 toxicity provides a compelling argument that mitochondrial dysfunction underlies the movement disorder. Secondly, this finding suggests that the Ndi1 gene may be an effective therapeutic target to counteract ALS symptoms. Previously, it has been shown that Ndi1 expression, mediated by a recombinant adeno-associated virus, can function in mammalian mitochondria in vivo and protect against mitochondrial insults (Barber-Singh et al., 2009; Marella et al., 2008; Marella et al., 2009). Our findings in flies lend further weight to the idea that that Ndi1 gene therapy as well as other approaches to enhancing mitochondrial ATP output in neurons hold promise for ameliorating the devastating effects of ALS.
Experimental Procedures
Fly stocks and maintenance
To ensure that female egg laying behavior did not interfere with survival and behavioral assays, male flies were used throughout the study. Fly food was prepared using the same protocol to that described previously (Bahadorani et al., 2010): 1 liter of stock medium consisted of 19 g of sucrose, 38 g of dextrose, 10 g of agar, 91 g of cornmeal, 30 g of yeast, 11 ml of propionic acid, and 1.5 g of tegosept. Metal-supplemented media were prepared by addition of iron (III) sulfate hydrate (10 mM), zinc chloride (10 mM) or copper (II) sulfate (1 mM) into normal fly food. The selected concentrations are partially lethal to developing Drosophila larvae (Bahadorani and Hilliker, 2009).
The GAL4 drivers Elav-GAL4, Repo-GAL4, and MHC-GAL4 are expressed in Drosophila neurons, glial cells, and muscles, respectively; da-GAL4 is expressed ubiquitously. UAS-SOD1-IR (or UAS-SOD1-RNAi), UAS-hSOD1, and UAS-Ndi1 transgenic flies were described previously (Bahadorani et al., 2010; Parkes et al., 1998; Wicks et al., 2009). UAS-GFP was obtained from the Bloomington Stock Center (stock 1521). UAS-D83S transgenic flies, also referred to as CuSOD or zinc-deficient SOD1, were constructed by cloning human D83S mutant SOD cDNA into the pUAST transformation vector, after which the pUAST transformation vector and the p(Δ2-3) helper plasmid were co-injected into w1118 embryos using standard procedures. To ensure that the genetic background was not a confounding factor in our study, UAS-hSOD1, UAS-D83S, UAS-GFP and UAS-Ndi1 transgenic flies were backcrossed into the w1118 background ten times.
Western blot analysis
Twelve adult flies from each genotype were homogenized in 1.5 ml polypropylene centrifuge tubes containing 100 μl of ice-cold 50 mM Tris-HCl, 1 mM EDTA, pH 7.2. The homogenate was centrifuged at 12,000 g for 5 min and then the protein concentration was determined using the Bio-Rad Protein Dye Reagent (Bio-Rad Canada, Mississauga, Ont) based on the method of Bradford (Bradford, 1976). The protein concentration was adjusted to 2 μg/μl in all samples, then mixed with an equal volume of SDS-PAGE sample loading buffer and boiled for 5 min. The samples were loaded into a 15% separating gel (10 μg/lane) and ran at 200 V. Following electrophoresis, the proteins were electroblotted onto a PVDF membrane and subsequently, the membrane was dried for 2 h at room temperature. The membrane was probed with a sheep anti-human SOD1 antibody (Stressgen, Victoria, BC) diluted 1:5000 and detected with a donkey anti-sheep HRP conjugate (Millipore, Mississauga, Ont) diluted 1:75,000 and finally visualized with the ECL Plus Detection Reagents (GE Life Sciences). Image was obtained with a digital camera (Minolta DiMAGE X).
SOD activity assay
SOD activity was measured using the nitroblue tetrazolium (NBT) in-gel assay. In brief, a total of 10 flies were homogenized in 100 μl of homogenizing buffer (1% Triton X-100), centrifuged at 10 000 g for 5 minutes at 4°C, and the supernatant was mixed at 1:1 ratio with 2× loading buffer (0.125 M Tris-HCl pH=6.8, 20% Glycerol, and 0.1% bromophenol blue). Afterward, samples were loaded on 10% acrylamide gels and run for 2 hours at 20 mA at 4°C. Each gel was soaked in 25 ml of NBT solution (50 mg of NBT dissolved in 25 ml of ddH2O) for few minutes and then transferred into 100 ml of stain solution (0.63 g of K2HPO4 pH 7.8, 1.3 mg of riboflavin, and 300 μl of TEMED dissolved in 100 ml of ddH2O) for 7 minutes while exposed to intense light. Thereafter, the gels were rinsed and soaked in water and were placed on a light box until the desired contrast was obtained. Image was obtained with a digital camera (Minolta DiMAGE X).
Lifespan assay
Lifespan studies were performed at 25°C on normal food on a 12h:12h light:dark cycle. A total of 150-180 newly-eclosed males (initially 30 per vial) were tested for each genotype, with survivors transferred to fresh food every 3-4 days. For each longevity assay, control and experimental lifespans were determined at the same time.
Physical Activity
Physical activity was measured using the same protocol to that described previously (Bahadorani et al., 2010). For measuring motor activity, 30 adult male flies were placed in a Drosophila activity meter (TriKinetics Inc., Massachusetts). Movements were recorded continuously under normal culturing conditions for about 24 hours on a 12:12-hour dark:light cycle. Triplicate or more samples were used for each activity measurement.
Electron and light microscopy
Flies were fixed in 1% glutaraldehyde, 1% paraformaldehyde in 0.1M phosphate buffer, 0.9% sodium chloride (PBS) overnight and washed. After postfixation in 1% OsO4 in PB for 1.5-2 hours, the tissues were dehydrated in a graded series of ethanol, treated with propylene oxide and embedded in Eponate 12 (Ted Pella). Sections of the dorsal IFM were cut on a Reichert-Jung Ultracut E ultramicrotome at approximately 60-70 nm thick and picked up on formvar coated copper grids. The sections were stained with uranyl acetate and Reynolds lead citrate and examined on a JEOL 100CX electron microscope at 80kV. Horizontal sections (1 um thick) of fly heads were mounted on glass slides and stained with 1% toluidine blue in 1% sodium borate, after which samples were examined with light microscope. For light microscopy, sections were 1.25 micrometer thick and were analyzed using the Nikon Microphot-FXA microscope.
ATP assay
Fly heads (10 to 20) were homogenized in 100 μl of 6 M guanidine-HCl in extraction buffer (100 mM Tris and 4 mM EDTA, pH 7.8) and boiled for 5 minutes. The samples were then centrifuged to collect the supernatant, which was then diluted (1/750) with extraction buffer. The luminescence was measured by a luminometer using autoinjection of the luminescent solution (Invitrogen: “ATP determination kit”), and the results were compared to the standards. The ATP level was then calculated by dividing the luminescence by the total protein concentration, which was determined by the Bradford method.
Stress resistance
Paraquat resistance: male flies were aged for 10 days, starved for 6 h and then transferred to vials containing a 2.4cm glass fiber filter circles (Whatman) wetted with 150 μl of 30 mM paraquat (Sigma) in 5% sucrose solution. Flies were kept in the dark at all times, except for scoring death and the daily addition of 100 μl 30mM paraquat solution.
Heavy metal resistance: w1118 control or UAS transgenic female flies were crossed to Elav-GAL4 males (GAL4 located on the X-chromosome), aged on normal food for 3-4 days, and then transferred to metal-supplemented medium for 2-3 days. Afterward, adult flies were removed from vials and eclosion rate was monitored by normalizing female eclosion rate to that of males.
Statistical analysis
Unless specified otherwise, all genotypes were compared to w1118 control flies. Bars are expressed as mean ± standard error of the mean (SEM) of triplicate or more samples. The significance of the difference between means was analyzed using one-way analysis of variance (ANOVA). For survival assays, the significance of the difference between longevity curves was analyzed using the log-rank test.
Acknowledgments
This work was partially supported by an operating grant from the Natural Science and Engineering Research Council of Canada to A.J.H. The authors would like to thanks Dr. David W. Walker for his support and contributions on the mitochondrial and rescue assays. The authors would also like to thank Marianne Cilluffo for help with electron microscopy and Peter Anderson for assisting with construction of the UAS-D83S construct, and the Drosophila Stock Center (Bloomington) for fly stocks.
Footnotes
Disclosure Statement: We declare that there are no actual or potential conflicts of interest related to this work.
References
- Bahadorani S, Cho J, Lo T, Contreras H, Lawal HO, Krantz DE, Bradley TJ, Walker DW. Neuronal expression of a single-subunit yeast NADH-ubiquinone oxidoreductase (Ndi1) extends Drosophila lifespan. Aging Cell. 2010;9:191–202. doi: 10.1111/j.1474-9726.2010.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadorani S, Hilliker AJ. Biological and Behavioral Effects of Heavy Metals in Drosophila melanogaster Adults and Larvae. J Insect Behavior. 2009;22:399–411. [Google Scholar]
- Bahadorani S, Hur JH, Lo T, Jr, Vu K, Walker DW. Perturbation of mitochondrial complex V alters the response to dietary restriction in Drosophila. Aging cell. 2010;9:100–103. doi: 10.1111/j.1474-9726.2009.00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber-Singh J, Seo BB, Nakamaru-Ogiso E, Lau YS, Matsuno-Yagi A, Yagi T. Neuroprotective effect of long-term NDI1 gene expression in a chronic mouse model of Parkinson disorder. Rejuvenation Res. 2009;12:259–267. doi: 10.1089/rej.2009.0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999;46:787–790. doi: 10.1002/1531-8249(199911)46:5<787::aid-ana17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Schölzke MN, Harrop A, Mathews E, Götz T, Han J, Ellisman MH, Perkins GA, et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de León A, Robinson KM, Mason RP, Beckman JS, Barbeito L, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28:4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Northington FJ, Martin LJ. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct Funct. 2010;214:219–234. doi: 10.1007/s00429-009-0226-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- Elia AJ, Parkes TL, Kirby K, St George-Hyslop P, Boulianne GL, Phillips JP, Hilliker AJ. Expression of human FALS SOD in motorneurons of Drosophila. Free Radic Biol Med. 1999;26:1332–1338. doi: 10.1016/s0891-5849(98)00333-5. [DOI] [PubMed] [Google Scholar]
- Estévez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS, Carrì MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field LS, Furukawa Y, O'Halloran TV, Culotta VC. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem. 2003;278:28052–28059. doi: 10.1074/jbc.M304296200. [DOI] [PubMed] [Google Scholar]
- Grover D, Ford D, Brown C, Hoe N, Erdem A, Tavaré S, Tower J. Hydrogen peroxide stimulates activity and alters behavior in Drosophila melanogaster. PLoS One. 2009;4:e7580. doi: 10.1371/journal.pone.0007580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervias I, Beal MF, Manfredi G. Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve. 2006;33:598–608. doi: 10.1002/mus.20489. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Lyons TJ, Liu H, Goto JJ, Nersissian A, Roe JA, Graden JA, Café C, Ellerby LM, Bredesen DE, Gralla EB, et al. Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc Natl Acad Sci U S A. 1996;93:12240–12244. doi: 10.1073/pnas.93.22.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M, Seo BB, Nakamaru-Ogiso E, Greenamyre JT, Matsuno-Yagi A, Yagi T. Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. PLoS One. 2008;3:e1433. doi: 10.1371/journal.pone.0001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M, Seo BB, Yagi T, Matsuno-Yagi A. Parkinson's disease and mitochondrial complex I: a perspective on the Ndi1 therapy. J Bioenerg Biomembr. 2009;41:493–497. doi: 10.1007/s10863-009-9249-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett RJ, Radyuk SN, Benes JJ, Orr WC, Sohal RS. Phenotypic effects of familial amyotrophic lateral sclerosis mutant Sod alleles in transgenic Drosophila. Proc Natl Acad Sci U S A. 2003;100:301–306. doi: 10.1073/pnas.0136976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Kim BH, Park SJ, Jin JK, Jeon YC, Wen GY, Shin HY, Carp RI, Kim YS. Association of endothelial nitric oxide synthase and mitochondrial dysfunction in the hippocampus of scrapie-infected mice. Hippocampus. 2011;21:319–333. doi: 10.1002/hipo.20753. [DOI] [PubMed] [Google Scholar]
- Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Roberts BR, Tainer JA, Getzoff ED, Malencik DA, Anderson SR, Bomben VC, Meyers KR, Karplus PA, Beckman JS. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol. 2007;373:877–890. doi: 10.1016/j.jmb.2007.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Tawara T, Fukushima T, Hojo N, Isobe A, Shiwaku K, Setogawa T, Yamane Y. Effects of paraquat on mitochondrial electron transport system and catecholamine contents in rat brain. Arch Toxicol. 1996;70:585–589. doi: 10.1007/s002040050316. [DOI] [PubMed] [Google Scholar]
- Trumbull KA, Beckman JS. A role for copper in the toxicity of zinc-deficient superoxide dismutase to motor neurons in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11:1627–1639. doi: 10.1089/ars.2009.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC. Pathophysiology of neurodegeneration in familial amyotrophic lateral sclerosis. Curr Mol Med. 2009;9:255–272. doi: 10.2174/156652409787847173. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MR, Lagow RD, Xu K, Zhang B, Bonini NM. A Drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem. 2008;283:24972–24981. doi: 10.1074/jbc.M804817200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicks S, Bain N, Duttaroy A, Hilliker AJ, Phillips JP. Hypoxia rescues early mortality conferred by superoxide dismutase deficiency. Free Radic Biol Med. 2009;46:176–181. doi: 10.1016/j.freeradbiomed.2008.09.036. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. 2002;80:616–625. doi: 10.1046/j.0022-3042.2001.00731.x. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Winkler K, Kuznetsov AV, Bartels C, Vielhaber S, Feistner H, Kunz WS. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci. 1998;156:65–72. doi: 10.1016/s0022-510x(98)00008-2. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Xu Z, Jung C, Higgins C, Levine J, Kong J. Mitochondrial degeneration in amyotrophic lateral sclerosis. J Bioenerg Biomembr. 2004;36:395–399. doi: 10.1023/B:JOBB.0000041774.12654.e1. [DOI] [PubMed] [Google Scholar]