

Abstract
Fluorine containing fluorochromes represent important validation agents for PET imaging agents as they can be easily rapidly validated in cells by fluorescence imaging. In particular, the 18F-labeled BODIPY-FL fluorophore has emerged as an important platform but little is known about alternative 18F-labeling strategies or labeling on red shifted fluorophores. Here we explore the acid-catalyzed 18F/19F exchange on a range of commercially available N-hydroxysuccinimidyl ester and maleimide BODIPY fluorophores. We show this method to be a simple and efficient 18F-labeling strategy for a diverse span of fluorescent compounds, including a BODIPY modified PARP-1 inhibitor, and amine- and thiol-reactive BODIPY fluorophores.
Keywords: 18F-Fluorine, Molecular Imaging, BODIPY, Fluorescence, Positron emission tomography (PET)
Fluorescence technology has revolutionized biological imaging through the development of biocompatible fluorescent small molecule, nanoparticles and proteins[1]. Despite these advances, translation into the clinic has proven more difficult and clinical trials remain scarce. Conversely, several thousand positron emission tomography (PET) imaging agents have been imaged in vivo in animals, but most of them have not been validated at the cellular level given the inherent spatial resolution limitations of PET.[2] Chemically and biologically equivalent bimodal, PET/fluorescence imaging agents circumvent this problem offering a number of advantages such as rapid screening, direct cellular validation (via microscopy or flow cytometry), and cost-effective testing of the stable isotope compound prior to rapid precursor scale-up and labeling of the optimized radiotracer.
Our and other groups have recently developed methods for labeling boron dipyrromethene fluorochromes (BODIPY) with the radionuclide fluorine-18.[3–5] Chemical removal of 19F yields a stable intermediate, which when treated with [18F]fluoride ion gives the chemically equivalent but is otopically distinct BODIPY, Scheme 1.
Scheme 1.
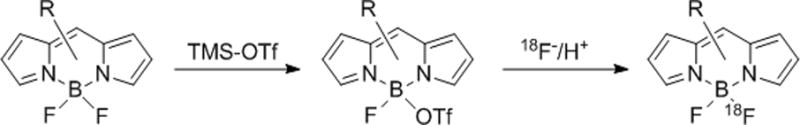
Two-step [18F]fluoride labeling of Bodipy dyes.
We reported the 18F-labeling of BODIPY dyes by a modification of the procedure of Hudnall[6] and during the course of our studies found acidic conditions were required for the labeling of the reactive intermediate[3]. Here, we describe and explore the relative reactivity of the acid-catalyzed exchange of 19F with 18F of a number of commercially available N-hydroxysuccinimidyl ester and maleimide BODIPY fluorophores, Scheme 2. Additionally, we show that the integrity of the N-hydroxysuccinimidyl (NHS) ester and maleimide (Mal) functional groups is conserved by demonstrating subsequent reaction with benzylamine and L-cysteine, respectively. Finally, using this method, we also describe the direct 18F-labeling small molecule, which is based on the PARP-1 inhibitor AZD2281 (Olaparib) modified with BODIPY-FL.
Scheme 2.
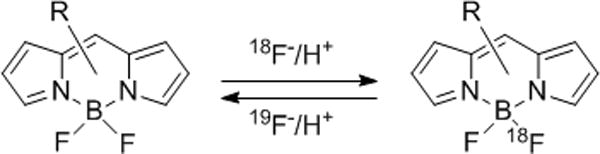
Acid-catalyzed fluoride exchange of Bodipy dyes.
Following our previously published procedure[3], 4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene-8-propionic succinimidyl ester (B493-NHS, 1) was electrophilically activated with trimethylsilyl triflate then treated with 2,6-lutidine to give a stable but reactive inter mediate. Addition of azeotropically dried 18F to the stable intermediate in the presence of acid gave the desired 18F-labeled BODIPY 18F-1 in high radiochemical yield in less than 2 minutes. We generat ed trifluorosulfonic acid (TfOH) in situ by the addition of Tf2O with tBuOH to the reaction mixture. These substances in the presence of residual water from the 18F were sufficient to generate the acidic conditions required for 18F fluorination of the stable intermediate. When these same conditions were applied to the labeling of BFL-NHS (2), we found the rate of the reaction significantly reduced. Upon further investigation, we found that structurally different BODIPY dyes (BODIPYs 1–6, Figure 1) display significant differences in their relative 18F/19F exchange rates.
Figure 1.
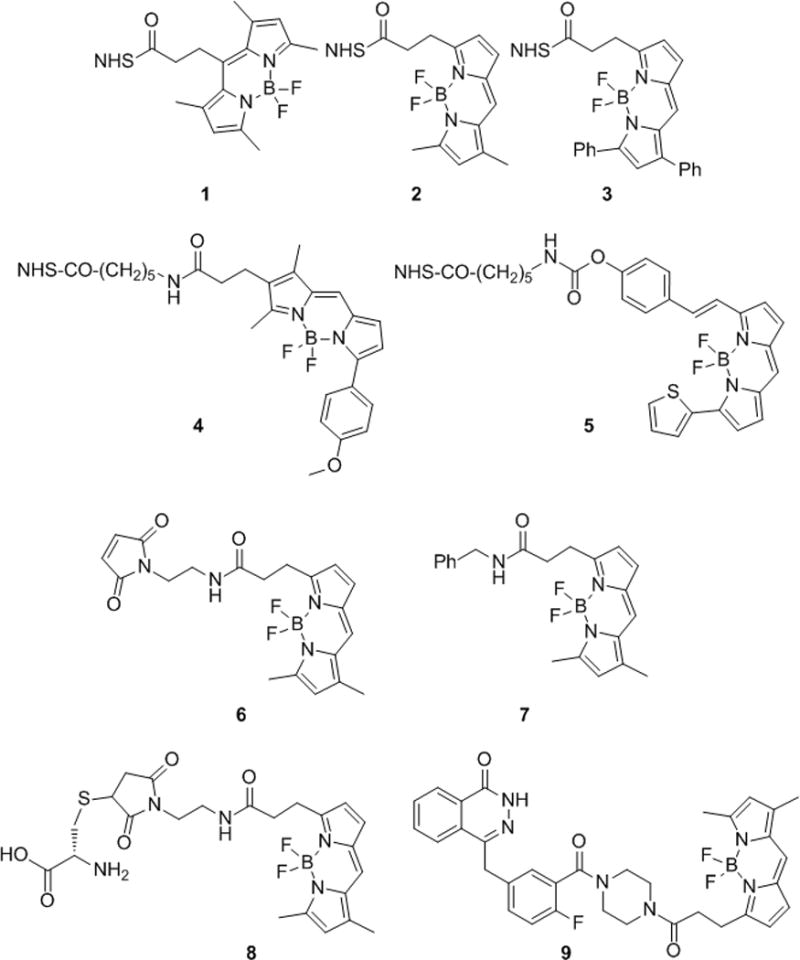
Chemical structures of B493-NHS (1), BFL-NHS (2), B530-NHS (3), BTMR-X-NHS (4), B630-X-NHS (5), BFL-Mal (6), BFL-benzyl (7), Cys-BFL (8), and PARPi (9).
Preliminary exchange experiments were conducted by treating NHS ester 2 with a mixture of Tf2O, tBuOH (40 mM final reaction concentration) and azeotropically dried 18F at 50 °C. Within minutes, radio-HPLC analysis showed 18F incorporation into 2. When 2 was treated with Tf2O or tBuOH separately only 1% exchange was observed after 2 h. With these observations we proceeded to study the exchange of 18F for 19F in the general BODIPY structure in more detail by varying the reagent concentrations, time, and temperature as well as the starting BODIPY fluorophore. The reaction rate was found to be dependent on both Tf2O/tBuOH and BODIPY.
Figure 2A and Table 1 show the dependence of the rate on the concentration of Tf2O/tBuOH concentrations (10, 14, and 40 mM) while all ot her concentrations were held constant. Linear regression of the data from these kinetic experiments provided a second order rate constant with respect to the acid concentration of (6.86 ± 1.01) × 10−5 M−1 s−1. Interestingly, we found that there was no incorporation of 18F at Tf2O/tBuOH concentrations that were below the concentration of the alkaline tetrabutylammonium bicarbonate (TBAB) phase transfer catalyst. We hypothesize that below this threshold, the reaction mixture is too basic for exchange to proceed. In addition to the second order dependence on Tf2O/tBuOH, we determined that there is a second order dependence on the concentration of the BODIPY substrate, as shown in Figure 2B. The BFL-NHS (2) concentrations were varied (0.5, 1.0 and 2.0 mM final reaction concentrations) while all other reagent concentrations were held constant. Linear regression of these observed rate constants provided a second order rate constant, (4.19 ± 0.15) × 10−4 M−1 s−1.
Figure 2.
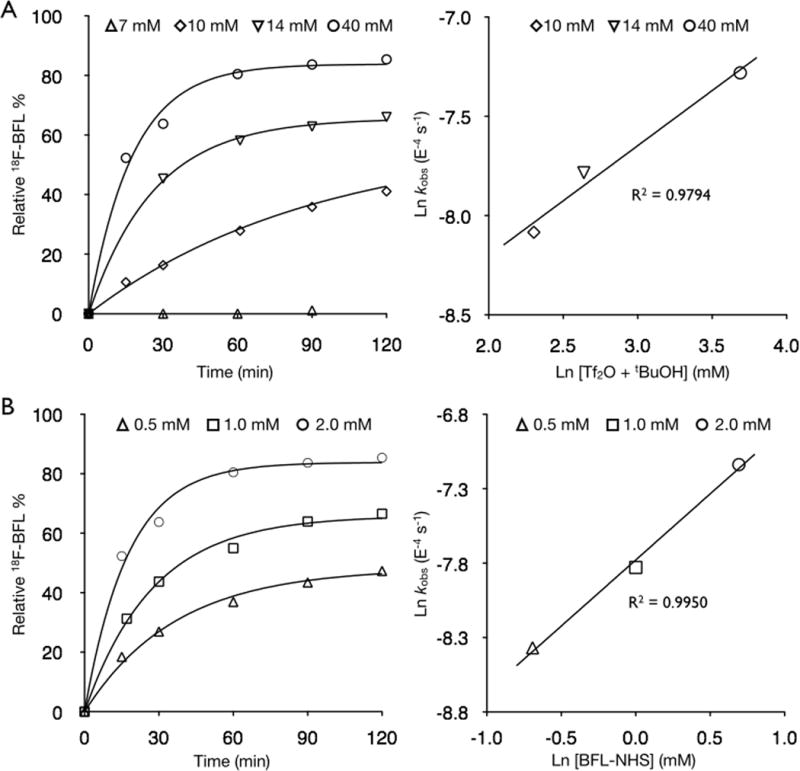
Concentration-versus-time curves and linear-regression analysis of the observed rate constants for BFL-NHS + 18F/Tf2O/tBuOH. A) Variation of [Tf2O/tBuOH] at 50 °C. B) Variation of [BFL-NHS] at 50 °C.
Table 1.
Observed and second order rate constants for the acid-catalyzed 18F/19F exchange of BFL-NHS (2)
| mM | kobs (10−4) | ±3σ (10−4) | % Error | |
|---|---|---|---|---|
| Tf2O/tBuOH | 40 | 7.95 | 1.01 | 12.7 |
| 14 | 4.16 | 0.26 | 6.3 | |
| 10 | 3.09 | 0.26 | 8.5 | |
|
| ||||
| BFL-NHS (2) | 2.0 | 7.95 | 1.01 | 12.7 |
| 1.0 | 3.98 | 0.40 | 10.0 | |
| 0.5 | 2.32 | 0.16 | 7.0 | |
|
| ||||
| k (10−4 M−1 s−1) | Error (10−4) | |||
| Tf2O/tBuOH | 0.69 | 0.10 | ||
| BFL-NHS (2) | 4.19 | 0.15 | ||
We also tested TfOH and methanesulfonic acid (MsOH) at the same final reaction concentration as the Tf2O/tBuOH mixture (40 mM). TfOH showed rapid 18F/19F exchange but also rapid decomposition of the fluorophore and thus the release of free 19F− into the reaction mixture and decreasing the incorporation of 18F. The MsOH experiment shows a significantly slower rate of 18F/19F exchange (Supplementary Information Figure S2A and S2B).
While the above kinetic experiments were conducted at 50 °C, two other temperatures, 0 and 23 °C, were also explored (Supplementary Information Figure S2C). The observed reaction rate at 50 °C (7.95 ± 1.01) × 10−4 s−1 was 11.7 times faster than that observed at 23 °C, (5.91 ± 0.89) × 10−5 s−1, and 33.1 faster than observed at 0 °C, (2.08 ± 0.38) × 10−5 s−1. Increasing the amount starting activity (2.0, 16.5 and 34.5 mCi) added to the reaction while maintaining a constant starting BFL-NHS (XYZ μmol) concentration resulted in a linear increase of specific activity of 6.5, 45.4 and 73.6 mCi/μmol, respectively.
In a series of separate experiments, individual exchange reactions on 2 were set up for each time point (15, 30, 60, 90, 120 min) in an effort to compare loss of activity due to evaporation, presumably in the form of [18F]-HF, while opening and closing the reaction tube for serial analysis of a single reaction. At each time point, the reaction activity was measured before and after removal of the aliquot for analysis. These data were decay corrected to the time of initial addition of activity and compared. Less than 5% loss of radioactivity to evaporation was observed during the course of running a single reaction with multiple analyses and less then 3% loss per reaction when running multiple reactions in parallel with single analysis per reaction.
Recognizing the potential utility of this method for the generation of dual PET-optical molecular imaging probes, we sought to broaden the scope of this labeling method therefore we tested these conditions (125 nmol BODIPY and 2.5 μmol Tf2O/tBuOH, 2 and 40 mM final reaction concentrations, respectively, at 50 °C) against a number of other commercially available BODIPY fluorochromes (B493-NHS (1), B530-NHS (3), BTMR-X-NHS (4), B630-X-NHS (5) and BFL-Mal (6)) as well as a biologically relevant BODIPY labeled small molecule PARPi (9)[7]. The structures of these compounds are shown in Figure 1 and the results of the exchange experiments are summarized in Figure 3 and Table 2. The previously studied B493-NHS (1) was the most reactive, showing over 87% 18F exchange within 15 min. Maleimide 6 and NHS esters 2 and 4 were found to be similar in reactivity, 60, 64, and 65% 18F exchange at 30 min. NHS esters 3 and 5 were considerably slower with only 16 and 24% exchange after 30 min, respectively. For the specific activity, each reaction has therefore is theoretical maximum, governed by the 18F activity incorporated and the amount intact radiolabeled plus cold BODIPY dye after the reaction. The specific activities range from 1.9 to 12.8 mCi/μmol (Table 2) correlating with the radiochemical yields, which range from 16.2 to 90.6% after 30 min reaction time.
Figure 3.
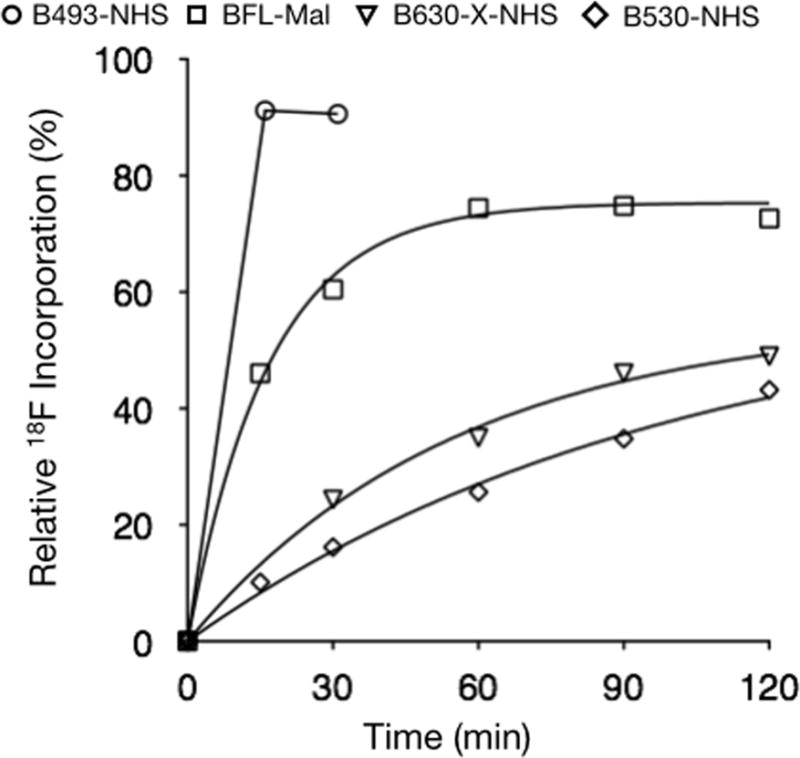
Concentration-versus-time curves for B493-NHS (2, ○), BFL-Mal (6, □), B630-X-NHS (5, ▽) and B530-NHS (3, ◇).
Table 2.
Radiochemical yields and specific activity of 18F/19F exchange of 6 commercially available BODIPY fluorophores.a
| Entry | BODIPY Dye | 18F− Incorporation (%) | Specific Activity (mCi/μmol) |
|---|---|---|---|
| 1 | B493-NHS | 90.6 | 12.8 |
| 2 | BFL-NHS | 63.8 | 6.5 |
| 3 | B530-NHS | 16.2 | 1.9 |
| 4 | BTMR-X-NHS | 65.1 | 8.5 |
| 5 | B630-X-NHS | 24.3 | 2.6 |
| 6 | BFL-Mal | 60.4 | 4.0 |
After 30 min reaction time at 50 °C (2.5 μmol Tf2O/tBuOH, 125 nmol BODIPY), yields determined by HPLC (Method C).
Following the labeling of 18F-2 and 18F-6 after 45 min at 50 °C, these two compounds were purified from unreacted [18F]fluoride ion by passage through a silica gel cartridge. Once loaded onto the cartridge, elution with DCM and EtOAc secured the radiochemically pure 18F-2 and 18F-6 in 75 and 66 % radiochemical yield (non decay-corrected), repectively. Addition of benzylamine to 18F-2 in the presence of triethylamine (Et3N) and L-cysteine to 18F-6, provided in 18F-7 and 18F-8, Figure 4A. Presence of the desired stable isotope conjugates was confirmed by detection of the correct mass during LC/MS analysis and the presence of radio-labeled conjugates was verified by HPLC co-elution with the stable isotope 7 and 8.
Figure 4.
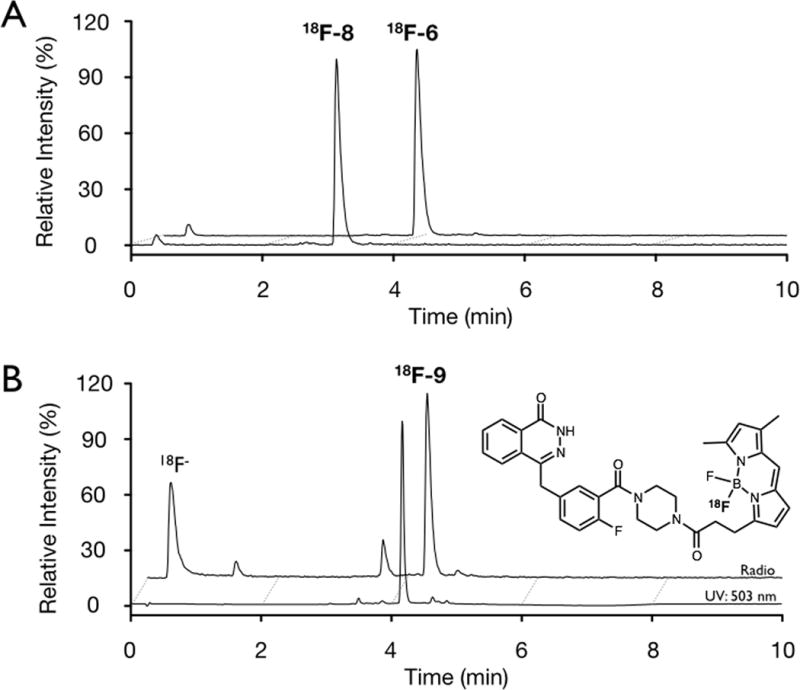
A) HPLC radio-chromatograms of conjugation of 18F-6 with L-cysteine to give 18F-8. B) HPLC chromatograms of 18F-PARPi (18F-9), blue: UV absorbance (503 nm); black: radio-chromatogram.
A BODIPY conjugate previously prepared in our laboratory, PARPi (9) was subjected to acid-catalyzed 18F/19F exchange (conditions: 125 nmol PARPi, 2.5 μmol Tf2O/tBuOH, 40 °C) with 49% 18F incorporation observed after 30 min, Figure 4B.
The acid-catalyzed exchange of 19F for 18F on BODIPY fluorophores was found to have second order kinetics with respect to the acid as well as the dye added. At 50 °C, the incorporation of 18F into BFL-NHS (2) achieved over 50% incorporation at 15 min and 64% at 30 min. The incorporation proceeded to equilibrium over the next 1.5 h, reaching a maximum of approximately 85% at 2 h total reaction time. An increase in the BODIPY concentration would not only increase the final concentration of the 18F-labeled species but also the rate at which this equilibrium would be achieved.
Exchange reactions on other commercial BODIPY fluorochromes (B493-NHS (1), B530-NHS (3), BTMR-X NHS (4), B630-X-NHS (5) and BFL-Mal (6)) all demonstrated 18F− incorporation, although differences in reactivity were observed. Our results indicate that for compound 2, increasing the concentration of Tf2O/tBuOH increases the 18F/19F exchange rate. We anticipate that the other compounds tested will follow this trend to approach the exchange rate of 1. Both amine-reactive N-hydroxysuccinimidyl esters and thiol-reactive maleimides are well tolerated under these reaction conditions and demonstrated reactivity in subsequent conjugation reactions. To our knowledge this is the first example of a direct 18F-labeling of a maleimide. Other reported 18F-labeling protocols require multiple steps to obtain the desired thiol-reactive maleimide prosthetic group.[8–11]
As a general labeling strategy this method does have a limitation in specific activity due to the degeneracy of the starting material and product and therefore an inability to chromatographically separate the desired labeled product from starting material. To address this, we have shown that increasing the amount of starting activity will increase the specific activity and that the relationship was found to be linear over the activity range studied, 2 – 35 mCi (74–1295 MBq), shown in Figure 5. Although the resulting activities of 70 mCi/μmol (2590 MBq/μmol) are lower than other commonly used radiotracers FES (2.5–5 Ci/μmol[12, 13]), the specific activity generated is likely sufficient even for clinical applications. For example, for a 18F-PARPi scan, only 2 μmol would have to be injected (35 mCi, 70 mCi/μmol), which represents less than 0.1% of a daily clinical dose of the parent compound, Olaparib (based on a 300 mg (390 μmol) bi-daily oral administration, ClinicalTrial.gov identifier: NCT01844986). We envision by varying the starting quantity of 18F activity and/or starting material, the radiochemical yield and specific activity may be optimized for a particular application.
Figure 5.
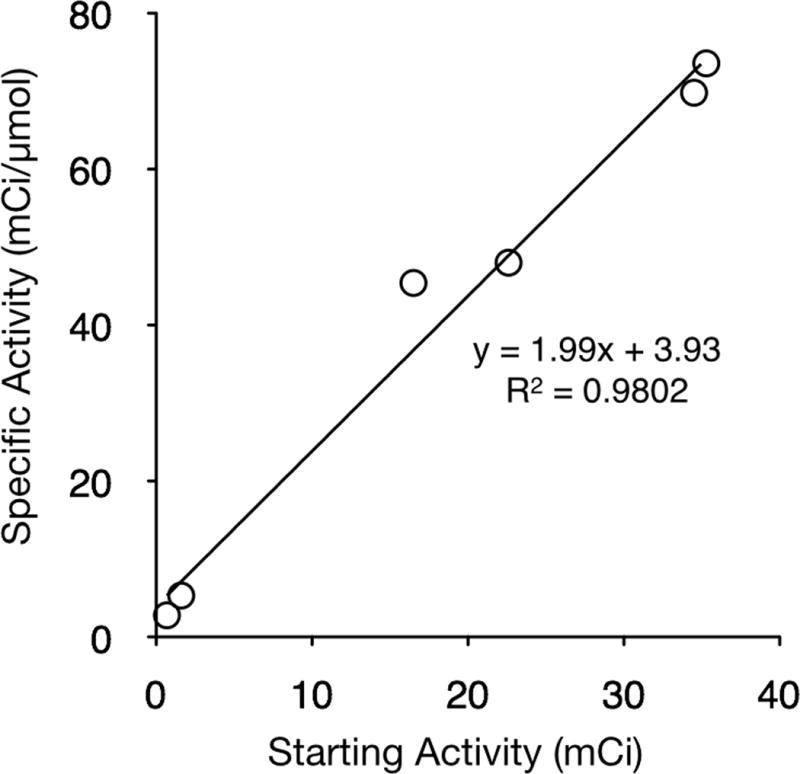
Specific activity (mCi/μmol) as a function of starting activity for the incorporation of 18F− in to BFL-NHS.
Experimental Section
General
Unless otherwise noted, solvents and reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. All NHS and maleimide BODIPY compounds were purchased from Invitrogen. Synthesis of 4-[[4-fluoro-3-(4-(5-oxopentanamide)piperazine-1-carbonyl)phenyl]methyl]-2H-phthalazin-1-one 9 was described earlier[14]. [18F]Fluoride ion (n.c.a.) in 18O-enriched water was purchased from PETNET (Woburn, MA) and dried by azeotropic distillation of water with acetonitrile (MeCN) in the presence of tetrabutylammonium bicarbonate (TBAB; ABX) using a Synthra RN Plus automated synthesizer (Synthra GmbH, Hamburg, Germany) operated by SynthraView software. The dried 18F/TBAB was reconstituted in MeCN, collected in a 2-mL vial, and diluted to achieve a 12 mM TBAB solution. For non-radioactive compounds, LC-ESI-MS analysis and HPLC-purifications were performed on a Waters (Milford, MA) LC-MS system. For LC-ESI-MS analyses, a Waters XTerra® C18 (4.6 × 50 mm, 5 μm) column was used (Method A: eluents 0.1% formic acid (v/v) in water (A) and MeCN (B); gradient: 0–1.5 min, 5–100% B; 1.5–2.0 min 100% B; 5 mL/min). Preparative high performance liquid chromatography (HPLC) runs for synthetic intermediates utilized an Atlantis® Prep T3 OBD™ (19 × 50 mm, 5 μm) column (Method B: eluents 0.1% TFA (v/v) in water (A) and MeCN (B); gradient: 0–1.5 min, 5–100% B; 1.5–2.0 min 100% B; 30 mL/min). Analytical HPLC of radiolabeled compounds was performed employing an Agilent 1200 Series HPLC and a Poroshell 120 EC-C18 (4.6 × 50 mm, 2.7 μm) reversed-phase column (Method C: eluents 0.1% TFA (v/v) in water (A) and MeCN (B); gradient: 0–0.3 min, 5% B; 0.3–7.5 min, 5–100% B; 7.5–10 min, 100% B; 2.5 mL/min) with a multichannel-wavelength UV-vis detector, fluorescence detector and a flow-through gamma detector connected in series. Solid-phase extraction cartridges used were Oasis C18 3-cc cartridge (60mg, 30 μm particle size, Waters, MA) and Sep-Pak Silica 3-cc Vac cartridge (500 mg, 55–105 μm particle size, (Waters, MA)). The two-step, one-pot 18F− labeling procedure employing TMS-OTf was previous described.[3]
Isotopic Exchange Reaction
To azeotropically dried 18F/TBAB (30 μL, 12 mM TBAB in MeCN) in a 1.5-mL centrifuge tube, triflic anhydride (Tf2O; 10 μL, 250 mM in MeCN), t-butanol (tBuOH; 10 μL, 250 mM in MeCN) and the BODIPY dye (12.5 μL, 10 mM in 1.5:1 DCM:MeCN, B493-NHS (1), BFL-NHS (2), B530-NHS (3), BTMR-X-NHS (4), B630-X-NHS (5), BFL-Mal (6), or PARPi (9)) were added sequentially in 2 minute inter vals. The radioactivity of the reaction tube was measured in a well counter then placed in 50 °C shaker. At 15, 30, 60, 90, 120, and 150 min (with exception of 1, only 15 and 30 min data points were obtained), the tubes were removed from heat, cooled in an ice bat h for 20 s, and the activity of the reaction tube measured. An aliquot (1–3 μL) was removed from the reaction tube, radioactivity measured in a well counter and analyzed by HPLC (Method C). Radioactivity of the reaction tube was measured and then returned to the heated shaker. Kinetic data points were obtained from the area of the HPLC radio-chromatograms and plotted as a percent fraction. Observed rate constants were generated from the data by the program KINETIC of Dr. R. Fink, a gift from the late Prof. William von Eggars Doering, which handles kinetic schemes containing up to seven components, and incorporates a calculation of Marquardt that generates error limits in the rate constants at the 95% confidence level.[15, 16] Second-order rate constants were calculated using Graph-Pad Prism 4.0c (GraphPad Software, Inc, San Diego, CA). Exchange experiments were repeated for 2 in the same manner as described above varying starting concentrations of 2 (final reaction concentrations of 1.0 and 0.5 mM) or Tf2O/tBuOH (final reaction concentrations of 14, 10, and 7 mM) while maintaining a constant final reaction volume. Additional experiments were conducted for 2 at 0 and 23 °C.
Conjugation Reactions
18F-Benzyl-BFL (18F-Bn-BFL, 18F-7)
The crude 18F-BFL-NHS mixture, prepared as described above in the acid-catalyzed exchange reaction after 45 min at 50 °C, was loaded on to a SPE silica gel cartridge (1.0 mCi) conditioned with pentane (500 μL). The cartridge was washed with pentane (2×150 μL) then the 18F-BFL-NHS eluted with dichloromethane (DCM, 3×150 μL) followed by DMSO (3×150 μL). Activity collected in the elution fractions was as follows: 1.7 and 2.7 μCi for pentane fractions 1 and 2; 179.5, 273.0 and 14.3 μCi for DCM fractions 1, 2 and 3; 48.6, 192.1, and 14.3 μCi for DMSO fractions 1, 2 and 3; and 229 μCi remaining on the silica cartridge. To the combined DCM fractions of 18F-BFL-NHS was added Et3N (10 μL, 250 mM in DCM) and benzylamine (37.5 μL, 25 mM in DCM) and stirred at rt for 40 min. HPLC analysis (10 μL aliquot) demonstrated full conversion of 18F-BFL-NHS to 18F-Bn-BFL. Bn-BFL, 7, LC-ESI-MS analysis found: 404.28 [M+Na+]+, 362.18 [M−F−]+; calculated: [M+Na+]+ = 404.17, [M−F−]+ = 362.18.
18F-Cysteine-BFL (18F-Cys-BFL, 18F-8)
The crude 18F-BFL-Mal mixture, prepared as described above in the acid-catalyzed exchange reaction after 45 min at 50 °C, was loaded on to a SPE silica gel cartridge (1.0 mCi) conditioned with pentane (300 μL). The cartridge was washed with pentane (2×150 μL) then the labeled compounds eluted with dichloromethane (EtOAc, 3×150 μL) and DMSO (3×150 μL). Activity collected in the elution fractions was as follows: 2.6 and 2.5 μCi for pentane fractions 1 and 2; 198.9, 291.9 and 95.9 μCi for EtOAc fractions 1, 2 and 3; 18.8, 30.5, and 5.6 μCi for DMSO fractions 1, 2 and 3; and 317 μCi remaining on the silica cartridge. To the combined EtOAc fractions of 18F-BFL-Mal was added Et3N (10 μL, 250 mM in DCM) and L-cysteine (37.5 μL, 25 mM in DCM) and stirred at rt for 40 min. HPLC analysis (10 μL aliquot) demonstrated full conversion of 18F-BFL-Mal to 18F-Cys-BFL. Cys-BFL, 8, LC-ESI-MS analysis found: 516.31 [M−F−]+, 534.25 [M−H+]− calculated: [M−F−]+ = 516.19, [M−H+]+ = 534.18.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health grants P50CA86355 and RO1EB010011 and the Department of Energy Training grant DE-SC0001781PO1.
References
- 1.Pittet MJ, Weissleder R. Cell. 2011;147:983–991. doi: 10.1016/j.cell.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallabhajosula S, Solnes L, Vallabhajosula B. Seminars in nuclear medicine. 2011;41(4):246–264. doi: 10.1053/j.semnuclmed.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Hendricks JA, Keliher EJ, Wan D, Hilderbrand SA, Weissleder R, Mazitschek R. Angew Chem Int Ed Engl. 2012;51:4603–4606. doi: 10.1002/anie.201107957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Lin TP, Liu S, Huang CW, Hudnall TW, Gabbai FP, Conti PS. Chem Commun (Camb) 2011;47:9324–9326. doi: 10.1039/c1cc13089g. [DOI] [PubMed] [Google Scholar]
- 5.Liu S, Lin TP, Li D, Leamer L, Shan H, Li Z, Gabbai FP, Conti PS. Theranostics. 2013;3:181–189. doi: 10.7150/thno.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hudnall TW, Gabbai FP. Chem Commun (Camb) 2008:4596–4597. doi: 10.1039/b808740g. [DOI] [PubMed] [Google Scholar]
- 7.Reiner T, Lacy J, Keliher EJ, Yang KS, Ullal A, Kohler RH, Vinegoni C, Weissleder R. Neoplasia. 2012;14:169–177. doi: 10.1593/neo.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berndt M, Pietzsch J, Wuest F. Nucl Med Biol. 2007;34:5–15. doi: 10.1016/j.nucmedbio.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Cai W, Zhang X, Wu Y, Chen X. J Nucl Med. 2006;47:1172–1180. [PMC free article] [PubMed] [Google Scholar]
- 10.Kiesewetter DO, Jacobson O, Lang L, Chen X. Appl Radiat Isot. 2011;69:410–414. doi: 10.1016/j.apradiso.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wuest F, Berndt M, Bergmann R, van den Hoff J, Pietzsch J. Bioconjug Chem. 2008;19:1202–1210. doi: 10.1021/bc8000112. [DOI] [PubMed] [Google Scholar]
- 12.Tsujikawa T, Yoshida Y, Kiyono Y, Kurokawa T, Kudo T, Fujibayashi Y, Kotsuji F, Okazawa H. Eur J Nucl Med Mol Imaging. 2011;38:37–45. doi: 10.1007/s00259-010-1589-8. [DOI] [PubMed] [Google Scholar]
- 13.van Kruchten M, Glaudemans AW, de Vries EF, Beets-Tan RG, Schroder CP, Dierckx RA, de Vries EG, Hospers GA. J Nucl Med. 2012;53:182–190. doi: 10.2967/jnumed.111.092734. [DOI] [PubMed] [Google Scholar]
- 14.Reiner T, Earley S, Turetsky A, Weissleder R. ChemBioChem. 2010;11:2374–2377. doi: 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doering WvE, Keliher EJ. J Am Chem Soc. 2007;129:2488–2495. doi: 10.1021/ja066018c. [DOI] [PubMed] [Google Scholar]
- 16.Marquardt DW. J Soc Ind Appl Math. 1963;11:431–441. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.