

Abstract
Substantial evidence suggests that soluble prefibrillar oligomers of the Aβ42 peptide associated with Alzheimer’s disease are the most cytotoxic aggregated Aβ isoform. Limited previous work has revealed that aromatic compounds capable of remodeling Aβ oligomers into non-toxic conformers typically do so by converting them into off-pathway aggregates instead of dissociating them into monomers. Towards identifying small molecule antagonists capable of selectively dissociating toxic Aβ oligomers into soluble peptide at substoichiometric concentrations, we have investigated the pathways used by polyphenol aglycones and their glycosides to remodel Aβ soluble oligomers. We find that eleven polyphenol aglycones of variable size and structure utilize the same remodeling pathway whereby Aβ oligomers are rapidly converted into large, off-pathway aggregates. Strikingly, we find that glycosides of these polyphenols all utilize a distinct remodeling pathway in which Aβ oligomers are rapidly dissociated into soluble, disaggregated peptide. This disaggregation activity is a synergistic combination of the aglycone and glycone moieties since combinations of polyphenols and sugars fail to disaggregate Aβ oligomers. We also find that polyphenolic glycosides and aglycones use the same opposing pathways to remodel Aβ fibrils. Importantly, both classes of polyphenols fail to remodel non-toxic Aβ oligomers (which are indistinguishable in size and morphology to Aβ soluble oligomers) or promote aggregation of freshly disaggregated Aβ peptide, revealing that they are specific for remodeling toxic Aβ conformers. We expect that these and related small molecules will be powerful chemical probes for investigating the conformational and cellular underpinnings of Aβ-mediated toxicity.
Keywords: protein misfolding, fibril, oligomer, disaggregation, beta amyloid
Introduction
Since abnormal protein folding and aggregation is the seminal event in several protein conformational disorders,[1] targeting and remodeling misfolded proteins with small molecules is an important therapeutic approach to treating such diseases.[2–4] Prefibrillar oligomeric intermediates are the most toxic species in several such disorders,[5–7] yet relatively little is known about how to remodel prefibrillar oligomers into alternative, non-toxic conformers using small molecule antagonists. Importantly, there are numerous challenges to targeting misfolded proteins with small molecules not encountered when targeting conventional protein targets.[8] First, proteins fail to misfold into a single folded structure, but rather form multiple misfolded conformers ranging from prefibrillar oligomers to fibrillar intermediates and mature amyloid fibrils that differ both in terms of size and structure.[7, 9–14] Thus, the specific aggregated conformers of biological interest (e.g., prefibrillar oligomers) must not only be defined, but also prepared in a homogeneous manner for systematic analysis. Second, defining the specificity of such small molecule antagonists necessitates not only the use of proteins of different sequence, but also multiple conformations of the specific amyloidogenic protein of interest. This requires that homogeneous aggregated conformers other than the target conformer be prepared and analyzed as well. A third challenge is that most misfolded conformers are not equilibrium structures, but rather kinetic intermediates that mature structurally over time scales similar to those commonly used in small molecule inhibition studies.[15–18] This requires that misfolded conformers must be formed and analyzed over time scales that are small relative to those required for aggregated species to mature conformationally in the absence of small molecules.[16, 19–21] A fourth challenge is that aggregated conformers are composed of several (identical) protein monomers that can be either disaggregated or remodeled into multiple types of on- and off-pathway aggregates by small molecule antagonists.[17, 18, 20] Thus, it is critical to analyze the size and structure of remodeled protein conformers to illuminate the pathways employed by small molecules to transform misfolded proteins into alternative conformers.
Despite these significant challenges, several recent studies are beginning to elucidate the remodeling activities and pathways used by multiple compounds to convert toxic prefibrillar oligomers into various conformers with low toxicity.[19–22] The most well characterized prefibrillar intermediates are soluble oligomers of the Aβ40 and Aβ42 peptides associated with Alzheimer’s disease (AD).[7, 23, 24] These intermediates are highly toxic to mammalian cells in vitro,[23, 25, 26] inhibit long-term potentiation[27, 28] and related cellular measures of learning and memory in rodents,[6, 29, 30] and are found in brains of humans with AD.[7, 31, 32] Importantly, conformation-specific antibodies have been developed that specifically recognize prefibrillar oligomers relative to monomers or fibrils,[31, 33, 34] which have greatly facilitated characterization and analysis of such oligomers ([7] and references therein). The most important aspect of these antibodies is that they enable characterization of toxic soluble oligomers of Aβ and other polypeptides in terms of structure instead of size. Structure-based classification of prefibrillar oligomers (and other aggregated protein conformers) of Aβ and related amyloidogenic polypeptides is critical since oligomers of similar size (and in some cases identical size[13, 20, 21]) can have dramatically different biochemical and biological properties due to differences in their structure instead of their size.[7, 12]
Using such conformation-specific antibodies, multiple aromatic small molecules have been identified that remodel toxic Aβ oligomers into alternative conformers with low toxicity.[18–22] A common finding of these studies is that aromatic small molecules capable of remodeling toxic Aβ oligomers most often do so by converting them into off-pathway aggregates instead of disaggregating them into soluble peptide.[18–21] In hindsight, this pathway is logical since it is simpler to promote alternative intermolecular contacts between hydrophobic Aβ peptides than to prevent all possible intermolecular contacts. Nevertheless, we and others have identified aromatic molecules that completely disaggregate toxic prefibrillar oligomers to soluble peptide.[20, 22] Some of these molecules are very large (e.g. tannic acid, MW 1700 Da) and their remodeling activity can be ascribed to their large size.[35, 36] However, we found that a smaller polyphenolic glycoside (piceid, MW 390 Da) completely disaggregates Aβ soluble oligomers at substoichiometric concentrations (as it does for aggregates of a small peptide fragment of Aβ[37]).[20] This finding is particularly striking since piceid is a glycoside of resveratrol, yet resveratrol remodels toxic Aβ oligomers into large, off-pathway aggregates.[21] Based on these findings, we hypothesize that polyphenol aglycones and their respective glycosides use two opposing pathways to remodel toxic Aβ oligomers into non-toxic conformers. More specifically, we posit that polyphenol aglycones that differ in size and structure employ a single remodeling pathway whereby toxic Aβ oligomers are converted into large off-pathway aggregates. In contrast, we posit that diverse polyphenol glycosides utilize a second unique remodeling pathway whereby toxic Aβ oligomers are completely dissolved into soluble peptide that is also non-toxic. Herein we test these hypotheses by interrogating the remodeling pathways utilized by a panel of polyphenol aglycones and their glycosides to remodel and inactivate toxic Aβ oligomers relative to other Aβ conformational variants.
Results
Activity of polyphenols and their glycosides for remodeling Aβ soluble oligomers
We first asked whether a relatively simple polyphenol (phloretin; Figure 1A) and its glycoside (phloretin 2′-β-D-glucoside; Figure 1A) would remodel mature Aβ oligomers via the same opposing pathways as employed by resveratrol and its glycoside piceid. To accomplish this, we formed Aβ42 soluble oligomers at 25°C in PBS (pH 7.4) without agitation, as we have described elsewhere.[20, 21] Such Aβ oligomers form after one day and do not mature structurally for an additional two days.[21] Thus, we examined the ability of phloretin and phloretin 2′-β-D-glucoside to rapidly remodel mature Aβ soluble oligomers over a short time interval (4 h) relative to the time required for such oligomers to mature structurally (48 h). Since Aβ toxic oligomers are selectively recognized by the conformation-specific antibody A11,[31] we examined the ability of each molecule to eliminate the oligomer-specific conformational epitope within Aβ oligomers using A11 dot blot analysis. We find that both phloretin and phloretin 2′-β-D-glucoside rapidly eliminate the A11 epitope with identical dose dependence relative to resveratrol and piceid (Figure 1B).
Figure 1. Characterization of remodeling pathways used by glycoside and aglycone forms of resveratrol and phloretin to inactivate mature Aβ42 soluble oligomers.

Aβ soluble oligomers (25 μM; formed after 1 d without agitation) were incubated with each small molecule shown in (A) for 4 h, and analyzed using (B) dot blots probed with conformation-specific (A11, soluble oligomer) and sequence-specific (6E10) antibodies, (C) PC12 cell toxicity assay (n=3), (D) AFM, and (E) ANS fluorescence. In (D), mature soluble oligomers were incubated with the vehicle (1% DMSO) or 200 μM compound, and each image is 3 × 3 μm (inset is 0.5 × 0.5 μm).
Since A11-positive Aβ oligomers are toxic when added to mammalian cells relative to Aβ monomers or off-pathway aggregates ([21, 31] and references therein), we posited that soluble oligomers remodeled by phloretin and its glycoside would also be non-toxic. Indeed, both compounds (as well as resveratrol and piceid) eliminate the toxicity of Aβ soluble oligomers relative to freshly disaggregated Aβ at the same concentrations (≥20 μM) required to eliminate the A11 epitope (Figure 1C). These findings not only reveal that the remodeled conformers possess indistinguishable toxicity, but also strongly suggest that elimination of the A11 epitope is linked to the elimination of oligomer-specific toxicity.
Importantly, our A11 dot blot and toxicity results do not reveal the remodeling pathways employed by phloretin and phloretin 2′-β-D-glucoside relative to resveratrol and piceid. Thus, we performed AFM imaging of Aβ soluble oligomers before and after addition of each polyphenol (4 h). We confirmed that soluble oligomers do not change size or shape when dosed with the vehicle (1% DMSO) over 4 h (Figure 1D). Strikingly, we find that phloretin 2′-β-D-glucoside disaggregates Aβ oligomers into soluble peptide not detectable by AFM in a manner indistinguishable from piceid. Conversely, phloretin remodels Aβ oligomers into large aggregates in manner indistinguishable from resveratrol (Figure 1D). We also confirmed the AFM results using SDS-PAGE analysis (Figure S1), which reveals that the aglycones promote large SDS-resistant aggregates, while the glycosides do not. The size and morphology of the phloretin-remodeled Aβ oligomers are consistent with off-pathway aggregates, as we previously observed for resveratrol-remodeled Aβ oligomers.[21] Indeed, we used a second conformation-specific antibody (OC) that selectively recognizes fibrillar intermediates and mature fibrils[38] to confirm that both phloretin- and resveratrol-remodeled Aβ oligomers lack fibrillar epitopes (Figure S1). Consistent with these results, we find that remodeled soluble oligomers possess random coil secondary structure regardless of whether they were disaggregated or converted in off-pathway aggregates (Figure S1).[20] Finally, we used the dye 8-anilino-1-naphthalene sulfonate (ANS) to evaluate the conformation of remodeled Aβ soluble oligomers relative to Aβ monomers and fibrils (Figure 1E and S1). As expected, we find that the ANS spectra for soluble oligomers remodeled with phloretin 2′-β-D-glucoside (λmax=452 nm) and piceid (λmax=454 nm) are similar to Aβ monomers (λmax=457 nm) and significantly different than Aβ soluble oligomers (λmax=484 nm) or fibrils (λmax=526 nm). In contrast, Aβ oligomers remodeled by resveratrol and phloretin possess ANS spectra (λmax=501 nm) intermediate to Aβ soluble oligomers (λmax=484 nm) and fibrils (λmax=526 nm). Collectively these results strongly suggest that simple polyphenols and their glycosides remodel Aβ soluble oligomers via unique pathways that result in off-pathway aggregates (aglycones) or soluble peptide (glycosides).
Since resveratrol and phloretin are relatively similar in structure, we next asked whether polyphenolic glycosides and their aglycones of different size and structure would also utilize the same opposing pathways to remodel Aβ oligomers. Thus, we investigated four pairs of polyphenolic glycoside-aglycone pairs (naringenin and naringin, apigenin and apigenin 7-glucoside, quercetin and quercetin 3-β-D-glucoside, and quercetin and rutin; Figure 2A). Naringin and rutin possess disaccharide glycones, while apigenin 7-glucoside and quercetin 3-β-D-glucoside possess monosaccharide glycones. Moreover, the sugars are connected to the phenolic rings in naringin and apigenin 7-glucoside, while they are connected to the central pyrone ring for quercetin 3-β-D-glucoside and rutin (Figure 2A).
Figure 2. Analysis of remodeling pathways used by diverse polyphenolic glycosides and aglycones to inactivate Aβ42 soluble oligomers.

Aβ soluble oligomers (25 μM) were incubated with each small molecule shown in (A) for 4 h, and analyzed using (B) dot blots probed with conformation-specific (A11, soluble oligomer) and sequence-specific (6E10) antibodies, (C) PC12 cell toxicity assay (n=3), (D) AFM, and (E) ANS fluorescence. In (D), soluble oligomers were incubated with the vehicle (1% DMSO) or 200 μM compound, and each image is 3 × 3 μm or (inset is 0.5 × 0.5 μm).
Using this panel of polyphenolic glycoside-aglycone pairs, we investigated their activity for remodeling Aβ oligomers using A11 dot blot analysis. As a control, we evaluated the remodeling activity of sugars of various size and structure (glucose, trehalose and heparin), and find that they are inactive for remodeling Aβ oligomers (Figure 2B). Moreover, we find that a glycosylated hydroquinone (arbutin) that possesses single phenolic and sugar moieties (Figure S2) is also inactivate (Figure 2B). However, we find that each polyphenol aglycone and glycoside rapidly eliminated the A11 epitope at substoichiometric concentrations (0.08–0.8 polyphenol:Aβ42 molar ratio). The disaccharide glycosides (naringin and rutin) were most active at eliminating the A11 epitope (≥ 2 μM), while the aglycones and monosaccharide variants (apigenin 7-glucoside and quercetin 3-βD-glucoside) were an order of magnitude less active (≥20 μM; 0.8:1 polyphenol:Aβ). We also evaluated whether the dose dependent remodeling activity of each compound for Aβ oligomers is accompanied by a loss of toxicity (Figure 2C). Indeed, we observe that the same concentrations required to eliminate the A11 conformational epitope in soluble Aβ oligomers correspond to those required to eliminate the toxicity of Aβ oligomers (Figures 2C and S2). Importantly, the remodeling activity of the polyphenol glycosides is insensitive to structural differences of the aglycones or location of the glycosidic linkage, but is dependent on the number of sugar groups per polyphenol.
Our findings that both polyphenolic glycosides and their aglycones are active for remodeling toxic Aβ oligomers enabled us to test our core hypothesis that glycosides and aglycones utilize two opposing remodeling pathways. Thus, we performed AFM analysis before and after addition of each polyphenol (Figure 2D). Strikingly, we find that each glycoside dissociates Aβ oligomers into disaggregated soluble peptide, which each aglycone variant remodels toxic oligomers into large aggregates. We confirmed the AFM results using SDS-PAGE analysis and observed similar striking differences between glycoside and aglycone variants (Figure S2). Moreover, we confirmed that none of these molecules converted Aβ soluble oligomers into structures that possess fibrillar epitopes (as detected via the OC antibody; Figure S2). Finally, we confirmed that the ANS spectra of soluble oligomers remodeled by rutin and naringin (λmax=454–455 nm) are similar to Aβ monomers (λmax=457 nm) and significantly different than Aβ soluble oligomers remodeled by their aglycones (λmax=497–498 nm), soluble oligomers (λmax=484 nm) and fibrils (λmax=526 nm; Figure 2E). Collectively, our findings reveal that polyphenol glycosides and aglycones remodel toxic Aβ oligomers through opposing pathways that generate remodeled conformers with low toxicity and highly dissimilar biochemical properties.
Structure-activity analysis of the Aβ oligomer remodeling activity of polyphenols
Interestingly, the five polyphenol aglycones evaluated in this work (resveratrol, phloretin, naringenin, apigenin and quercetin) remodel toxic Aβ oligomers using the same remodeling pathway and with identical dose dependence (Figures 1 and 2). This led us to investigate the minimal determinants of the remodeling activity of polyphenol aglycones, as well as whether simpler or more complex aglycones would also employ that same remodeling pathway whereby Aβ oligomers are remodeled into large off-pathway aggregates. Using A11 dot blot analysis, we find that a single phenolic ring compound (resorcinol) and two polyaromatic, non-phenolic compounds (4,4′-dimethoxybenzophenone and meso-hydrobenzoin; Figures 3A and S3) are inactive for remodeling Aβ oligomers (Figure 3B). Pterostilbene, which is similar to resveratrol except that two of three hydroxyl groups are methylated (Figure 3A), is active at 200 μM (8:1 molar ratio of pterostibene:Aβ; Figure 3B) relative to resveratrol which is active at ≥20 μM (Figure 1B). Interestingly, three compounds (4′-hydroxyl-2,4-dimethylbenzophenone, 2,2′-dihydroxybenophenone and 2,4-dihydroxybenzophenone) with two aromatic rings (of which at least one is phenolic) that are separated by a single carbon linker (Figures 3A and S3) are also active at 200 μM (Figure 3B). These findings suggest that the minimal structural requirement for phenolic aglycones to remodel Aβ oligomers is two aromatic rings, at least one of which must possess a hydroxyl moiety.
Figure 3. Structure-activity analysis of the pathways used by polyphenol aglycones to remodel Aβ42 soluble oligomers.

Aβ soluble oligomers (25 μM) were incubated with each small molecule shown in (A) for 4 h, and analyzed using (B) dot blots probed with conformation-specific (A11, soluble oligomer) and sequence-specific (6E10) antibodies, (C) AFM, and (D) PC12 cell toxicity assay (n=3). In (C), soluble oligomers were incubated with the vehicle (1% DMSO) or 200 μM compound, and each image is 3 × 3 μm (inset is 0.5 × 0.5 μm).
We next investigated the structural requirements for polyphenol aglycones that remodel Aβ oligomers at even lower (substoichiometric) concentrations than resveratrol and closely related polyphenols (which do so at ≥20 μM; Figures 1–3). We hypothesized that substoichiometric remodeling activity of Aβ oligomers at concentrations ≤2 μM would require molecules with three or more phenolic rings. Indeed, we find that addition of a hydroxyl moiety to resveratrol (piceatannol; Figure 3A) did not improve its activity for remodeling A11-positive Aβ oligomers (Figure 3B). However, we find that the three phenolic ring compound epigallocatechin gallate (EGCG) remodels Aβ oligomers at an order of magnitude lower concentration (≥2 μM; Figure 3B) than several two ring polyphenols (Figures 1–2).
Since the enhanced remodeling activity may be due to the specific structure of EGCG instead of a more general property of three ring polyphenols, we enzymatically synthesized a dimeric form of resveratrol (resveratrol-trans-dehydrodimer; Figure 3A) to evaluate the remodeling activity of a polyphenol with three phenolic rings and a unique molecular architecture relative to EGCG. We find that this resveratrol dimer displays indistinguishable activity for eliminating the oligomer-specific conformational epitope relative to EGCG (Figure 3B), despite that the resveratrol dimer possesses five hydroxyls while EGCG possess eight hydroxyl moieties (Figure S3). Our findings in Figure 3, as well as those in Figures 1 and 2, further suggest that the number of phenolic rings is the primary determinant of the remodeling activity of polyphenols.
We next evaluated whether these polyphenol aglycones of diverse size and structure use a single or multiple remodeling pathways to neutralize the A11-epitope in Aβ soluble oligomers. Each polyphenol aglycone active for remodeling Aβ oligomers does so by converting them into large, off-pathway aggregates, as judged by AFM (Figures 3C and S3) and SDS-PAGE analysis (Figure S3), that are devoid of β-sheet fibrillar epitopes (Figure S3)). Moreover, the polyphenol concentrations required to eliminate the A11-epitope are precisely those required to eliminate the toxicity of Aβ oligomers relative to Aβ monomers (Figures 3D and S4). Our results strongly suggest that polyphenol aglycones that remodel Aβ soluble oligomers do so through a single remodeling pathway that yields large, non-toxic assemblies regardless of the molecular architecture of the aglycone variants.
Activity of polyphenol glycosides and aglycones for remodeling Aβ fibrils
Are polyphenolic glycosides and their aglycones specific for remodeling Aβ oligomers relative to other Aβ conformational variants such as fibrils and non-toxic oligomers? Aβ fibrils possess a combination of biochemical properties that are “opposite” from those of Aβ soluble oligomers. Aβ fibrils are SDS-insoluble, immunoreactive with the OC antibody but not with the A11 antibody and rich in β-sheet secondary structure, while oligomers are SDS-soluble, immunoreactive with the A11 antibody but not with the OC antibody, and devoid of β-sheet secondary structure.[7, 20, 21, 31] These highly dissimilar biochemical properties suggest that polyphenolic glycosides and their aglycones may either fail to remodel Aβ fibrils or do so through a unique remodeling pathway. To test this hypothesis, we first evaluated the activity of six pairs of polyphenolic glycosides-aglycones for rapidly eliminating fibrillar conformational epitopes in Aβ fibrils using OC antibody dot blot analysis. We find that multiple sugars (glucose and trehalose), a polysaccharide (heparin) and a simple phenolic glycoside (arbutin) fail to eliminate the OC epitope in fibrils (Figure 4A). However, six polyphenolic glycosides and five aglycone counterparts are active for rapidly remodeling Aβ fibrils (4 h; Figure 4A). Each glycoside is an order of magnitude less effective at doing so than for remodeling Aβ oligomers (Figures 1B, 2B and 4A), while the aglycones display identical effectiveness for remodeling fibrils and oligomers (Figures 1B, 2B and 4A). Importantly, both polyphenols and their glycosides convert β-sheet rich fibrils into conformers with random coil secondary structure (Figure 4B).
Figure 4. Characterization of remodeling pathways used by polyphenolic glycosides and aglycones to transform Aβ42 fibrils in alternative, non-toxic conformers.

Mature Aβ fibrils (25 μM) were incubated with polyphenols (4 h), and analyzed using (A) dot blots probed with conformation-specific (OC, fibrils) and sequence-specific (6E10) antibodies, (B) circular dichroism, (C) AFM, (D) ANS fluorescence, and (E) PC12 cell toxicity assay (n=3). In (C), soluble oligomers were incubated with the vehicle (1% DMSO) or 200 μM compound, and each image is 3 × 3 μm.
To evaluate whether polyphenols and their glycosides utilize the same opposing pathways for remodeling fibrils as they do for soluble oligomers, we performed AFM and SDS-PAGE analysis on the remodeled fibrils. Without exception, we find that each polyphenol aglycone remodels Aβ fibrils into large, off-pathway aggregates, while their respective glycosides dissolve fibrils into non-aggregated peptide (Figure 4C and S5). The ANS fluorescence spectra of fibrils remodeled with glycosides (λmax=452–454 nm) are similar to monomers (λmax=457 nm), while fibrils remodeled with their aglycone counterparts (λmax=497–504 nm) are intermediate to soluble oligomers (λmax=484 nm) and fibrils (λmax=526 nm; Figure 4D). Finally, the dose dependence of both classes of polyphenols for eliminating the OC-epitope (Figure 4A) and the toxicity of fibrils (Figures 4E and S5) are indistinguishable, revealing that remodeling fibrils into alternative conformers eliminates their toxicity relative to freshly disaggregated Aβ.
Based on our findings that multiple polyphenol aglycones transform Aβ soluble oligomers and fibrils into off-pathway aggregates via a single remodeling pathway and with identical dose dependence, we investigated whether other polyphenol aglycones that are simpler or more complex would also display such similar activity for remodeling Aβ oligomers and fibrils. As shown in Figure S6, we find that seven polyphenol aglycones active for remodeling Aβ fibrils do so with identical dose dependence as they do for Aβ oligomers (Figure 3). Moreover, each polyphenol remodels Aβ fibrils by converting them in large, off-pathway aggregates indistinguishable from remodeled Aβ oligomers (Figure S6) that are non-toxic (Figure S7).
We also investigated whether the activity of polyphenolic glycosides to disaggregate Aβ oligomers and fibrils is a synergistic combination of the aglycone and glycone moieties. Thus, we evaluated if mixtures of mono- and disaccharides with polyphenol aglycones would disaggregate Aβ oligomers and fibrils. In each case, we find that combinations of polyphenols and glycone moieties remodel Aβ oligomers and fibrils into large, off-pathway aggregates (Figure 5) in a manner identical to polyphenol aglycones in the absence of sugars (Figures 2D, 4C and S5).
Figure 5. Impact of glucose and trehalose on the pathways employed by polyphenol aglycones to remodel Aβ42 soluble oligomers and fibrils.

(A) Aβ soluble oligomers and (B) fibrils (25 μM) were incubated with glucose and trehalose with and without polyphenol aglycones (4 h), and analyzed using AFM. Each image is 3 × 3 μm (inset is 0.5 × 0.5 μm).
Polyphenol glycosides and aglycones fail to remodel Aβ non-toxic oligomers and monomers
Finally, we performed two other important control experiments. First, we evaluated the activity of polyphenolic glycosides and their aglycones for remodeling non-toxic Aβ oligomers that are the same size and morphology as toxic Aβ oligomers.[20, 21, 39] Interestingly, non-toxic oligomers are SDS-resistant, yet they possess random coil secondary structure and are negative for ThT as well for the A11- and OC-conformational epitopes.[20, 21] Importantly, both glycosides and their respective aglycones fail to remodel non-toxic oligomers over the same time interval (4 h) in which they are active for remodeling Aβ oligomers and fibrils (Figure 6). Moreover, we find that none of the polyphenol variants accelerate the aggregation of freshly disaggregated Aβ (Figure S8). Collectively, our findings reveal that both classes of polyphenols selectively remodel toxic Aβ conformers relative to non-toxic ones, and they do so through two opposing pathways (Figure 7).
Figure 6. Impact of polyphenolic glycosides and aglycones on mature Aβ42 non-toxic oligomers.
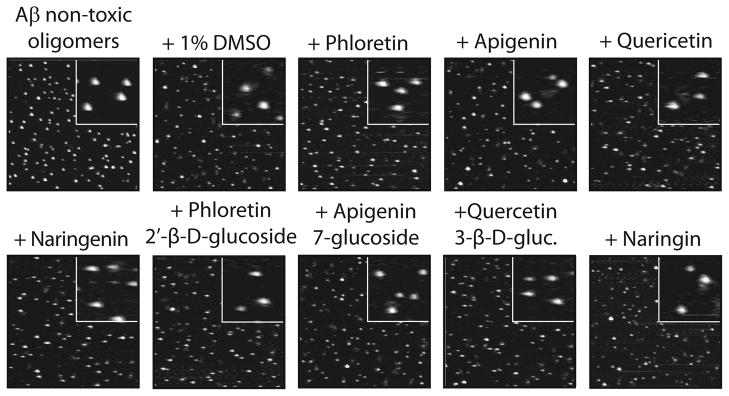
Aβ non-toxic oligomers (25 μM; formed after 5 d without agitation) were incubated with each small molecule (200 μM; 4 h) and analyzed using AFM. Each image is 3 × 3 μm (inset is 0.5 × 0.5 μm).
Figure 7. Summary of pathways used by polyphenolic glycosides and aglycones to selectively remodel Aβ soluble oligomers and fibrils into alternative, non-toxic conformers.

Polyphenol aglycones rapidly remodel Aβ soluble oligomers and fibrils into large aggregates that are SDS-insoluble, negative for multiple conformation-specific antibodies (fibril-specific antibody OC and oligomer-specific antibody A11), and non-toxic relative to freshly disaggregated Aβ. In contrast, polyphenolic glycosides remodel soluble oligomers and fibrils into low molecular species that are negative for multiple conformation-specific antibodies (OC and A11), SDS-soluble and non-toxic relative to freshly disaggregated Aβ. Both polyphenolic glycosides and aglycones fail to remodel non-toxic oligomers or accelerate aggregation of freshly dissolved Aβ.
Discussion
Our findings illuminate the structural determinants of polyphenolic glycosides and their aglycones to remodel Aβ oligomers and fibrils, as well as key mechanistic details about the opposing remodeling pathways these molecules employ. Importantly, we found that the remodeling activity of polyphenolic glycosides requires at least two phenolic rings (as do their aglycone counterparts), and that the aglycone and glycone moieties of such glycosides synergistically disaggregate Aβ oligomers and fibrils. Since polyphenols are suspected to target π-stacking interactions in aggregated conformers of Aβ and other amyloidogenic polypeptides,[40] we hypothesize that the aglycone portion of both classes of polyphenols initiate the remodeling process by disrupting intermolecular contacts between Aβ peptides in oligomers and fibrils. Disrupting such intermolecular contacts does not require per se that Aβ peptides are dissociated into monomers. Indeed, previous work reveals that EGCG remodels Aβ fibrils into off-pathway structures without dissociating them into monomers or small oligomers.[19]
Analogous to π-stacking interactions between aromatic residues, sugars are well known to interact with aromatic residues via so-called “CH-π” stacking interactions that are likely driven by association between aliphatic protons of sugar rings with theπ-electron cloud of aromatic rings.[41, 42] Thus, we posit that once the aglycone portion of polyphenols disrupts intermolecular contacts involving aromatic residues in Aβ oligomers and fibrils, the sugar moiety of polyphenol glycosides associates with newly exposed aromatic residues and prevents their promiscuous association with other Aβ residues. Importantly, the association of aromatic residues with sugars must be mediated by polyphenol aglycones since combinations of sugars and polyphenols fail to disaggregate Aβ oligomers and fibrils (Figure 5). Conversely, we posit that once polyphenol aglycones destabilize π-stacking interactions, they are incapable of preventing non-specific interactions involving such aromatic residues without a sugar moiety, leading to alternative aggregated conformers that lack secondary structure (e.g., β-sheets) and specific conformational epitopes recognized by the A11 and OC antibodies.
The failure of polyphenols and their glycosides to remodel Aβ non-toxic oligomers (Figure 6) is puzzling. Since Aβ non-toxic oligomers possess sizes and morphologies indistinguishable to soluble oligomers and are significantly smaller than fibrils,[20, 21] it is surprising that polyphenolic glycosides selectively target toxic Aβ conformers relative to non-toxic ones. These findings suggest that both classes of polyphenols recognize one or more structural attributes common to Aβ soluble oligomers and fibrils that are absent in non-toxic oligomers (as well as in monomers; Figure S8). The structural features shared by Aβ prefibrillar oligomers and fibrils are unknown since these two conformers possess highly dissimilar biochemical properties.[20, 21, 31, 38] The toxic nature of Aβ soluble oligomers and fibrils is the only common property between these conformers we identified relative to monomers and non-toxic oligomers. Thus, it is possible that polyphenolic glycosides and their aglycones recognize a common structural feature in oligomers and fibrils that also mediates the cellular toxicity of these conformers. One possible structural feature is π-stacking interactions known to occur in Aβ fibrils[43] and which we posit occurs in toxic oligomers but not in non-toxic oligomers. While speculative, this hypothesis would explain the differential remodeling activity of both classes of polyphenols for toxic and non-toxic Aβ conformers.
Conclusions
Recent studies have revealed that cells in diverse organisms ranging from worms to mice minimize the proteotoxicity of Aβ oligomers by related opposing pathways.[44, 45] The complex cellular pathways used to remodel toxic Aβ conformers into soluble peptide or large aggregates are poorly understood (see ref. [46] for recent progress). We expect that our identification of small molecules with well defined remodeling activities for toxic Aβ oligomers will facilitate studies of the cellular consequences of remodeling toxic Aβ oligomers into off-pathway aggregates relative to soluble peptide. Moreover, we expect that these molecules will be valuable chemical probes for identifying the elusive conformational epitopes that mediate cellular toxicity for Aβ oligomers and fibrils relative to non-toxic oligomers and monomers.
Experimental Section
Preparation of Aβ conformers
Aβ42 (American Peptide) was dissolved in an aqueous, 50% acetonitrile solution (1 mg/mL), aliquoted, dried under vacuum and lyophilized, and then stored at −20°C. The preparation of Aβ soluble oligomers, non-toxic oligomers and fibrils is described elsewhere.[21] Briefly, Aβ soluble oligomers and non-toxic oligomers were prepared by dissolving the peptide in 100% hexafluoroisopropanol (Fluka). Once dried, the peptide was reconstituted in 50 mM NaOH (1 mg/mL Aβ) and diluted in PBS (25 μM Aβ; pH 7.4). The peptide was then centrifuged (22,000xg for 30 min), and the pelleted fraction (5% of starting volume) was discarded. The supernatant was incubated at 25°C for 0–6 d without agitation. For preparing amyloid fibrils, aliquoted Aβ was solubilized as described above, diluted into PBS (25 μM), and mixed with pre-existing fibrils (10–20 wt% seed) without mixing for 24 h at 25°C. For the small molecule remodeling experiments, Aβ conformers were dosed with polyphenol aglycones and glycosides (all of which were obtained from Sigma) or the vehicle (1% DMSO), incubated at 25°C without agitation, and analyzed after 4 h.
AFM
Aβ samples (25 μM) were spotted on cut mica mounted on glass slides. The samples were adsorbed (30 min), and then washed with water and dried overnight. Images were taken using an Asylum Research MFP 3D AFM system with Olympus AC240TS cantilevers.
Cell toxicity assay
The procedure for assaying the toxicity of Aβ conformers is reported elsewhere.[21] Briefly, rat adrenal medulla cells (PC12, ATCC) were cultured in Dulbecco’s Modified Eagle Media (5% Fetal Bovine Serum, 10% Horse Serum and 1% Penicillin-Streptomycin). The cell suspension was aliquoted (90 μL) into 96 well microtiter plates (CellBIND, Corning) and allowed to adhere for 24 h. Afterward, Aβ or control samples (10 μL) were added to microtiter plates, and the cells were further incubated for 48 h at 37°C. The media was then removed, and fresh media (200 μL) and Thiazolyl Blue Tetrazolium Bromide (Sigma; 50 μL of 2.5 mg/ml) were added to each well (3 h, 37°C. Finally, these solutions were discarded, DMSO (250 μL) was added, and the absorbance was measured at 562 nm. The toxicity values were normalized to the PBS control without Aβ.
Gel electrophoresis and silver staining
Aβ samples (25 μM) were diluted into sample buffer (Novex LDS, Invitrogen), sonicated, analyzed using 10% Bis-Tris gels (Invitrogen), and silver stained (SilverXpress kit, Invitrogen).
Antibody dot blot analysis
Dot blot analysis of Aβ conformers (25 μM) was conducted as reported elsewhere.[21] Briefly, each Aβ sample was spotted (1 μL) on nitrocellulose membranes (Hybond ECL, GE Healthcare). The blots were probed with A11 (Invitrogen), OC or 6E10 (Millipore) antibodies, along with the appropriate horseradish peroxidase-conjugated secondary antibodies, and developed.
ANS fluorescence
8-anilino-1-naphthalene sulfonate (ANS, Sigma-Aldrich) was used at 75–125μM to assay the conformation of Aβ (25 μM). The ANS fluorescence spectra (λex=380 nm) was measured using a Tecan Safire2 plate reader.
Circular dichroism
The secondary structures of Aβ conformers (25 μM in 1% PBS; 300 μL) were evaluated using a Jasco 815 Spectrometer (1 mm path length cuvette) at 25°C. Each sample spectra is the average of at least fifteen readings.
Acknowledgments
We thank Dr. Charles Glabe for providing the OC antibody, Dr. Michel Weiwer for performing NMR analysis, and Dr. Robert Linhardt and Dr. Fuming Zhang for providing heparin. This work was funded by grants from the Alzheimer’s Association (NIRG-08-90967) and NIH (NS065333) to P.M.T.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Bulic B, Pickhardt M, Schmidt B, Mandelkow EM, Waldmann H, Mandelkow E. Angew Chem Int Ed Engl. 2009;48:1740. doi: 10.1002/anie.200802621. [DOI] [PubMed] [Google Scholar]
- 3.Fecke W, Gianfriddo M, Gaviraghi G, Terstappen GC, Heitz F. Drug Discov Today. 2009;14:453. doi: 10.1016/j.drudis.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Rochet JC. Expert Rev Mol Med. 2007;9:1. doi: 10.1017/S1462399407000385. [DOI] [PubMed] [Google Scholar]
- 5.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Nature. 2002;416:507. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 6.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Nat Neurosci. 2005;8:79. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 7.Glabe CG. J Biol Chem. 2008;283:29639. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts BE, Shorter J. Nat Struct Mol Biol. 2008;15:544. doi: 10.1038/nsmb0608-544. [DOI] [PubMed] [Google Scholar]
- 9.Frost B, Ollesch J, Wille H, Diamond MI. J Biol Chem. 2009;284:3546. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chien P, Weissman JS. Nature. 2001;410:223. doi: 10.1038/35065632. [DOI] [PubMed] [Google Scholar]
- 11.Tessier PM, Lindquist S. Nature. 2007;447:556. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Science. 2005;307:262. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 13.Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, Chiti F. Nat Chem Biol. 2010;6:140. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 14.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. J Biol Chem. 1997;272:22364. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 15.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. Nat Struct Mol Biol. 2008;15:558. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Duennwald ML, Roberts BE, Rozeboom LM, Zhang YL, Steele AD, Krishnan R, Su LJ, Griffin D, Mukhopadhyay S, Hennessy EJ, Weigele P, Blanchard BJ, King J, Deniz AA, Buchwald SL, Ingram VM, Lindquist S, Shorter J. Proc Natl Acad Sci U S A. 2008;105:7159. doi: 10.1073/pnas.0801934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Necula M, Kayed R, Milton S, Glabe CG. J Biol Chem. 2007;282:10311. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- 18.Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, Glabe CG. Biochemistry. 2007;46:8850. doi: 10.1021/bi700411k. [DOI] [PubMed] [Google Scholar]
- 19.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. Proc Natl Acad Sci U S A. 2010;107:7710. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladiwala AR, Dordick JS, Tessier PM. J Biol Chem. 2011;286:3209. doi: 10.1074/jbc.M110.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ladiwala AR, Lin JC, Bale SS, Marcelino-Cruz AM, Bhattacharya M, Dordick JS, Tessier PM. J Biol Chem. 2010;285:24228. doi: 10.1074/jbc.M110.133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong HS, Rana S, Barrigan L, Shi A, Zhang Y, Zhou F, Jin LW, Hua DH. J Neurochem. 2009;108:1097. doi: 10.1111/j.1471-4159.2008.05866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono K, Condron MM, Teplow DB. Proc Natl Acad Sci U S A. 2009;106:14745. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bitan G, Lomakin A, Teplow DB. J Biol Chem. 2001;276:35176. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- 25.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Proc Natl Acad Sci U S A. 1998;95:6448. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Agnaf OM, Mahil DS, Patel BP, Austen BM. Biochem Biophys Res Commun. 2000;273:1003. doi: 10.1006/bbrc.2000.3051. [DOI] [PubMed] [Google Scholar]
- 27.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Nature. 2002;416:535. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 28.Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. J Neurosci. 2005;25:2455. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Nat Med. 2008;14:837. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. Nature. 2006;440:352. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 31.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Science. 2003;300:486. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 32.Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. J Neurochem. 2007;100:23. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- 33.Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL. J Neurochem. 2001;79:595. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- 34.Zameer A, Kasturirangan S, Emadi S, Nimmagadda SV, Sierks MR. J Mol Biol. 2008;384:917. doi: 10.1016/j.jmb.2008.09.068. [DOI] [PubMed] [Google Scholar]
- 35.Ono K, Hasegawa K, Naiki H, Yamada M. Biochim Biophys Acta. 2004;1690:193. doi: 10.1016/j.bbadis.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Ono K, Condron MM, Ho L, Wang J, Zhao W, Pasinetti GM, Teplow DB. J Biol Chem. 2008;283:32176. doi: 10.1074/jbc.M806154200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riviere C, Delaunay JC, Immel F, Cullin C, Monti JP. Neurochem Res. 2009;34:1120. doi: 10.1007/s11064-008-9883-6. [DOI] [PubMed] [Google Scholar]
- 38.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Biochemistry. 2003;42:12749. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- 40.Gazit E. FASEB J. 2002;16:77. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- 41.Fantini J. Cell Mol Life Sci. 2003;60:1027. doi: 10.1007/s00018-003-3003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishio M, Umezawa Y, Hirota M, Takeuchi Y. Tetrahedron. 1995;51:8665. [Google Scholar]
- 43.Margittai M, Langen R. Q Rev Biophys. 2008;41:265. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 44.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Science. 2006;313:1604. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 45.Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Cell. 2009;139:1157. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bieschke J, Cohen E, Murray A, Dillin A, Kelly JW. Protein Sci. 2009;18:2231. doi: 10.1002/pro.234. [DOI] [PMC free article] [PubMed] [Google Scholar]