

Abstract
Introduction
Oncolytic virus therapy is a promising therapy for numerous tumor types. Edmonston-strain measles virus (MV) has been tested in clinical trials for ovarian cancer, glioma, and myeloma. Therefore, the antitumor activity of MV against NSCLC was assessed.
Methods
Human NSCLC cells and immortalized lung epithelial cell lines, Beas2B, were infected with either MV producing GFP (MV-GFP) or MV producing carcinoembryonic antigen (MV-CEA). Cells were assessed for viability, induction of apoptosis by caspase and PARP cleavage, and for viral transgene production. The dependency of MV entry on CD46 and nectin-4 were determined using blocking antibodies. The role of host translational activity on viral replication was assessed by overexpression of eIF4E and translation inhibition. Antitumor activity was assessed by measuring treated NSCLC xenografts from flanks of nude mice.
Results
MV infection of NSCLC cells results in potent cell killing in most of the cell lines compared to immortalized Beas2B cells and induces apoptosis. MV infection was prevented by blocking of CD46, however was independent of nectin-4 blockade. Tumor weights are diminished following intratumoral injections of MV-CEA in 1 of 2 cell lines and result in detectable viral transgene in serum of mice.
Conclusions
These data indicate that MV is oncolytic for human NSCLC and this was independent of nectin-4 expression. Dysregulated protein translational machinery may play a role in determining tumor tropism in NSCLC. MV combined with gemcitabine could be explored further as chemovirotherapy for NSCLC.
Keywords: non-small cell lung cancer, measles virus, translation
INTRODUCTION
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related death worldwide 1. Despite recent advances in targeted therapy, long-term survival of patients with NSCLC remains dismal. Novel treatment approaches are needed to extend survival of these patients and improve control of this disease.
Utilizing the tumor tropic properties of certain live-replicating viruses has garnered attention recently 2, 3. Edmonston strains of measles virus (MV) are promising oncolytic viruses as the MV receptor, CD46, is often overexpressed on many tumor tissues, resulting in more selective infection in cancer cells 4. Moreover, abnormalities in the innate antiviral response in cancer cells allow for viral replication and spread selectively within tumors 5. At doses used for vaccination, MV has a strong track record of safe use in humans making it an excellent candidate virus for clinical translation. Furthermore, the MV genome can accommodate insertion or deletion of genetic material allowing for MV to serve as a platform for novel recombinant viral gene therapy.
Previous work has demonstrated clear oncolytic effects of MV in many cancer types. A phase I trial was recently completed in heavily pre-treated patients with ovarian cancer with promising results6. Importantly, there was minimal toxicity seen. All patients on the study were previously immunized against MV; however, the neutralizing antibody titer remained relatively stable throughout the study.
Though there are some prior data on the effects of different strains of MV on NSCLC cells7, 8, there are little data regarding the MV strains used in clinical trials on NSCLC. Expression of CD46 has been previously documented in as high as 40% of NSCLC tumors 9, therefore MV could be a therapeutic agent appropriate for clinical translation in NSCLC. In the present manuscript, data are presented that show the anticancer activity of an Edmonston strain MV virus against NSCLC. These data show that MV replicates within NSCLC cells and in human xenografts and induces apoptosis. These results also show that part of the tumor tropism of MV for cancer cells may be determined by constitutively activated protein translation in NSCLC.
MATERIALS AND METHODS
Cell lines and reagents
Immortalized Beas2B cells, and NSCLC cells, H460, A549, H838, H520, H522, H2009, H2030 and murine Lewis lung cancer (LLC) were obtained from the ATCC. Human bronchial epithelial cells (HBECs) immortalized with CDK4 and hTERT were provided by John Minna, MD, PhD (UT Southwestern). HBECs were cultured with keratinocyte serum-free medium (KSFM; Life Technologies Inc.) media containing 50 μg/mL of bovine pituitary extract (BPE; Life Technologies Inc.) and 5 ng/mL of EGF (Life Technologies Inc.). Beas2B cells were grown in keratinocyte serum free media with 5 pg/mL human EGF and 0.05 mg/mL bovine pituitary extract (Gibco, Invitrogen). NSCLC cells were grown in RPMI 1640 supplemented with 10% calf serum (Biofluids) (R10). H838 cells were grown with R10 supplemented with 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/L glucose, and 1.5 g/L sodium bicarbonate. LLC cells were grown with DMEM media supplemented with 10% calf serum. The authenticity of these cell lines were verified by short tandem repeat analysis (Johns Hopkins). Rapamycin (Calbiochem) and 4EGI-1 (Chembridge Corp., San Diego, CA) were dissolved in 100% DMSO, aliquoted, and stored at -20°C until used. Gemcitabine (Eli Lilly and Co.) was dissolved in 1X PBS and stored at room temperature.
Viral Vectors
MV bearing the gene that encodes green fluorescent protein (MV-GFP) or human CEA (carcinoembryonic antigen) (MV-CEA) were constructed and propagated as described 10. The titer was determined by 50% end-point dilution assays [(TCID50)/mL] on Vero cells and stored at -80°C as described 11.
Cell Transfection
Beas2B were transfected with previously described pMSCV-M1GR1 constructs 12, either encoding or not encoding HA-tagged translational regulator eIF4E from mouse, linked to green fluorescent protein (GFP). GFP-positive cells were sorted on a fluorescence-activated cell sorting (FACS) DiVA (BD Biosciences). Immunoblots for HA-tagged eIF4E were performed on cell lysates to confirm the presence of the transgene.
In vitro cap-affinity assay
The strength of cap-mediated complex formation was measured using cap-affinity assay as described 13. Lysate from Beas2B cells that either do or do not produce exogeneous HA-tagged mouse eIF4E were diluted in lysis buffer and combined with 7-methyl GTP-Sepharose™4B (Amersham Biosciences) and incubated while mixing for two hours at 4°C. 100 μM 7-methylguanosine 5’-triphosphate (Sigma-Aldrich) was employed to elute eIF4E from the 7-methyl GTP-Sepharose™4B beads. The eluted samples were then prepared for immunoblot analysis.
Immunoblot analysis
Protein samples for the cap-affinity assay were separated by 8-15% gradient SDS-PAGE (polyacrylamide gel electrophoresis). Ten, twelve, or fifteen percent SDS-PAGE was used for other immunoblots as previously described 13. The primary antibodies employed were rabbit α-eIF4E, rabbit α-4E-BP1, rabbit α-PARP, α-eIF2α, α-phospho-eIF2α(Ser51), α-PKR, α-phospho-PKR, all from Cell Signaling at a 1:1000 dilution, mouse α-b-actin (Sigma) at a 1:10,000 dilution and rabbit α-eIF4GI (kindly provided by Nahum Sonenberg, McGill University Montreal, Quebec, Canada) at a 1:2500 dilution. Detection was performed utilizing ECL Plus Western Blotting System (Pierce) to visualize the bands of interest.
Viral infection and cell viability assays
Beas2B and NSCLC cells were seeded at 105 cells/well in 6 well plates. Following overnight incubation the cells were washed with Opti-MEM (Gibco) and then infected with MV-GFP or MV-CEA at different multiplicities of infection (MOI) (0.1, 0.5, 1.0) in 0.4 mL of Opti-MEM for 2 h at 37 °C. Complete medium for each cell line was then added to the wells for the duration of the treatment. For cells treated with MV-GFP, viral syncytia were visualized and photographed using light and fluorescence microscopy.
For combination treatments, cells (105 cells/well) were infected with MV-GFP at an MOI of 0.01. Rapamycin (Sigma) was used at a concentration of 50 nM. Gemcitabine was used at a concentration of 25 nM. 4EGI-1 was used at a concentration of 15μM. Controls were treated with equal volumes of the drug’s vehicle (DMSO or PBS, respectively). Cells were counted by trypan blue exclusion employing a hemacytometer 72 hours following infection for all cell viability assays. Cell survival is expressed as cell number normalized to untreated cells. Each experiment was performed in triplicate. For experiments employed to determine combination indices, 7500 cells/well were seeded onto 96 well plates. Following overnight incubation cells were treated with MV-GFP at an MOI of 0.1, and 0.25 and treated with varying concentrations of rapamaycin or 4EGI-1 alone and with different combinations for 72 h. All treatments were done in triplicate and analyzed for viable cells by CCK-8 assay (Dojindo Molecular Technologies, Inc.).
CEA level determination
Beas2B and NSCLC cells were treated with the indicated MOI of MV-CEA. Seventy-two hours later conditioned medium was collected from cells and analyzed for CEA. The CEA levels were measured using CEA ELISA kit (Bio-Quant, Inc.) following manufacturer’s instructions. Samples were analyzed in triplicate with both positive and negative controls included.
Apoptosis Assays
Apoptosis was assessed by immunoblot assay for PARP cleavage. Additionally, caspase-3 and -7 activities were measured using the Caspase-Glo 3/7 assay kit (Promega) following instructions provided by manufacturer. Cells were treated with MV-GFP at an MOI of 1.0 in Opti-MEM. 2 hours later, complete growth medium was added to the cells and cells were incubated at 37°C. After 48 hours of incubation, luminescence for each sample was measured after 1 hour incubation in Caspase-Glo reagent using a plate-reading luminometer (FLUOstar Omega from BMG Labtech). Background luminescence of blank wells was subtracted from the luminescence of sample wells. Caspase activity was normalized to untreated cells. These experiments were performed in triplicate.
Flow Cytometry
NSCLC cells were washed in PBS containing 1% BSA (PBS-BSA), centrifuged and resuspended in PBS-BSA. Cells were then incubated for 30 minutes at 4°C with phycoerythrin (PE) CD46 antibody (Abcam), anti-Nectin-4-PE (R & D Systems, FAB2659) or control isotype-IgG-PE antibody (Invitrogen), centrifuged and resuspended in PBS-BSA. Cells were analyzed for CD46 and Nectin-4 levels on a Bectin Dickinson FACSCalibur cytometer. Analysis was done using FlowJo software (Treestar).
Blocking of CD46 and Nectin-4
104 H2009 cells were seeded onto 96-well plates in Opti-MEM. The next day, cells were treated with Isotype IgG (R & D systems, AB-108-C), Nectin-4 IgG (R & D systems, AF2659) 14, CD46 IgG (Santa Cruz, sc-5267-clone m177), or both Nectin-4 and CD46 Abs at a concentration of 100 μg/mL. 1 hour after addition of the antibodies, MV-GFP was added to the wells at an MOI of 1.0. After 2 hours of infection, R10 media were added to the wells and cells were incubated for 48 hours. Cells were analyzed by fluorescence microscopy at 48 hours and photomicrographs were taken. In parallel, fluorescence was quantified using a 96-well plate reader. Cells were treated in triplicate. Uninfected cells and MV-GFP infected cells without blocking antibodies were used as negative and positive controls, respectively. Experiment was repeated two times.
Animal Experiments
All animal experiments were performed in concordance with Institutional Animal Care and Use Committee (IACUC) approved protocols at the University of Minnesota (Protocol #: 1103A97372). Nude mice were injected in the flank with 2.5 × 106 A549 or 3.5 × 106 H2009 cells. When tumors were 0.5 cm3, tumors were injected with 8.4 × 105 MV-CEA twice weekly for 3 weeks for a total of 6 intratumoral treatments. Tumors were measured in 2 dimensions weekly and tumor volume was estimated using the following equation: [long diameter × (short diameter2)]/2. Mice were weighed weekly. Blood was collected from mice for CEA determination. Whole blood was collected in EDTA-treated tubes (BD Bioscience), inverted to mix, centrifuged (1500 × g) and plasma was collected and stored at -80°C until further analysis. Animals were sacrificed one week following the last dose of MV-CEA using CO2 euthanasia. Tumors were excised and weighed. Ten mice were treated per cohort.
Statistical Analysis
Data are presented as means and error bars are standard deviation or standard error of the mean as indicated. Differences between groups were compared using two-sided t tests. A P-value of less than 0.05 was significant. Synergy of MV with 4EGI-1 or rapamycin was analyzed by the method of Chou and Talalay 15 using Compusyn (ComboSyn, Inc.).
RESULTS
The expression of the MV receptor, CD46, on NSCLC cell lines were compared to immortalized lung epithelial cells (Beas2B). While all the cells express CD46, the levels of CD46 were higher in H838, H2030, A549 and H460 cells than Beas2B while levels were comparable for the other cell lines tested (Figure 1A & B). Nectin-4 is expressed in up to 58% of NSCLC tumors and its expression is associated with a poorer prognosis 16. Nectin-4 has recently been identified as a receptor for MV, and therefore, we were interested to further evaluate the importance of nectin-4 expression in determining MV infection14, 17. Of the cell lines tested, nectin-4 was highly expressed only in H838, and H2009 cells. (Figure 1C & D). To further analyze the importance of nectin-4 and CD46 in mediating viral entry, H2009 cells were treated with blocking antibodies against nectin-4, CD46, or both. Isotype IgG antibodies were used as a control. Cells were then infected with MV-GFP. Blocking nectin-4 had virtually no effect on viral infection in H2009. On the contrary, blocking CD46 resulted in near complete inhibition of MV-GFP infection (Figure 1E & F). Blocking of both receptors did not yield different results from blocking of CD46. These results suggest that on H2009 cells that express Nectin-4, MV-GFP viral entry depends upon CD46.
Figure 1. A&B. CD46 expression on NSCLC cells.
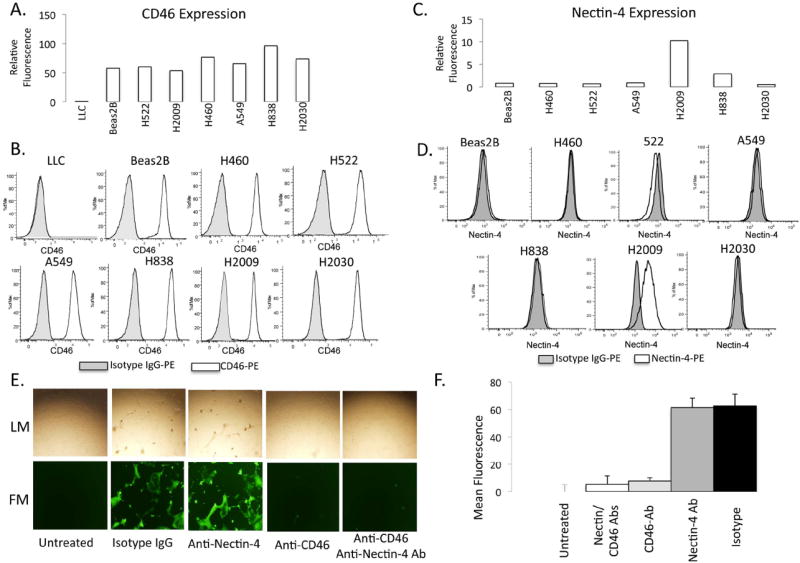
A) Plot of fluorescence intensity (relative to isotype) of cells stained with phycoerythrin-labeled anti-CD46 and assessed by flow cytometry. B) Representative histograms. LLC = murine Lewis Lung Carcinoma cell line as a negative control. C) Plot of fluorescence intensity (relative to isotype) of cells stained with phycoerythrin-labeled anti-Nectin-4 and assessed by flow cytometry. D) Representative histograms. E) Light (LM-top) and fluorescence (FM-bottom) microscopy of H2009 cells infected with MV-GFP after treatment with blocking antibodies against CD-46 and Nectin-4 or the combination. F) Quantification of fluorescence after blocking CD-46 and Nectin-4. Isotype IgG and H2009 cells untreated with MV-GFP were used as positive and negative controls, respectively.
Following treatment of cells with MV-GFP, growth inhibitory effects were seen in all cell lines tested in a dose-dependent manner (Figure 2A). Beas2B and H838 cells were relatively resistant to the cytotoxic effects of MV at all MOI tested. The rest of the cell lines tested were inhibited by MV treatment in vitro. Syncytia were visible by fluorescence microscopy at 24 hours and visibly increased daily at all MOI tested (Figure 2B) demonstrating that MV is able to enter and replicate within NSCLC cells. Separate experiments were performed using MV-CEA and the CEA levels detected by ELISA from treated supernatants 72 hours after infection. Each of the infected NSCLC cell lines robustly produced CEA several thousand fold greater than untreated cells, indicating that the CEA measured was representative of viral infection (Figure 2C).
Figure 2. A-C. MV infects and lyses NSCLC cells in vitro.
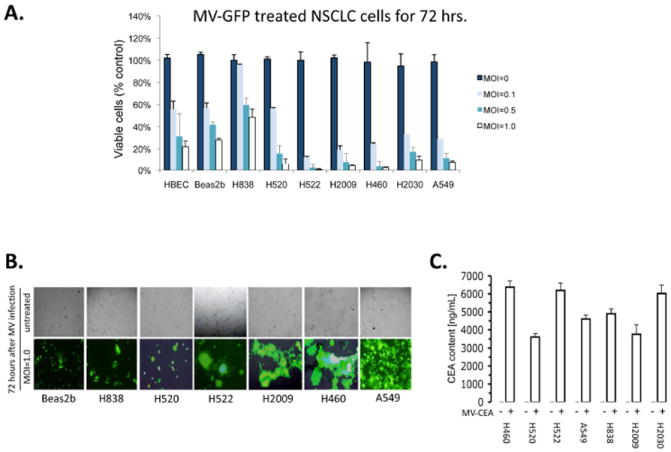
A) Cell viability assay of cells treated with MV-GFP after 72 hours of treatment. Immortalized bronchial epithelial cells (HBEC and Beas2B) are used as a non-malignant control. (MOI = multiplicity of infection). Data are expressed as a percentage of untreated cells. B) Fluorescence microscopy of cells 72 hours following treatment with MV-GFP at MOI of 1.0 demonstrating fluorescent syncitia. Corresponding light microscopy photographs are shown. C) Human CEA measurement from supernatants of MV-infected NSCLC cells. Supernatants were collected 48 hours after infection of cells and measured by ELISA assay (CEA=carcinoembryonic antigen). Error bars indicate standard deviation of the mean.
We next examined the mechanism by which MV induces death of the NSCLC cells. Viral infection often induces the cellular apoptotic response 18. Poly-ADP Ribose Polymerase (PARP) cleavage was assessed in NSCLC cells treated with MV or control as an indicator for induction of apoptosis (Figure 3A). PARP cleavage was induced for all of the NSCLC cell lines tested by 48 hours post-treatment. PARP cleavage was most pronounced in H2009 and A549 and to a lesser degree in H2030 and H838 cells. The same cells were assayed for Caspase 3/7 cleavage, which is an earlier indicator of apoptosis. Similar results were obtained for the cell lines tested (Figure 3B). Despite being relatively resistant to MV, H838 cells did undergo a 3-fold increase in caspase 3/7 activity after MV infection, however PARP cleavage was fairly minimal. It is unclear why there was this discrepancy, however PARP cleavage is downstream of caspase 3/7 activation 19-21. Therefore, it is conceivable that these cells harbor a defect in the execution phase of the apoptotic program, however this is speculative.
Figure 3. A & B. MV infection of NSCLC induces apoptosis.
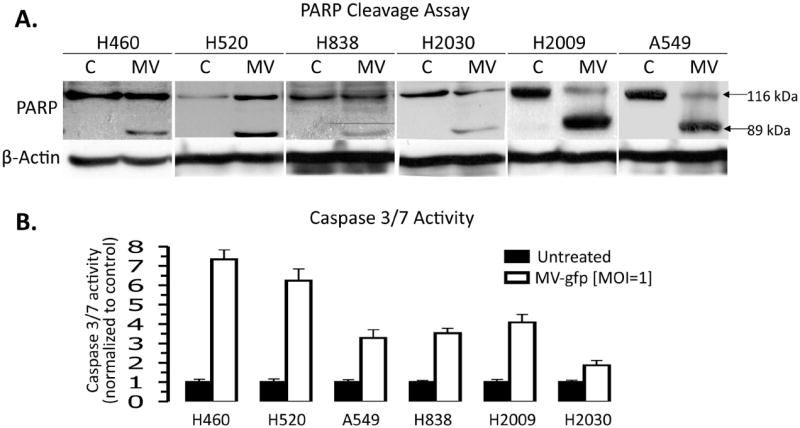
A) Immunoblot of PARP and PARP cleavage products following 48 hours of treatment with MV-GFP at an MOI=1.0. Presence of the 89 kDa fragement is indicative of apoptosis induction. B) Caspase 3/7 cleavage assay done on NSCLC cells 48 hours after treatment with MV-GFP at an MOI=1.0. Data are normalized to untreated NSCLC cells. Error bars indicate standard deviation of the mean.
Next, to determine the activity of MV against human NSCLC, we tested the effects of MV in nude mouse xenografts. NSCLC xenografts were grown in the flanks of nude mice and given twice weekly intratumoral injections of MV-CEA for 3 weeks. All mice were sacrificed 4 weeks following initial treatment and tumors were excised and weighed (Figure 4 A, B). Intratumoral injections of MV-CEA resulted in substantial activity against H2009 tumors resulting in markedly decreased tumor growth compared to controls (p<0.0001). H2009 tumor weights were substantially decreased by MV treatment. The results with the A549 xenografts were modest and there was no significant difference in measured tumor volumes. However, there was separation of the growth curves that was evident by the end of treatment and excised tumors were smaller (p<0.05). Since mice are not hosts for MV infection and do not make CEA, the sera of mice was assayed for CEA as an indicator of viral infection. In A549, the CEA in the blood quantitatively increased with each week of treatment suggesting that the virus was able to replicate and was sustained throughout the treatment period (Figure 4C). H2009 cells also had detectable CEA in the serum at the time of sacrifice one week after completing treatment with MV-CEA (data not shown). Human NSCLC express CEA as well 22. At later time, points, there was some CEA detectable in the serum of control mice, however, it was >3000 fold higher in MV-CEA treated mice suggesting that the majority of CEA detected was representative of viral transgene.
Figure 4. A-C. MV oncolysis in NSCLC xenografts.
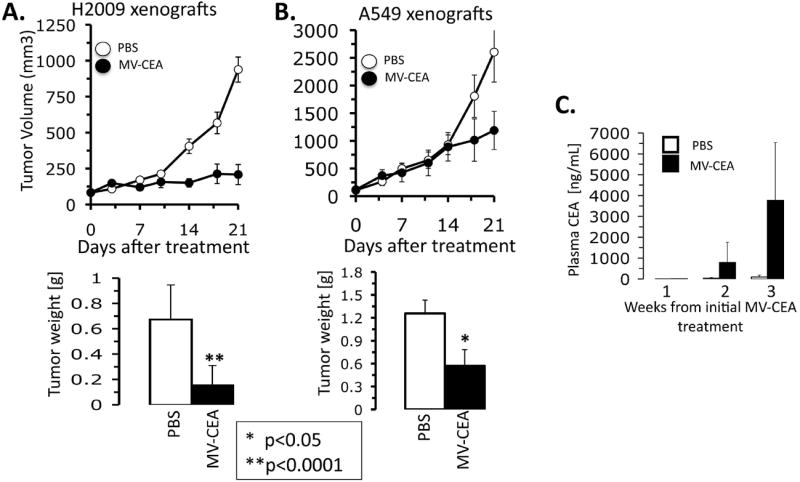
A) H2009 tumors treated for 3 weeks with twice weekly MV-CEA intratumoral injections. Tumor volumes were estimated by caliper measurements in two dimensions. 4 weeks after initial treatment, all mice were sacrificed and tumors excised and weighed. N=10 per group. B) A549 tumors were treated as above. C) CEA ELISA performed on blood samples collected weekly after treatment of mice bearing A549 xenografts. Error bars depict the standard error of the mean. P-value <0.05 was taken as significant.
One of the hallmarks of innate antiviral defenses is to inhibit viral protein synthesis by disabling translational initiation pathways. Because NSCLC is characterized by deregulated, constitutively active translation, this defect may promote viral replication and account for the tropism of oncolytic viruses for NSCLC 23. First, the effect of MV infection on factors regulating protein translation was assayed by immunoblot. The levels of eukaryotic initiation factor 4G (eIF4G), 4E (eIF4E), and its inhibitory binding protein (4E-BP1) were measured to assess the effects on key proteins regulating cap-dependent protein translation. Double-stranded RNA protein kinase (PKR) and its substrate, eukaryotic initiation factor 2α (eIF2α), were assessed by immunoblot to determine the effects on cap-independent translational initiation (Figure 5A). Expression and phosphorylation of 4E-BP1 was unchanged by MV infection compared to untreated cells. eIF4E was decreased in A549 cells after MV infection, however, it was unchanged in H838 and H2009. Notably, the phosphorylation of eIF2α was decreased in A549 and H2009 cells in response to MV infection. In H838 cells, the phosphorylation level of eIF2α was unchanged upon MV infection. Since phosphorylation of eIF2α results in blockade of ribosomal recruitment and inhibition of translation initiation, the decreased phosphorylation of eIF2α upon MV infection likely aids in allowing persistent viral translation. To test the potential effect of constitutively active eIF4F cap-dependent translational complex, Beas2B cells were engineered to overexpress eIF4E. Beas2B overexpressing eIF4E (Beas2B-E) and Beas2B transfected with empty vector (Beas2B-V) were treated with MV and effects on viability of the cells were assayed (Fig 5B). Beas2B-E was not statistically different from Beas2B-V with regards to cytotoxic effects of MV-GFP though there were numerically less viable cells at each MOI tested. Conversely, pretreatment of cells with rapamycin, an mTOR inhibitor and known inhibitor of translation initiation decreased virally-mediated CEA production and resulted in less than additive effects compared to either treatment alone (Fig 5C). In H2009, the combination of rapamycin and MV was less cytotoxic than rapamycin alone (p<0.05). At MOI of 0.01, MV had little cytotoxic effect on H522 cells. While the cytotoxicity data was not significant in H522, virally mediated CEA production was markedly reduced as a result of prior rapamycin treatment. Using MV-GFP in combination with rapamycin resulted in qualitatively decreased syncitia formation after 48 hours of infection compared to MV-GFP treatment alone in 4 NSCLC cell lines (Fig 5E). To confirm these results, a small molecule inhibitor of cap-dependent translation, 4EGI-1, was used alone and in combination with MV to assess the effect of translation initiation on MV-mediated cytotoxicity 24. Similarly, the combination of 4EGI-1 and MV was not statistically different from either drug alone. To expand on these results, 4 NSCLC cell lines were treated with MV-GFP, rapamycin, or 4EGI-1 alone along with the combination of MV with rapamycin and MV with 4EGI-1 to formally analyze for synergy/antagonism using the method of Chou and Talalay 15. MV-GFP was moderate to strongly antagonistic with both translation inhibitors particularly at higher MOI of MV-GFP used. One exception was H2030 with 4EGI-1 at the low dose range, which demonstrated synergy. Though translation inhibitors do not completely block viral replication, the above data suggest that veracity of viral oncolysis is dependent upon active protein synthesis machinery.
Figure 5. A-D. Effects of MV infection on protein translation signaling pathways.
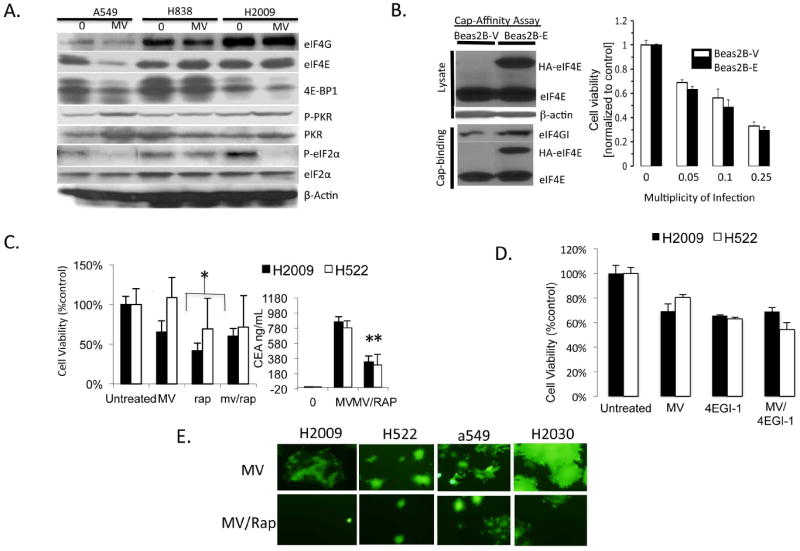
A) H2009, A549, and H838 cell lysates were assayed by immunoblot before and 48 hours after infection with MV-GFP for key proteins involved in cap-dependent and independent translation initiation. B) Beas2B expressing empty vector (Beas2B-V) and Beas2B expressing eIF4E (Beas2B-E) were infected with MV-CEA at indicated MOI and assayed for cell viability 72 hours after infection. 5’ cap-affinity assay of untreated Beas2B-V and Beas2B-E are shown. Relative increases in eIF4G binding in the cap-affinity assay correspond to enhanced eIF4F cap-complex formation. C) H2009 and H522 NSCLC cells were treated with MV-CEA (MOI=0.01), rapamycin (Rap), or the combination and assayed for cell viability 72 hours after infection. Supernatants were collected from cells and assayed for CEA production as a measure of viral replication. Error bars indicate standard deviation of the mean. * indicates statistical significance. D) H2009 and H522 cells were treated with MV-CEA (MOI=0.01), 4EGI-1 (15μM), or the combination and cell viability assayed 72 hours after infection. E) NSCLC cell lines were treated with MV-GFP at MOI of 0.25 either alone or in combination with rapamycin. Representative fluorescence micrographs are shown.
Perhaps the major limitation to MV for clinical translation in lung cancer is the fact that the majority of people in the developed world are immunized to measles by vaccination or previous infection. The presence of neutralizing antibodies could limit the ability of MV to reach sites of tumors, particularly for systemic diseases such as lung cancer. Therefore, suppression of the humoral immune response might be a reasonable way to counteract the elaboration of antibodies against measles. Cyclophosphamide has been successfully used to increase MV replication in animal toxicity studies in primates 25. Cyclophosphamide is not a standard therapy for lung cancer and therefore, it may not be an ideal drug to use in combination with MV for clinical translation in NSCLC. Gemcitabine, a nucleoside analogue, is a commonly used chemotherapeutic agent for NSCLC. In animal models, gemcitabine has been observed to selectively deplete Blymphocytes and as a result, may decrease the humoral immune response and increase MV-mediated oncolysis 26. Therefore, the combination of gemcitabine and MV was assessed in vitro to indicate whether or not this combination was one that could be pursued in future clinical trials of MV for lung cancer. The combination of gemcitabine and MV resulted in additional cytotoxicity compared to either treatment alone (Fig. 6). While these data do not imply drug synergy between the MV and gemcitabine, it does indicate that in vitro, gemcitabine does not have a negative impact upon viral oncolysis and support the notion that this combination could be pursued for further testing in NSCLC.
Figure 6. MV combined with gemcitabine results in enhanced cytotoxicity.
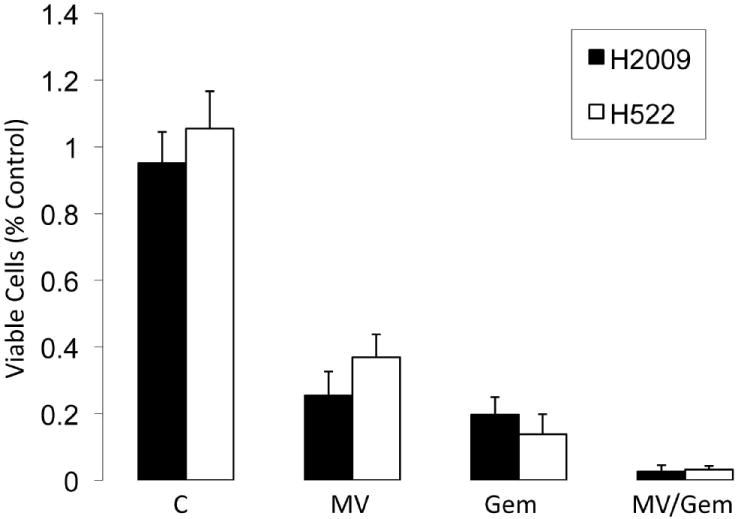
H522 and H2009 cells were treated with MV (MOI=0.1), gemcitabine (25nM), or the combination. Cells were assayed 72 hours after MV infection for viability by trypan blue exclusion. Error bars indicate standard deviation of the mean.
DISCUSSION
Data presented here demonstrate that MV has potential as a therapeutic agent for patients with non-small cell lung cancer. While the NSCLC cell lines tested all strongly express the MV receptor, CD46, there is also evidence in the literature that up to 40% of human NSCLC specimens strongly express CD46 by immunohistochemistry 9. In H2009 cells, CD46 was required for viral entry. Blocking of CD46 resulted in near complete inhibition of MV-GFP infection in H2009 cells. Though Edmonston-strain MV has been shown to utilize nectin-4 as a receptor for viral entry, the above data show that MV entry is not dependent upon nectin-4 expression in these NSCLC cells. There are potentially other mechanisms to allow viral entry. Recently, it has been demonstrated that MV can infect neuroblastoma cells independently of CD46 and nectin-4 expression, though the cellular receptor in that example remains undefined 27. Murine cells do not express CD46 and we could not demonstrate evidence of infection in murine LLC cells (data not shown). This is at odds with recently published literature, which suggests antitumor activity of a different MV strain (Hu-191) in LLC tumors in C57/Bl6 mice 28. Though this data needs to be reproduced, perhaps there is an immunologic explanation for the antitumor activity separate from viral replication. Though CD46 expression was substantially greater in the cancer cells than the immortalized Beas2B cells, CD46 is unlikely to fully explain the tropism for cancer cells. That H838 cells are relatively resistant to MV oncolysis despite possessing relatively high CD46 expression suggest that other mechanisms play a role as well.
PKR-induced eIF2α phosphorylation is one of the major innate cellular mechanisms to inhibit viral replication 29. Phosphorylation of eIF2α results in failure to recycle eukaryotic translation initiation factors and results in a global decrease in protein synthesis 30. H2009 and A549 cells that are quite sensitive to measles oncolysis in vitro show near complete dephosphorylation of eIF2α in response to MV infection. There was virtually no change in eIF2α phosphorylation in the relatively resistant H838. This may explain, in part, the relative resistance of H838 cells to MV-mediated lysis. On the other hand, the apoptosis data suggest that H838 cells may not fully be able to execute the apoptotic program after MV infection, which also may be playing a role in development of resistance to this therapy. These conclusions are purely speculative and further work will be required to determine if these mechanisms are truly playing a role in resistance to MV therapy.
Previous literature indicates that NSCLC have constitutive activation of cap-dependent translation 23. Overexpression of eIF4E, and constitutive activation of eIF4F complex, had an insignificant impact upon MV oncolysis of Beas2B cells. However, the combination of MV with rapamycin and 4EGI-1, resulted in moderate to strong antagonism. The combination of MV and rapamycin resulted in inhibition of viral expression of CEA, and decreased syncitia when infected with MV-GFP. Though the data presented cannot exclude the role of other defects in innate antiviral defenses, they are suggestive that translational control plays a role in MV oncolysis. These results are consistent with observed effects in mesothelioma cells31.
Based on the exquisite sensitivity of A549 cells in vitro to MV infection, the A549 xenograft results were somewhat unexpected. There was a trend toward the end of the experiment toward smaller tumors in the treated animals, and therefore, it is possible that longer observation may have been required to see a larger effect. Analysis of CEA in the serum indicates that MV was able to continue to replicate throughout the experiment. It has been observed that viral spread within A549 tumors might be limited by murine stromal elements when treated by intratumoral injection of oncolytic adenovirus 32. Like adenovirus, MV does not infect murine tissues and therefore, could be inhibited by a similar mechanism. Interestingly, recent work has delineated a potentially important role for tumor stromal elements in limiting viral spread that can be modified using angiotensin receptor blockers 33.
There are several caveats to the translation of these findings to the clinical setting. First, the majority of patients in the developed world has been vaccinated against MV, and therefore has circulating antibodies that can limit therapy with MV. The in vitro and immune deficient mouse models do not take this potential obstacle into account. Since mice lack the MV receptor and are not natural hosts to MV infection, use of known immune competent mouse models of lung cancer are precluded. For lung cancer, in particular, intratumoral injection may not be feasible in clinical practice for a disease that is hallmarked by bloodborne metastasis. Experience with systemic delivery indicates that immune suppression to decrease neutralizing antibodies may be more efficacious. Cyclophosphamide treatment blocks the humoral immune response and results in higher titer and longer duration of MV viremia in primates, and cyclophosphamide treatment with MV therapy is being tested for patients with multiple myeloma 34. Gemcitabine has been observed to result in similar inhibition of the humoral immune response, and therefore is a potentially useful drug to use in combination, as it is a clinically active drug against NSCLC. In support of this hypothesis, a recent clinical trial of oncolytic reovirus in combination with gemcitabine resulted in a muted neutralizing antibody response 35. In vitro, MV cytotoxicity is not inhibited by concomitant treatment of NSCLC cells with gemcitabine. Thus our results support the clinical translation of MV in combination with gemcitabine for patients with NSCLC. Due to the limited host range of MV, this question will be difficult to study further in lung cancer animal models.
In summary, these data suggest a potential role of MV in treatment of NSCLC. Further investigation will be required to evaluate the potential of systemic administration of MV and strategies to enhance viral delivery to tumors in the setting of neutralizing antibodies. A phase I study utilizing MV as a cancer therapy has now been completed and 3 others are ongoing, including by intravenous delivery for patients with myeloma. Thus far the treatment has been exceedingly safe. NSCLC is the leading cause of cancer death worldwide. Further development of this novel form of therapy for NSCLC is warranted.
Table 1.
Combination Index for MV-GFP and translation inhibitors.
| H2030 | H2009 | ||||
|---|---|---|---|---|---|
|
| |||||
| MV (MOI) | Rapamycin (nM) | Combination Index | MV (MOI) | Rapamycin (nM) | Combination Index |
| 0.1 | 1 | 5.48 | 0.1 | 1 | 3.96 |
| 0.1 | 10 | 8.41 | 0.1 | 10 | 7.99 |
| 0.1 | 50 | 8.5 | 0.1 | 50 | 5.03 |
| 0.25 | 1 | 236 | 0.25 | 1 | 57.6 |
| 0.25 | 10 | 46.7 | 0.25 | 10 | 35.9 |
| 0.25 | 50 | 49952 | 0.25 | 50 | 540.7 |
|
| |||||
| MV (MOI) | 4E-GI-1(μm) | Combination Index | MV (MOI) | 4E-GI-1 (μM) | Combination Index |
|
| |||||
| 0.1 | 1 | 17 | 0.1 | 1 | 9.419 |
| 0.1 | 10 | 1.07 | 0.1 | 10 | 12.299 |
| 0.1 | 50 | 2.41 | 0.1 | 50 | 460918 |
| 0.25 | 1 | 0.11 | 0.25 | 1 | 1.72 |
| 0.25 | 10 | 0.54 | 0.25 | 10 | 173 |
| 0.25 | 50 | 2.67 | 0.25 | 50 | 2.73E+14 |
|
| |||||
| A549 | H522 | ||||
|
| |||||
| MV (MOI) | Rapamycin (nM) | Combination Index | MV (MOI) | Rapamycin (nM) | Combination Index |
|
| |||||
| 0.1 | 1 | 1.55 | 0.1 | 1 | 0.79 |
| 0.1 | 10 | 0.59 | 0.1 | 10 | 0.77 |
| 0.1 | 50 | 3.08 | 0.1 | 50 | 1.15 |
| 0.25 | 1 | 0.957 | 0.25 | 1 | 1.49 |
| 0.25 | 10 | 1.22 | 0.25 | 10 | 1.29 |
| 0.25 | 50 | 1.93 | 0.25 | 50 | 1.53 |
|
| |||||
| MV (MOI) | 4E-GI-1(μM) | Combination Index | MV (MOI) | 4E-GI-1 (μM) | Combination Index |
|
| |||||
| 0.1 | 1 | 1.02 | 0.1 | 1 | 1.22 |
| 0.1 | 10 | 1.89 | 0.1 | 10 | 2.26 |
| 0.1 | 50 | 1.79 | 0.1 | 50 | 1.1 |
| 0.25 | 1 | 1.23 | 0.25 | 1 | 1.2 |
| 0.25 | 10 | 1.72 | 0.25 | 10 | 2.17 |
| 0.25 | 50 | 1.67 | 0.25 | 50 | 1.36 |
Acknowledgments
Funding sources: This work was funded, in part, by the NIH (5 T32 HL07062).
Footnotes
The authors have no relevant conflicts of interest to disclose
References
- 1.Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Blechacz B, Russell SJ. Measles virus as an oncolytic vector platform. Curr Gene Ther. 2008;8:162–175. doi: 10.2174/156652308784746459. [DOI] [PubMed] [Google Scholar]
- 3.Liu TC, Kirn D. Systemic efficacy with oncolytic virus therapeutics: clinical proof-of-concept and future directions. Cancer Res. 2007;67:429–432. doi: 10.1158/0008-5472.CAN-06-2871. [DOI] [PubMed] [Google Scholar]
- 4.Anderson BD, Nakamura T, Russell SJ, et al. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Peng KW, Dingli D, et al. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010;17:550–558. doi: 10.1038/cgt.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galanis E, Hartmann LC, Cliby WA, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boisgerault N, Guillerme JB, Pouliquen D, et al. Natural oncolytic activity of live-attenuated measles virus against human lung and colorectal adenocarcinomas. Biomed Res Int. 2013;2013:387362. doi: 10.1155/2013/387362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda M, Tahara M, Hashiguchi T, et al. A human lung carcinoma cell line supports efficient measles virus growth and syncytium formation via a SLAM- and CD46-independent mechanism. J Virol. 2007;81:12091–12096. doi: 10.1128/JVI.01264-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niehans GA, Cherwitz DL, Staley NA, et al. Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin) Am J Pathol. 1996;149:129–142. [PMC free article] [PubMed] [Google Scholar]
- 10.Peng KW, Facteau S, Wegman T, et al. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat Med. 2002;8:527–531. doi: 10.1038/nm0502-527. [DOI] [PubMed] [Google Scholar]
- 11.Langfield KK, Walker HJ, Gregory LC, et al. Manufacture of measles viruses. Methods Mol Biol. 2011;737:345–366. doi: 10.1007/978-1-61779-095-9_14. [DOI] [PubMed] [Google Scholar]
- 12.Avdulov S, Li S, Michalek V, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell. 2004;5:553–563. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 13.Patel MR, Jacobson BA, De A, et al. Ras pathway activation in malignant mesothelioma. J Thorac Oncol. 2007;2:789–795. doi: 10.1097/JTO.0b013e31811f3aab. [DOI] [PubMed] [Google Scholar]
- 14.Noyce RS, Bondre DG, Ha MN, et al. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011;7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 16.Takano A, Ishikawa N, Nishino R, et al. Identification of nectin-4 oncoprotein as a diagnostic and therapeutic target for lung cancer. Cancer Res. 2009;69:6694–6703. doi: 10.1158/0008-5472.CAN-09-0016. [DOI] [PubMed] [Google Scholar]
- 17.Muhlebach MD, Mateo M, Sinn PL, et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature. 2011;480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDonald CJ, Erlichman C, Ingle JN, et al. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast Cancer Res Treat. 2006;99:177–184. doi: 10.1007/s10549-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 19.Herceg Z, Wang ZQ. Failure of poly(ADP-ribose) polymerase cleavage by caspases leads to induction of necrosis and enhanced apoptosis. Mol Cell Biol. 1999;19:5124–5133. doi: 10.1128/mcb.19.7.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Los M, Mozoluk M, Ferrari D, et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Amours D, Sallmann FR, Dixit VM, et al. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J Cell Sci. 2001;114:3771–3778. doi: 10.1242/jcs.114.20.3771. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Kaye FJ, Henslee JG, et al. Expression of carcinoembryonic antigen and related genes in lung and gastrointestinal cancers. Int J Cancer. 1992;52:718–725. doi: 10.1002/ijc.2910520509. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson BA, Alter MD, Kratzke MG, et al. Repression of cap-dependent translation attenuates the transformed phenotype in non-small cell lung cancer both in vitro and in vivo. Cancer Res. 2006;66:4256–4262. doi: 10.1158/0008-5472.CAN-05-2879. [DOI] [PubMed] [Google Scholar]
- 24.Moerke NJ, Aktas H, Chen H, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 25.Peng KW, Myers R, Greenslade A, et al. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2012 doi: 10.1038/gt.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowak AK, Robinson BW, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemoimmunotherapy. Cancer Res. 2002;62:2353–2358. [PubMed] [Google Scholar]
- 27.Zhang SC, Cai WS, Zhang Y, et al. Engineered measles virus Edmonston strain used as a novel oncolytic viral system against human neuroblastoma through a CD46 and nectin 4-independent pathway. Cancer Lett. 2012;325:227–237. doi: 10.1016/j.canlet.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Zhao D, Chen P, Yang H, et al. Live attenuated measles virus vaccine induces apoptosis and promotes tumor regression in lung cancer. Oncol Rep. 2013;29:199–204. doi: 10.3892/or.2012.2109. [DOI] [PubMed] [Google Scholar]
- 29.Barber GN. Host defense, viruses and apoptosis. Cell Death Differ. 2001;8:113–126. doi: 10.1038/sj.cdd.4400823. [DOI] [PubMed] [Google Scholar]
- 30.Balachandran S, Barber GN. Defective translational control facilitates vesicular stomatitis virus oncolysis. Cancer Cell. 2004;5:51–65. doi: 10.1016/s1535-6108(03)00330-1. [DOI] [PubMed] [Google Scholar]
- 31.Sadiq AA, J BA, Jay-Dixon J, Patel MR, Russell SJ, Kratzke RA. Cap-dependent translational control of oncolytic measles virus in malignant mesothelioma. Proceedings of the 13th World Conference on Lung Cancer; San Francisco, CA. July, 2009. [Google Scholar]
- 32.Sauthoff H, Hu J, Maca C, et al. Intratumoral spread of wild-type adenovirus is limited after local injection of human xenograft tumors: virus persists and spreads systemically at late time points. Hum Gene Ther. 2003;14:425–433. doi: 10.1089/104303403321467199. [DOI] [PubMed] [Google Scholar]
- 33.Diop-Frimpong B, Chauhan VP, Krane S, et al. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A. 2011;108:2909–2914. doi: 10.1073/pnas.1018892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myers RM, Greiner SM, Harvey ME, et al. Preclinical pharmacology and toxicology of intravenous MV-NIS, an oncolytic measles virus administered with or without cyclophosphamide. Clin Pharmacol Ther. 2007;82:700–710. doi: 10.1038/sj.clpt.6100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lolkema MP, Arkenau HT, Harrington K, et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res. 2011;17:581–588. doi: 10.1158/1078-0432.CCR-10-2159. [DOI] [PubMed] [Google Scholar]