

Abstract

Cervical cancer is the sixth most common cancer in women worldwide and the leading cause of women’s death in developing countries. Nearly all cervical cancers are associated with infection of the human papillomavirus (HPV). This sexually transmitted pathogen disrupts the cell cycle via two oncoproteins: E6 and E7. Cells respond to E7-mediated degradation of pRB by upregulating the p53 tumor suppressor pathway. However, E6 thwarts this response by binding to the cellular E6-Associating Protein (E6AP) and targeting p53 for degradation. These two virus-facilitated processes pave the way for cellular transformation. Prophylactic HPV vaccines are available, but individuals already infected with HPV lack drug-based therapeutic options. To fill this void, we sought to identify small molecule inhibitors of the E6–E6AP interaction. We designed an ELISA-based high throughput assay to rapidly screen compound libraries, and hits were confirmed in several orthogonal biochemical and cell-based assays. Over 88,000 compounds were screened; 30 had in vitro potencies in the mid-nanomolar to mid-micromolar range and were classified as validated hits. Seven of these hits inhibited p53 degradation in cell lines with HPV-integrated genomes. Two compounds of similar scaffold successfully blocked p53 degradation and inhibited cell proliferation in cells stably transfected with E6. Together, these studies suggest that small molecules can successfully block E6-dependent p53 degradation and restore p53 activity. The compounds identified here constitute attractive starting points for further medicinal chemistry efforts and development into beneficial therapeutics.
More than 130 different types of mucosal and cutaneous human papillomaviruses (HPVs) have been identified.1 The alpha genus of mucosal HPVs is composed of both low risk (e.g., HPV6, 11) and high risk (e.g., HPV16, 18) forms.2 While low risk HPVs cause benign cellular proliferations, high risk HPVs are associated predominantly with cervical cancer,2,3 one of the leading causes of women’s deaths in developing countries and the sixth most common cancer in women worldwide.4 HPV infection has also been implicated in having a causative role in about 20% of head and neck cancers5 and is thought to be responsible for the majority of anal and vaginal cancers6 as well as approximately 35% of penile cancers.7
The E6 and E7 proteins from high risk HPVs mediate the oncogenic properties of the virus, in large part, by perturbing the cell cycle regulatory functions of the p53 and Retinoblastoma (pRB) tumor suppressor proteins, respectively. HPV E7 has also been suggested to mediate the degradation of pRb.8 In healthy cells, pRb cooperates with E2F/DP transcription factors to coordinate the transcription of S phase genes,9 while HPV E7 binds to pRb to perturb the progression into S phase.10 In response to deregulated entrance into S phase, p53 is normally modified post-translationally and stabilized, causing cells to undergo cell cycle arrest or apoptosis.11 However, HPV E6 forms a complex with the cellular E3 ligase E6-Associating Protein (E6AP) and targets p53 for degradation via the ubiquitin-proteasome pathway.12,13
In addition to their pRb and p53 inhibitor activities, the E6 and E7 proteins from high-risk HPVs perturb normal cell function in other ways. For example, the C terminal PDZ binding motif of HPV E6 targets the cytoplasmic membrane proteins hDLG, Scribble, MUPP1, and MAG1-3 for degradation.14−17 E6 can also bind four-way DNA Holliday junctions and can inhibit p300/CBP acetylation to disrupt p53-dependent gene activation.18,19
The HPV vaccines Gardasil (Merck) and Cervarix (GlaxoSmithKline) offer preventative care for millions of uninfected young adults.20 However, these vaccines are costly and were not designed to therapeutically treat those who are already infected with HPV. While the overall 5-year cure rate for cervical cancer is approximately 90%, it is considerably worse for cases where the cancer has spread to other organs (down to 15% according to the American Cancer Society). Since HPV-containing tumors have low levels of nonmutated p53 that is unable to function due to degradation, several therapeutic strategies have focused on p53 stabilization through blocking of E6 function, either with RNAi or antisense oligodeoxynucleotides.21,22 Such studies have resulted in increased p53 levels and inhibition of tumor growth in both tissue culture and animal models. Spurred by this success, the development of inhibitors to the E6–E6AP interaction, a prerequisite to p53 degradation,23 presents an opportunity to stabilize p53 levels and bring about cell cycle arrest or apoptosis in infected cells. Several specific inhibitors of the E6–E6AP interaction have been developed including the Pitx2a protein inhibitor,24 intrabodies,25 and alpha helical peptides;26,27 however, all show modest activity.
E6 is a small, monomeric protein of 19 kDa,28 and since the region of E6AP that is necessary and sufficient for E6 binding is a 20 amino acid α helix,29−31 this suggests that it is feasible to inhibit this interaction with a small molecule compound. Ten small molecule inhibitors were also identified after pharmacophore development and limited in silico screening;32 however, only one compound proved to be active in cells and only at high-micromolar concentrations. A more recently reported follow-up in silico screen from this earlier study produced a family of flavonoid compounds with IC50 values in the mid- to low-micromolar range in vitro. Two of these compounds (luteolin and CAF024) showed an increase in p53 and p21Cip1/Waf1 protein and decreased viability of HPV-positive cell lines.33
Given that the pharmacophore-based screening campaign described above resulted in E6 inhibitors with favorable properties, albeit low potency, we carried out a solution screen for inhibitors of the E6–E6AP interaction to obtain more novel and potent small molecule inhibitors. Toward this end, we developed a high throughput solution assay to screen for small molecule inhibitors of the E6–E6AP interaction. Through the screening of ∼88,000 diverse compounds, we identified seven inhibitors with IC50 values in the low-micromolar to mid-nanomolar range that were able to specifically block p53 degradation in HPV-derived tumor cell lines and two of the seven inhibitors of similar scaffold that specifically inhibited cell proliferation in cells stably transfected with E6. These HPV E6 inhibitors provide a framework for developing HPV inhibitors with possible therapeutic applications.
Results and Discussion
Development of a High Throughput Screen and Inhibitor Identification
A high throughput screen (HTS) for small molecule inhibitors of the E6–E6AP interaction was designed around a modified sandwich enzyme-linked immunosorbance assay (ELISA) (Figure 1). Full length HPV16 E6 was fused C terminally to glutathione-S-transferase (GST). To improve E6 solubility and decrease aggregation, all nonconserved, surface-exposed cysteine residues of the HPV16 E6 sequence were substituted to the analogous residues found in HPV1A E6, and the result is referred to as E6M. A similarly mutated E6 protein containing analogous cysteine to serine mutations in the context of the full length protein was shown to properly fold and to display in vitro and in vivo p53 degradation properties that were comparable to those of wild-type E6.28,34 To confirm the integrity of E6M, we performed in vitro pulldowns with E6M and E6AP versus other proteins to demonstrate that E6M pulled down E6AP but not other proteins (Supplementary Figure S1).
Figure 1.
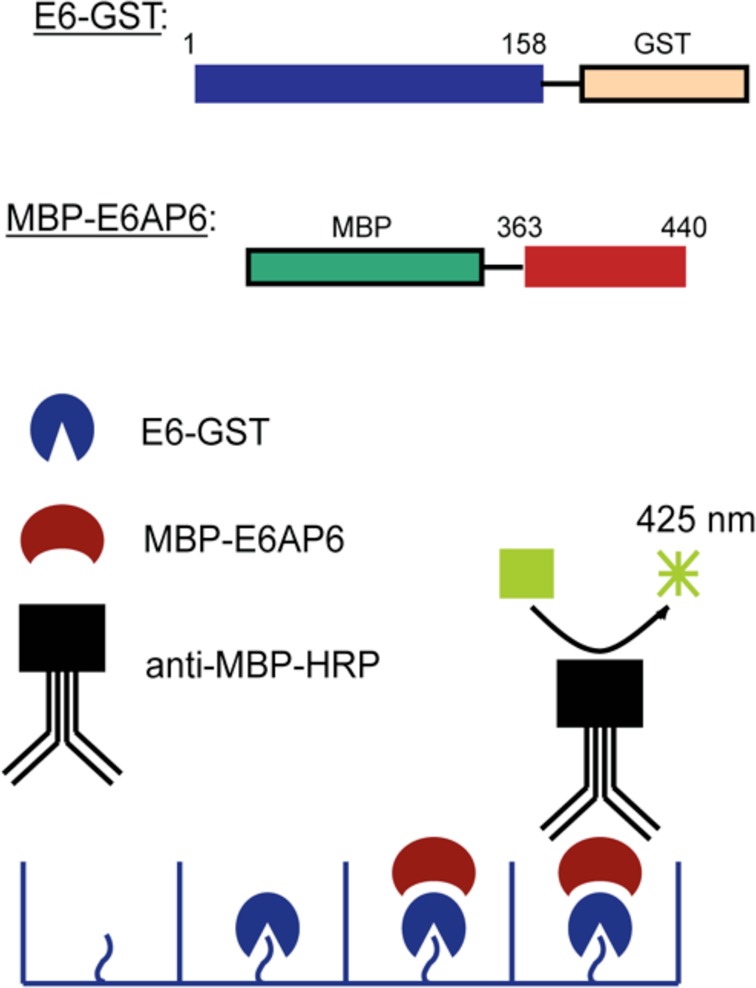
Scheme for the high throughput screening assay. The ELISA assay to detect compound inhibition (compound not shown) is represented schematically along with the fusion constructs used for screening.
Residues 363–440 of E6AP (isoform II), which contain the LQELLGE motif necessary for E6 binding,30 were fused N terminally to the maltose binding protein (MBP). Following binding of 30 nM E6M-GST to glutathione-coated wells, MBP-E6AP was added to the wells at a concentration of 6 μM (the reported Kd of the E6–E6AP interaction35) in the presence of a small molecule or a DMSO control. The extent of binding between the two proteins was assessed by incubation with the anti-MBP-HRP antibody and chemiluminescence.
The robustness of our assay was substantiated with Z′ factors36 of 0.784 (positive control: E6M-GST + MBP-E6AP; negative control: GST + MBP-E6AP) and 0.624 (positive control: E6M-GST + MBP-E6AP; negative control: E6M-GST + MBP) in 96-well format; miniaturization to a 384-well format and automation optimization resulted in Z′ factors consistently above 0.60 (Supplementary Figure S2).
Three different chemically diverse compound libraries were screened: Spectrum (Microsource; 2000 compounds), HitFinder (Maybridge; 14,000 compounds), and the Orthogonally Pooled Screening libraries (OPS, Lankenau Institute for Medical Research; 72,000 compounds), resulting in ∼88,000 total compounds. The initial screening resulted in the selection of 201 potential inhibitors of the E6–E6AP interaction (a 0.23% primary hit rate) that were further investigated in follow-up studies. All assigned hits were cherry picked and retested at the screening concentration in the ELISA assay. Following retesting, the number of potential inhibitors dropped to 54, producing a confirmed hit rate from the entire screen of 0.061% (Table 1).
Table 1. Screening Summary.
| library name | library size (no. of compounds) | hitsa | primary screenb | secondary assaysc |
|---|---|---|---|---|
| Spectrum | 2,000 | 7 | 4 | 4 |
| Maybridge HitFinder | 14,400 | 56* | 13 | 12 |
| OPS | 72,000 | 138* | 37 | 14 |
| totals | 88,400 | 201 | 54 | 30 |
Compounds that showed a signal 3 standard deviations from the mean were considered hits. Asterisks show which libraries included ambiguous and orphan hits to the original hit number.
Primary screen hits are the number of potential inhibitors as confirmed in the ELISA assay at the original screen concentration following cherry picking from their original stock solutions kept frozen.
Secondary assays hits are compounds that, following purchase of powders, passed all secondary assays for inhibition (thermostability and HPV-mediated in vitro p53 degradation).
Identification of Compounds That Reduce Association of E6 with E6AP in Vitro
We ranked the potency of the inhibitors by determining the compound concentration that reduced association between E6M-GST and MBP-E6AP by 50% (IC50 value). For each of the inhibitors tested, fresh powders were ordered directly from commercial suppliers, and 42 of the 54 compounds had IC50 values of 30 μM or less, with more than half of the inhibitors having IC50 values in the low- to mid-nanomolar range (Table 2, Supplementary Table 1). The IC50 curves from the ELISA-based assay of six of the compounds, to be discussed in more detail below, are shown in Figure 2a. To eliminate any artifacts from the ELISA-based format, these six inhibitors were retested by performing pulldowns on amylose beads onto which MBP-E6AP was immobilized. After incubation with inhibitor, the ability of E6M-GST to bind to E6AP was assessed by Western blot (Figure 2b). Each inhibitor was able to reduce association between E6M-GST to E6AP in this format, confirming that the inhibitors reduce this interaction in vitro.
Table 2. Secondary Assay Results Summary for Six Representative Inhibitor Compounds.
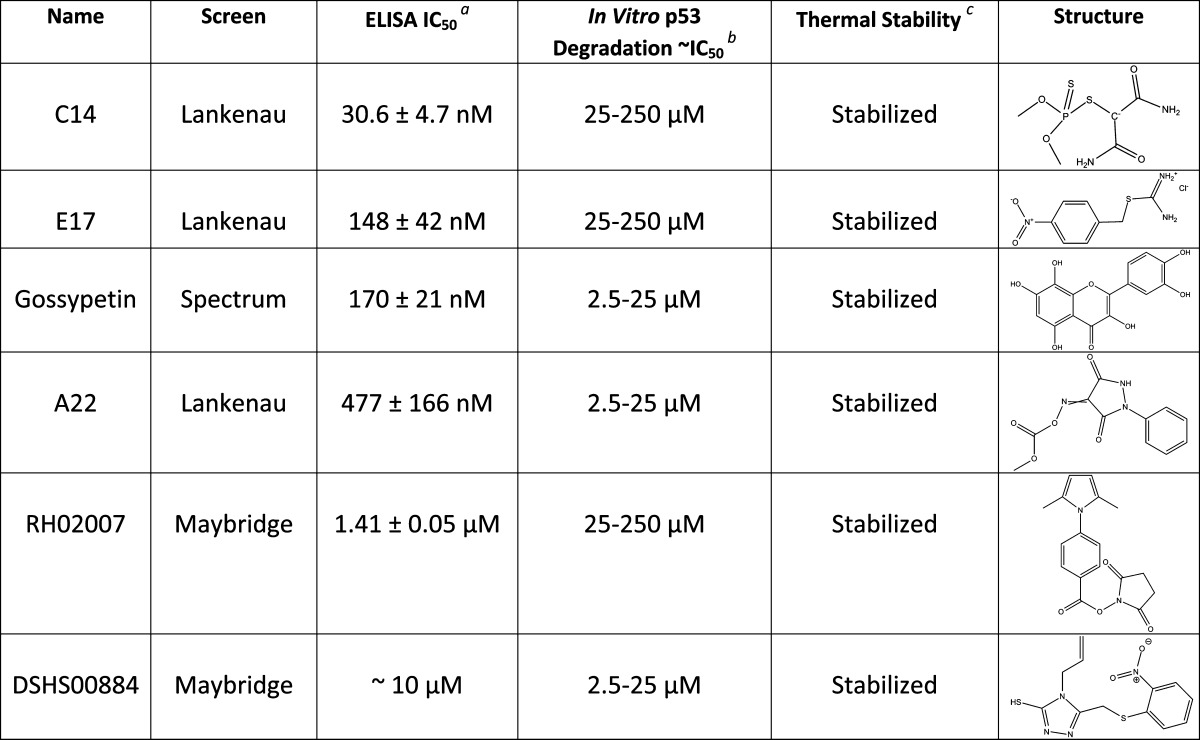
The ELISA IC50 values were determined by performing the assay in duplicate for each compound, followed by normalization of data and subsequent data fitting in GraphPad with the aid of log(inhibitor) vs response (variable slope) plots and nonlinear regression fit.
The degradation of p53, as determined by the in vitro experiments with both HPV 16 and HPV 18 E6, is reported as an approximate IC50 over a concentration range.
The thermal stability result is as described in the text.
Figure 2.

Reduction of E6–E6AP complex formation by small molecule inhibitors. (a) ELISA-based assay IC50 curves for the association of E6 with E6AP in the presence of compound. Calculated IC50 values indicating the concentration of compound needed to reduce binding by 50% are reported. (b) Pull-down-based assay Western blot results for the same compound in an orthogonal solution assay. The degree of GST-E6 association with MBP-E6AP at each compound concentration was assessed by Western blot.
To ensure that these 42 compounds were specifically acting on the E6–E6AP interaction before moving forward, a parallel screen using the same assay format for inhibitors against HPV E7’s ability to reduce pRb/E2F complex formation did not identify any of these compounds as hits (data not shown). Five of these compounds caused E6M precipitation and so were eliminated from further screening. We demonstrated that compound 9, a previously reported E6/E6AP binding inhibitor,32 had a dose–response inhibition of E6–E6AP binding using our ELISA assay (Supplementary Figure S3), albeit with a 10-fold lower IC50 value (1.59 μM) compared to the value reported by Baleja et al. (17 μM). We believe this difference is likely due to the lower protein concentrations used in our assay as compared to those used by Baleja et al. These combined data support the premise that the compounds that we identified in our high-throughput screen specifically reduce the level of E6–E6AP interaction.
Effects of Compounds on E6 Stability
To determine if the remaining 37 compounds interact directly with E6, a fluorescent E6 thermal stability assay was employed to monitor protein unfolding in the presence of a given inhibitor. Full length, untagged E6M was incubated at increasing temperatures (20–80 °C) with either DMSO control or with compound dissolved in DMSO and the reporter dye, SYPRO orange. The control reaction with DMSO showed that E6M was mildly unstable from 20 °C to approximately 37 °C (as indicated by the decrease in fluorescence signal), after which the protein melted in a standard sigmoidal fashion (Figure 3A). Overall, two different types of curves were observed from this assay with added compound. Fourteen compounds resulted in E6M melting curves that were indistinguishable from the protein melting curves without added compound (Figure 3B, Supplementary Table 1). Unchanged melting curves could indicate that these compounds were binding E6M but not significantly stabilizing its structure or that the compound mediated its function in other ways, most likely by binding to E6AP. Eighteen compounds showed significant stabilization of E6M in the region of 20–37 °C (as indicated by the sigmoidal fluorescence signal in the temperature range of 20–60 °C), which suggested that these compounds were binding to E6M (Figure 3C, Table 2, Supplementary Table 1). Five compounds resulted in flat melting curves that were difficult to interpret (data not shown) and ultimately were eliminated from potential hits following subsequent in vitro p53 degradation studies.
Figure 3.
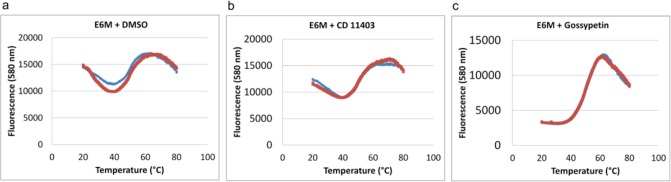
Effect of inhibitors on E6 thermal stability. Replicate data (blue and red) for fluorescence at 580 nm (SYPRO Orange bound to protein) vs temperature for each compound are shown as plots. (a) E6M alone illustrating inherent protein instability as indicated by the initial decrease and fluctuation of the fluorescence signal with increasing temperature over the range of 20–60 °C. (b) E6M with CD11403, representative of a compound that does not significantly stabilize E6M, as there is a modest initial decrease in fluorescence signal (compared to A) with a small but significant increase in fluorescence signal from 20 to 60 °C. (c) E6M with gossypetin, representative of a compound that stabilizes E6M (and presumably directly binds to the protein) as the fluorescence signal is stable between 20 and 37 °C and increases linearly between 37 and 60 °C to give an overall sigmoidal fluorescence signal. This type of thermal denaturation pattern is generally observed for the thermal unfolding of stable proteins.
Identification of Compounds That Reduce HPV16 E6-Mediated p53 Degradation in Vitro and in Cells
To establish if the E6 inhibitors could inhibit the ability of E6 to mediate p53 degradation, we assayed the same 37 compounds in a p53 degradation assay. Full length, wild-type HPV16 E6 and full length, wild-type human p53 were translated in separate reactions using the TNT T7 Coupling Reticulocyte System. The reactions were mixed together with compound and excess lysate containing E6AP and the machinery for ubiquitin-mediated degradation. The amount of p53 present in each sample was determined by a Western blot against the N terminus of human p53.
These studies revealed that seven compounds had no effect on p53 degradation in the cell-free system. Of the 30 compounds that did modulate p53 degradation (six are shown in Figure 4A), they increased p53 levels by about 2-fold relative to p53 levels in the absence of compound over a range of concentrations from mid-nanomolar to high-micromolar (Table 2, Supplementary Table 1, Figure 4A).
Figure 4.

Effect of compounds on p53 degradation in vitro using reticulocyte lysate. Representative Western blots of p53 degradation reactions treated with increasing concentrations of compound are shown with positive (E6 + p53 + DMSO) and negative (p53 + blank reaction + DMSO) controls and either (a) HPV 16 E6 added, (b) HPV 18 E6 added, or (c) in the absence of HPV E6.
These 30 compounds were then tested for their ability to inhibit p53 degradation in HPV-positive cells. Two different HPV-positive, tumor-derived cell lines were used: SiHa (HPV16 integrated) and HeLa (HPV18 integrated).38 Forty-eight hours after the addition of compound, cells were lysed and the level of p53 in each sample was assessed by Western blot using an anti-p53 antibody. Four compounds (DSHS 00884, RH02007, A22, and gossypetin) showed a significant increase in p53 levels in both cell lines over a compound concentration range of 1–10 μM, whereas E17 and C14 more modestly protected against p53 degradation in both cell lines (Figure 5A and B). The increases in p53 levels upon increasing compound concentration appear to be more substantial in HeLa cells compared to SiHa cells, and we attribute this to the inherently lower steady-state level of p53 protein in HeLa cells.39 Notably, these results suggest that our compounds are effective not only against HPV16 E6 but also against HPV18 E6 in reducing E6-mediated p53 degradation, further supporting the potential clinical utility of these findings.
Figure 5.

Effect of E6 inhibitors on p53 degradation in SiHa (HPV16-positive), HeLa (HPV16-positive), and HCT116 (HPV-negative) cells. p53 Western blots of cells treated with increasing concentrations of compound or DMSO control for 48 h in (a) HeLa, (b) SiHa, and (c) HCT116 cells.
To determine if the compounds’ effects on p53 were due to nonspecific cell stress, HCT116 cells (colon cancer cells bearing wild-type p53) were treated with these six compounds at the identical concentrations, and p53 levels were assessed by Western blotting following 48 h of treatment. Ethidium bromide was used as a positive control compound to show that endogenous p53 in these cells could respond appropriately to DNA damage. All compounds failed to increase p53 levels in this cell line; in contrast, ethidium bromide effectively stabilized p53 (Figure 5C). These results show that these compounds increased p53 expression by an E6-dependent mechanism.
The remaining six compounds (DSHS00884, A22, RH02007, gossypetin, C14, and E17) were then retested with the MTS assay in HCT116 cells to confirm their lack of cytotoxic effects. Staurosporine, a nonspecific kinase inhibitor, was used as a positive control.40 DSHS00884, A22, RH02007, and gossypetin were not toxic at all concentrations tested (1.6–25 μM), and C14 and E17 were not toxic at 12.5 μM or lower. None of these compounds exhibited significant toxicity in HCT116 cells at concentrations as high as 25 μM (Supplementary Figure S4). On the basis of these results, we conclude that these six compounds stabilize p53 levels in HPV-positive cells by blocking E6-mediated p53 degradation.
E6 Inhibitors Protect against p53 Degradation by E6 from HPV16 and HPV18
Since the six compounds exhibited some inhibitory activity in HeLa cells, which contain HPV18 E6, we tested whether these compounds could reduce p53 degradation in the cell-free system in the presence of HPV18 E6. As before, full length, wild-type HPV18 E6 and full length, wild-type human p53 were translated using the TNT T7 Coupling Reticulocyte System. As shown with HPV16 E6, the E6 inhibitors protected against p53 degradation by HPV18 E6 as well, and to a similar extent (Figure 4B). To ensure that the compounds were not resulting in increased p53 levels by some other means, such as an increase of p53 expression, the levels of p53 were determined in the absence of HPV E6. We found that the compounds alone did not affect p53 levels, leading to the conclusion that the six compounds reduce the ability of HPV16 E6, as well as HPV18 E6, from degrading p53 in a dose dependent manner (Figure 4C). Interestingly, the compounds were effective in the cell-free system at higher concentrations than in cells and in the ELISA assay. We attribute this to the fact that the levels of E6 and p53 are higher in our cell-free system, thereby requiring more compound to achieve the same effect.
Selected E6 Inhibitors Protect against p53 Degradation and Inhibit Cell Proliferation in Cells Stably Transfected with E6
While HPV expresses several viral proteins in an infected cell, only the continual expression of E6 and E7 are necessary for cellular transformation.41 E6 has several functions, but its role in p53 degradation is thought to be critical for cellular transformation. Actively disabling the cell’s p53 defense mechanism ensures host survival and propagation of deleterious mutations. We wanted to test the ability of these compounds to reactivate p53 function in an E6-expressing cell line where the p53 pathway was otherwise functional. For this purpose, we employed PA-1 cells (human ovarian teratocarcinoma), which have an otherwise functional p53 pathway and are stably transfected with HPV16 E6 (PA-1/E642). We used the compounds A22, RH02007, DSHS00884, gossypetin and a related compound, baicalein, for these studies (Supplementary Table 2).
PA-1 parental cells, PA-1/E6, and HCT116 cells were treated with gossypetin, baicalein, and puromycin for 24 h or A22, RH02007 and DSHS00884 for 72 h. Resazurin was then added to a final concentration of 5 μM, incubated for 1 h, and fluorescence emission of the resulting resofurin was read at 590 nm, where fluorescence of resofurin was a surrogate for cell viability. Puromycin was equally cytotoxic in all three cell lines, but only PA-1/E6 cells were sensitive to gossypetin and the related compound baicalein (Figure 6A). Cells began to visibly darken and shrink by 6 h post treatment with these two compounds and greater than 50% were nonviable at 24 h. Notably, neither PA-1 nor HCT116 cells responded in this manner to compound treatment. In contrast, the structurally unrelated E6 inhibitors, A22, RH02007 and DSHS00884, did not induce specific PA-1/E6 cell death. We noted that, in general, higher concentrations of these two compounds were required to kill the PA-1/E6 cells. We reasoned that this may be due to the higher levels of E6 in these cells as a result of the stable transfection as opposed to the normal, low levels induced by viral expression. As such, the compound concentrations tested in this assay may not have been sufficiently high for A22, RH02007 or DSHS00884. This conclusion is further supported by the ELISA assay IC50 values obtained for these compounds. Gossypetin and baicalein are more potent, as indicated by their sub-micromolar IC50 values, while both RH02007 and DSHS00884 are weaker effectors, with IC50 values of 1.41 and 10 μM, respectively. Interestingly, A22 has a sub-micromolar IC50 that is similar to gossypetin and baicalen, but it failed to inhibit PA-1/E6 cell proliferation. This suggests that there is something unique about the related structural scaffold of gossypetin and baicalen that allow them to be effective E6 inhibitors in cells.
Figure 6.

Effect of E6 Inhibitors on cells stabling expressing HPV16E6. (a) HCT116, PA-1, and PA-1/E6 cells were treated with different concentrations of compound and tested for viability. Puromycin (10 mg mL–1), gossypetin, and baicalein were tested after 24 h; RH02007 and DSHS00884 were tested after 72 h. (b) The same cells as in panel a were treated with the same concentrations of compounds, but cells were lysed and blotted for p53 levels.
To further confirm that gossypetin and baicalein are working specifically to block E6-mediated p53 degradation, we treated all three cell lines with compound, then lysed the remaining cells and blotted for p53 levels. As expected, p53 levels were unchanged in PA-1 and HCT116 cells, but these compounds induced a concentration dependent increase on p53 levels in PA-1/E6 cells (Figure 6B).
Conclusions
The purpose of this high throughput screen was to identify novel and potent inhibitors of the E6–E6AP interaction with the downstream effect of protecting against p53 degradation in HPV-infected systems. We identified 30 compounds that are able to reduce E6 binding to E6AP and subsequently block p53 degradation in a cell-free system. The activity of these compounds in HeLa and SiHa cells relied on their ability to traverse the plasma membrane of both cell types and to not become degraded over the time course of the experiment. Seven compounds demonstrated protection against p53 degradation in both SiHa and HeLa cell lines, although two of these (C14 and E17) had a modest effect. Significantly, these compounds did not increase p53 levels in the absence of E6, as shown in HCT 116 cells and in our cell-free system. This suggests that these compounds specifically block E6-mediated degradation and do not increase p53 levels by some other mechanism. Two flavonoid compounds, gossypetin and baicalein, were specifically able to inhibit cell proliferation of E6-expressing PA-1 cells, but not parental PA-1 cells or in other cancer cells lacking E6.
We did not observe an inhibition of cell proliferation by gossypetin and baicalein in HeLa and SiHa cells (data not shown), likely because the growth arrest and apoptotic pathways are largely disabled in these cell lines through other genetic alterations, in part because of the extensive time in which these cell lines have been in culture.43,44 In support of this premise, a failure of HeLa cells to stabilize p53 and induce p53-mediated growth arrest/apoptosis has been seen by others.45 Interesting, the recently reported pharmocaphore-based in silico screen for inhibitors of the E6–E6AP interaction also reported the identification of a family of flavonoid compounds as E6 inhibitors both in vitro and in E6-expressing cells33 albeit with lower potency than gossypetin and baicalein.
Although the remaining 23 compounds did not show positive results in cells, the cell-based liabilities of these compounds could be addressed through additional medicinal chemistry efforts. It is also likely that some of the inhibitors identified in the ELISA assay work through binding to E6AP, consistent with the observation that some of these compounds do not affect the thermostability of E6 (Figure 3).
The results described here also provide useful structure–activity relationship (SAR) information. For example, gossypetin, brazilin and baicalein share a chromenone scaffold and give similar results in all secondary assays. Together with recent identification of flavonoid compounds as E6 inhibitors33 suggests that this scaffold may be a particularly promising lead for further E6 inhibitor development. Another related pair of compounds is HTS13545 and HTS10308, which differ only in an amino for carbonyl substitution at the 2 position of the pyridine ring. Both compounds have very similar potencies, but HTS13545 stabilized E6M in the thermostability assay while HTS 10308 did not. This difference might be due to the ability of the amine group of HTS13545 to donate two additional hydrogen bonds to E6 thus leading to greater stabilization of the complex.
While E6 binds p53, the other oncogenic protein of HPV, E7, targets pRB to prematurely release E2F and bring about pRB degradation. Both pathways must be inhibited in order for HPV to replicate. Studies have shown that continual expression of these two genes is necessary to maintain malignancy, while suppression of both genes results in cellular death.46 Furthermore, others have reported that knocking down both E6 and E7 simultaneously can be more effective at inducing apoptosis in HeLa cells than knocking down E6 alone.47 To our knowledge, the E6 inhibitors reported here are the most potent HPV-E6 inhibitors reported to date and represent several different potential scaffolds that might be further developed into molecular probes or therapeutic agents that might be used in combination with other agents that block pRB binding to E7.48 Such combination studies could experimentally evaluate their utility as synergistic agents in a therapeutic setting to block HPV transformation in HPV-infected cells.
Methods
Expression and Purification of Recombinant Proteins
All E6M and E6AP fusion proteins were expressed recombinantly in bacteria and purified to homogeneity using affinity chromatography via the respective tags. Free E6M was produced by cleaving the 6xHis-SUMO-E6M fusion protein with SUMO protease and removing the SUMO fusion partner with Ni-NTA resin.
ELISA Assay
E6M-GST was added to each well in a 384-well glutathione-coated plate, incubated with shaking, and washed with buffer. MBP-E6AP was then added to each well followed by DMSO/compound. After incubation with shaking, all wells were washed with buffer, anti-MBP-HRP was added, plates were incubated and washed with buffer, and then ELISA Pico chemiluminescence substrate was added for analysis with a plate reader. GST or MBP only were used for negative controls.
High Throughput Screening and Data Processing
Spectrum, HitFinder, and OPS library compounds were screened at final concentrations of 6–10 μM. The percent E6–E6AP binding was calculated from raw luminescent values for each test compound, and compounds that displayed 3 standard deviation units (∼50% inhibition) from the average of E6–E6AP binding in the presence of DMSO were assigned as active. Software was used to deconvolute the orthogonally compressed data for both HitFinder and OPS37 and to group the compounds into the four categories of actives, ambiguous, orphan, and inactive.
E6 Thermal Stability Assay
Reactions contained E6M, SYPRO orange, and compound in buffer. Thermal melt curves were obtained by heating the protein from 20 to 80 °C and monitoring fluorescence using a Real Time PCR System. Control reactions contained protein or compound only.
In Vitro Pull-Down Assays
For the pulldown assays with or without inhibitor, E6M and MBP-E6AP were incubated with amylose resin (in the absence or presence of the indicated concentrations of compound), the resin was washed with buffer, and proteins were resolved on SDS-PAGE gel followed by Western analyses to determine extent of protein binding. Negative controls were performed with tag alone plus the partner protein or DMSO.
In Vitro p53 Degradation Assay
For in vitro translation, full length, wild-type human p53 and wild-type human papillomavirus type 16 or 18 E6 was cloned into expression vectors, and each protein was translated in separate reactions using the TNT T7 coupled rabbit reticulocyte lysate systems. For p53 degradation assays, the translation reactions were combined in the absence or presence of the respective compound at the indicated concentrations or a DMSO control. Following incubation, reactions were analyzed on SDS-PAGE followed by p53 Western blotting.
p53 Level Assay/Western Blots/Resazurin
On day 1, SiHa, HeLa, HCT116, PA-1, or PA-1/E6 cells were plated with media into a 6-well, clear tissue culture plate. On day 2, 2 μL of DMSO/compound was added to each well, and cells were incubated for the indicated amount of time. For the p53 level assays, cells were washed twice with buffer and then harvested with radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors. Protein sample concentrations were normalized with the bicinchoninic acid (BCA) protein assay, and samples were run on SDS-PAGE gel and transferred to a PVDF membrane for Western analysis.
For resazurin assay, 5 μM final concentration of resazurin was added and incubated for 1 h. Fluorescence was read at 590 nm.
Acknowledgments
This work was supported by NIH grants CA094165 and CA114046 and a Hiliary Koprowski, M.D. Professorship awarded to R.M., NIH grant R43EB009626 awarded to M.R., and NIH grant CA102184 to M.E.M. K.A.M. was supported by NIH training grant GM008275, and D.F. was supported by NIH training grant GM071339. We would like to acknowledge support of the Molecular Screening, Protein Expression and Libraries and Flow Cytometry core facilities at the Wistar Institute (supported by NIH grant CA010815) for the studies presented here.
Supporting Information Available
Supporting information and methods, supplementary figures, and supplementary table. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- de Villiers E. M.; Fauquet C.; Broker T. R.; Bernard H. U.; Hausen H. z. (2004) Classification of papillomaviruses. Virology 324, 17–27. [DOI] [PubMed] [Google Scholar]
- Hausen H. z. (2002) Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer 2, 342–350. [DOI] [PubMed] [Google Scholar]
- Hausen H. z. (1996) Papillomavirus infections - a major cause of human cancers. Biochim. Biophys. Acta 1288, F55–78. [DOI] [PubMed] [Google Scholar]
- Baseman J.; Koutsky L. (2005) The epidemiology of human papillomavirus infections. J. Clin. Virol. 32(suppl1), S16–S24. [DOI] [PubMed] [Google Scholar]
- Dufour X.; Beby-Defaux A.; Agius G.; Lacau St Guily J. (2011) HPV and head and neck cancer. Eur. Ann. Otorhinolaryngol. Head Neck Dis 129, 26–31. [DOI] [PubMed] [Google Scholar]
- De Vuyst H.; Clifford G. M.; Nascimento M. C.; Madeleine M. M.; Franceschi S. (2009) Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: a meta-analysis. Int. J. Cancer 124, 1626–1636. [DOI] [PubMed] [Google Scholar]
- Kreimer A. R.; Clifford G. M.; Snijders P. J.; Castellsague X.; Meijer C. J.; Pawlita M.; Viscidi R.; Herrero R.; Franceschi S. (2005) HPV16 semiquantitative viral load and serologic biomarkers in oral and oropharyngeal squamous cell carcinomas. Int. J. Cancer 115, 329–332. [DOI] [PubMed] [Google Scholar]
- Boyer S. N.; Wazer D. E.; Band V. (1996) E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56, 4620–4624. [PubMed] [Google Scholar]
- Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- Munger K.; Basile J. R.; Duensing S.; Eichten A.; Gonzalez S. L.; Grace M.; Zacny V. L. (2001) Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 20, 7888–7898. [DOI] [PubMed] [Google Scholar]
- Amundson S. A.; Myers T. G.; Fornace A. J. Jr. (1998) Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene 17, 3287–3299. [DOI] [PubMed] [Google Scholar]
- Scheffner M. (1998) Ubiquitin, E6-AP, and their role in p53 inactivation. Pharmacol. Ther. 78, 129–139. [DOI] [PubMed] [Google Scholar]
- Scheffner M.; Huibregste J. M.; Viestra R. D.; Howley P. M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505. [DOI] [PubMed] [Google Scholar]
- Gardiol D.; Kuhne C.; Glaunsinger B.; Lee S. S.; Javier R.; Banks L. (1999) Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 18, 5487–5496. [DOI] [PubMed] [Google Scholar]
- Thomas M.; Laura R.; Hepner K.; Guccione E.; Sawyers C.; Lasky L.; Banks L. (2002) Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene 21, 5088–5096. [DOI] [PubMed] [Google Scholar]
- Grm H. S.; Banks L. (2004) Degradation of hDlg and MAGIs by human papillomavirus E6 is E6-AP-independent. J. Gen. Virol. 85, 2815–2819. [DOI] [PubMed] [Google Scholar]
- Nakagawa S.; Huibregtse J. M. (2000) Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 20, 8244–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristriani T.; Masson M.; Nomine Y.; Laurent C.; Lefevre J. F.; Weiss E.; Trave G. (2000) HPV oncoprotein E6 is a structure-dependent DNA-binding protein that recognizes four-way junctions. J. Mol. Biol. 296, 1189–1203. [DOI] [PubMed] [Google Scholar]
- Thomas M. C.; Chiang C. M. (2005) E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol. Cell 17, 251–264. [DOI] [PubMed] [Google Scholar]
- Harper D. M. (2009) Currently Approved prophylactic HPV vaccines. Expert Rev. Vaccines 8, 1663–1679. [DOI] [PubMed] [Google Scholar]
- Niu X. Y.; Peng Z. L.; Duan W. Q.; Wang H.; Wang P. (2006) Inhibition of HPV 16 E6 oncogene expression by RNA interference in vitro and in vivo. Int. J. Gynecol. Cancer 16, 743–751. [DOI] [PubMed] [Google Scholar]
- Marquez-Gutierrez M. A.; Benitez-Hess M. L.; DiPaolo J. A.; Alvarez-Salas L. M. (2007) Effect of combined antisense oligodeoxynucleotides directed against the human papillomavirus type 16 on cervical carcinoma cells. Arch. Med. Res. 38, 730–738. [DOI] [PubMed] [Google Scholar]
- Cooper B.; Schneider S.; Bohl J.; Jiang Y.; Beaudet A.; Vande Pol S. (2003) Requirement of E6AP and the features of human papillomavirus E6 necessary to support degradation of p53. Virology 306, 87–99. [DOI] [PubMed] [Google Scholar]
- Wei Q. (2005) Pitx2a binds to human papillomavirus type 18 E6 protein and inhibits E6-mediated P53 degradation in HeLa cells. J. Biol. Chem. 280, 37790–37797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin H.; Elston R.; Jackson D.; Ansell K.; Coleman M.; Winter G.; Doorbar J. (2006) Inhibition of papillomavirus protein function in cervical cancer cells by intrabody targeting. J. Mol. Biol. 355, 360–378. [DOI] [PubMed] [Google Scholar]
- Butz K.; Denk C.; Ullmann A.; Scheffner M.; Hoppe-Seyler F. (2000) Induction of apoptosis in human papillomavirus-positive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proc. Natl. Acad. Sci. U.S.A. 97, 6693–6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Liu Z.; Androphy E.; Chen J.; Baleja J. (2004) Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus. Biochemistry 43, 7421–7431. [DOI] [PubMed] [Google Scholar]
- Nomine Y.; Charbonnier S.; Ristriani T.; Stier G.; Masson M.; Cavusglu N.; Dorsselaer A. V.; Weiss E.; Kieffer B.; Trave G. (2003) Domain substructure of HPV E6 oncoprotein: Biophysical characterization of the E6 C-terminal DNA-binding domain. Biochemistry 42, 4909–4917. [DOI] [PubMed] [Google Scholar]
- Cooper B.; Schneider S.; Bohl J.; Jiang Y.-h.; Beaudet A.; Pol S. V. (2003) Requirement of E6AP and the features of human papillomavirus E6 necessary to support degradation of p53. Virology 306, 87–99. [DOI] [PubMed] [Google Scholar]
- Huibregste J. M.; Scheffner M.; Howley P. M. (1993) Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol. 13, 4918–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Be X.; Hong Y.; Wei J.; Androphy E. J.; Chen J. J.; Baleja J. D. (2001) Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP. Biochemistry 40, 1293–1299. [DOI] [PubMed] [Google Scholar]
- Baleja J.; Cherry J.; Liu Z.; Gao H.; Nicklaus M.; Voigt J.; Chen J.; Androphy E. (2006) Indentification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins. Antiviral Res. 72, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J. J.; Rietz A.; Malinkevich A.; Liu Y.; Xie M.; Bartolowits M.; Davisson V. J.; Baleja J. D.; Androphy E. J. (2013) Structure based identification and characterization of flavanoids that disrupt human papillomavirus-16 E6 function. PLoS One 8, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomine Y.; Masson M.; Charbonnier S.; Zanier K.; Ristriani T.; Deryckere F.; Sibler A.-P.; Desplancq D.; Atkinson R. A.; Weiss E.; Orfanoudakis G.; Kieffer B.; Gilles T. (2006) Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 21, 665–678. [DOI] [PubMed] [Google Scholar]
- Zanier K.; Charbonnier S.; Baltzinger Mireille; Nomine Y.; Alstschuh D.; Trave G. (2005) Kinetic analysis of the interactions of human papillomavirus E6 oncoproteins with the ubiquitin ligase E6AP using surface plasmon resonance. J. Mol. Biol. 349, 401–412. [DOI] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Devlin J. J.; Liang A.; Trinh L.; Polokoff M. A.; Senator D.; Zheng W.; Kondracki J.; Kretschmer P. J.; Morser J.; Lipson S. E.; Spann R.; Loughlin J. A.; Dunn K. V.; Morrissey M. M. (1996) High capacity screening of pooled compounds: identification of the active compound without re-assay of pool members. Drug Dev. Res. 37, 80–85. [Google Scholar]
- Yee C.; Krishnan-Hewlett I.; Baker C. C.; Schlegel R.; Howley P. M. (1985) Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 119, 361–366. [PMC free article] [PubMed] [Google Scholar]
- Scheffner M.; Munger K.; Byrne J. C.; Howley P. M. (1991) The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc. Natl. Acad. Sci. U.S.A. 88, 5523–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg U. T.; Burgess G. M. (1989) Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol. Sci. 10, 218–220. [DOI] [PubMed] [Google Scholar]
- Alvarez-Salas L.; diPaolo J. (2007) Molecular approaches to cervican cancer therapy. Curr. Drug Discovery Technol. 4, 208–219. [DOI] [PubMed] [Google Scholar]
- Wu G. S.; El-Diery W. (1996) p53 and chemosensitivity. Nat. Med. 2, 255. [DOI] [PubMed] [Google Scholar]
- Friedl F.; Kimura I.; Osato T.; Ito Y. (1970) Studies on a new human cell line (SiHa) derived from carcinoma of uterus. I. Its establishment and morphology. Proc. Soc. Exp. Biol. Med. 135, 543–545. [DOI] [PubMed] [Google Scholar]
- Scherer W.; Syverton J.; Gey G. (1953) Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J. Exp. Med. 97, 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C.; Lu D.; Huang J.; Basu A. (2002) Regulation of p53 stabilization by DNA damage and protein kinase C. Mol. Cancer Therapy 1, 861–867. [PubMed] [Google Scholar]
- Alvarez-Salas L.; diPaolo J. (2007) Molecular approaches to cervical cancer therapy. Curr. Drug Discovery Technol. 4, 208–219. [DOI] [PubMed] [Google Scholar]
- Qi Z.; Xu X.; Zhang B.; Li Y.; Liu J.; Chen S.; Chen G.; Huo X. (2010) Effect of simultaneous silencing of HPV-18 E6 and E7 on inducing apoptosis in HeLa cells. Biochem. Cell Biol. 88, 697–704. [DOI] [PubMed] [Google Scholar]
- Fera D.; Schultz D. C.; Hodawadekar S.; Reichman M.; Donover P. S.; Melvin J.; Troutman S.; Kissil J. L.; Huryn D. M.; Marmorstein R. (2012) Identification and characterization of small molecule antagonists of pRb inactivation by viral oncoproteins. ACS Chem. Biol. 19, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.