

Abstract
The use of genome wide RNA interference (RNAi) screens to investigate host-viral interactions has revealed unexpected connections that have improved our understanding of viral pathogenesis and cell biology. This work describes the use of an RNAi screening method employing an immunofluorescence image-based strategy and influenza A virus. We find this approach to be readily implemented, scalable and amenable to the direct evaluation of a variety of viral lifecycles.
Keywords: siRNA screening, automated siRNA transfection, reverse transfection, functional genomics, host virus interactions, influenza A virus, whole genome screen
1. Introduction
1.1 Background
The Nobel prize winning discovery that double-stranded RNA can regulate gene expression by specifically degrading mRNA, combined with seminal work showing a similar strategy could be used in mammalian systems, has permitted researchers to deplete the expression of individual human genes using RNAi technology [1–2]. Together with the information provided by the sequencing of the human genome and the expressed sequence tagged data base, small interfering RNAs (siRNAs) have been designed against each human gene to create siRNA libraries with which to conduct genome wide loss-of-function screens. Thus, these combined technological breakthroughs allow genetic studies to be carried out in human systems on a level only possible before in simpler model organisms. Many groups, within both the academic and commercial sectors have contributed substantially to developing the tools and applications which have enabled the use of RNAi in high throughput mammalian genetic screens; this methodology report is indebted to those collective efforts and focuses on the use of siRNA technology to discover host factors required for influenza A virus replication [3].
1.2 Overview
Previous efforts to find viral-host interactions have been successful in discovering important steps in RNA virus biology, yet many basic processes in viral lifecycles remain uncharacterized. The advent of siRNA screens now permits an expanded approach for defining how RNA viruses replicate. Using RNAi-based genetics, we and others have completed genome-wide screens to find host factors required by influenza A virus infection [3–7]. In combination these efforts identified previously unappreciated, as well as known host factors involved in viral replication. Indeed, our screen “rediscovered” 11 of the 56 (19.6%) human orthologs found in an elegantly designed screen using a recombinant influenza A virus and dipteran cells [7]. These proteins participate in a broad array of cellular functions and implicate potential new pathways in the respective viral lifecycles. Example of newly discovered host factors that modulate influenza A viral replication using this approach include the IFITM3 protein, the COP1 complex, SART1 and multiple mRNA splicing factors, and CALCOCO2 among others (3).
2. Material and Methods
2.1 Suggested Equipment
The availability of the following equipment is suggested to perform this method: sterile tissue culture hood, automated plate filler with small bore dispensing cassette for volumes of 5 μl to 30 μl (i.e. Wellmate or Multidrop, Thermo Electron), a 12-channel pipette (Rainin), a liquid handling robot with 96 and/or 384 well plate pipetting capabilities (i.e. Bravo, Velocity 11), a screening microscope, (i.e. Image Xpress Micro, Molecular Devices, or In Cell 2000 General Electric), a benchtop centrifuge with 96 or 384 well plate holders, a tissue culture incubator, and an aspiration wand (i.e. 12 channel VP-185L-1 or 16 channel VP-186L-1, V & P Scientific)
2.2 siRNAs
To optimize conditions for siRNA transfection the depletion of polo kinase 1 (PLK1, Dharmacon SMARTpool M-003290-01) is useful because its reduction produces an easily monitored “rounded-up” cellular phenotype apparent at 18–24h post-transfection due to a G2/M cell cycle arrest. Positive control siRNAs for the influenza A virus experiments included a custom siRNA against the viral nucleoprotein (NP, GGAUCUUAUUUCCUUCGGAGUU produced by Dharmacon, [8]) and a SMARTpool directed against NFX1, a host mRNA exporter, previously found be required by influenza A virus in an elegant screen using drosophila cells (SMARTpool M-013680-01, [7]). In subsequent work based on the candidates found in the initial screens, a SMARTpool against the host vacuolar ATPase subunit, ATP6V0B (SMARTpool M-010907-02), reduced endosomal and lysosomal acidification in transfected cells with minimal toxicity, and strongly inhibited pH-dependent events such as HA’s acid-induced conformational change and viral infection. A non-targeting negative control from Dharmacon (NT, siCONTROL Non-Targeting siRNA #2, Dharmacon D-001210-02) has worked well in these assays, with the absence of increased toxicity and/or any impact on the lifecycles of multiple viruses.
2.3 siRNA Screen Library
To find host factors that modulate influenza A virus replication, we performed a whole genome siRNA screen using the Dharmacon siARRAY siRNA Library (Human Genome, G-005000-05, [3], Fig. 1). This library contains SMARTpools comprised of four sequence-unique siRNAs targeting each of 17,877 genes. The remaining 3,244 pools of the original 21,121 gene library were not screened because the majority were replaced by a new sub-library of 4,506 SMARTpools (Dharmacon Human 5: RefSeq27 Reversion Pools).
Fig. 1.
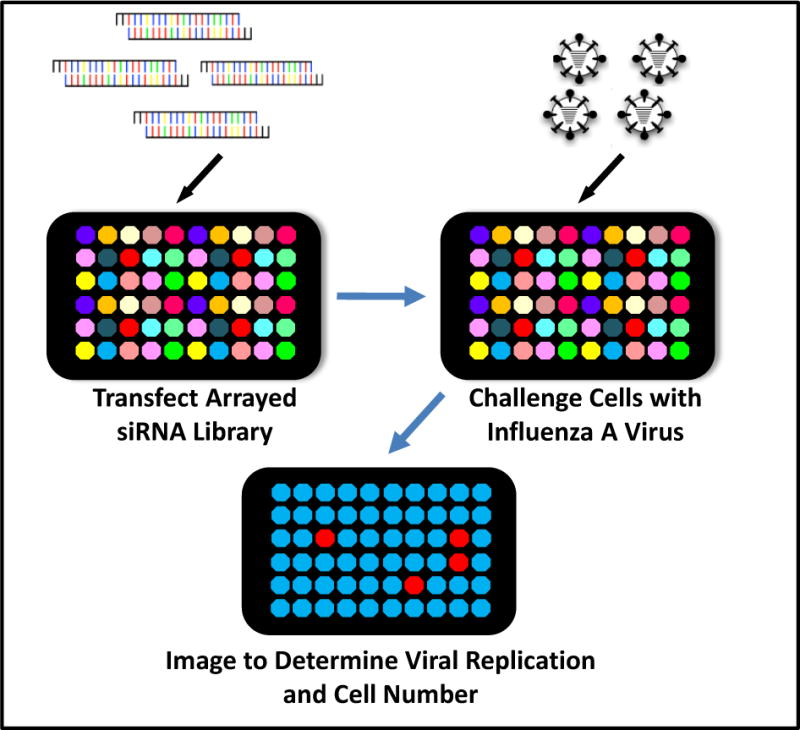
Loss-of-function assay to find host proteins involved in influenza A virus infection. The schematic depicts an image-based genetic screening method whereby individual host factor expression is depleted by an arrayed siRNA whole genome library. The siRNA transfected cells are then challenged with infectious influenza A virus. Twelve hr post-infection, the infected cells are stained for the expression of the influenza A viral protein (HA) to determine the role of the siRNA-targeted host factor in viral replication.
2.4 siRNA Transfection Reaction
For this screen we chose U2OS cells (ATCC) because they met criteria discussed in section 3.2. U2OS Cells were grown in DMEM (Invitrogen 11965) with 10% FBS (Invitrogen). Cells were kept at 50–80% confluency and were split the day before transfection. After 4 to 6 weeks in culture, the cells being used for screening were replaced with fresh cells that had been thawed and cultured for two weeks prior to use. A reverse transfection method using 384 well clear bottom and black wall plates (Corning 3712) was used for the genome wide screen. All procedures are carried out in sterile manner in a tissue culture hood. A master mix of Oligofectamine transfection lipid and Opti-MEM serum free media (both allowed to equilibrate to room temperature prior to use) is prepared first using the ratio of 0.1 μl lipid to 9.9 μl media for each well to be transfected. Of note, with different users performing optimizations using different lots of lipid, we have observed final concentrations of Oligofectamine in the transfection mixture ranging between 0.3 and 0.4% v/v.
2.5 siRNA Transfection Assay
A Wellmate is employed to dispense 9 μl of the lipid mixture per well. The filled plates are then centrifuged at 1000 rpm for one minute to drive all of the lipid mixture to the bottom of the well. The plates are now transfected using the Bravo Velocity 11 and dispensing a desired volume of 1.5 μl of a 1 μM siRNA stock into three Corning 3712 plates containing 9 μl matured lipid-Opti-MEM mix as follows: Tips (Fluotics P30-V11.NS) on; then aspirate 5.2 μl volume from the siRNA library plate. In series, dispense 1.6 μl into the first plate, 1.55 μl into the second plate, and the remaining solution into the third plate. Then beginning with the last plate receiving the siRNA solution, perform seven sequential mixing steps of 6 μl each.
While the transfection reactions are incubating for 20 min, trypsinize and resuspend the U2OS cells in DMEM with 15% FBS. We have found that using this concentration of FBS has decreased transfection-induced toxicity. Maintaining a consistent cell count and plating density is a key step and therefore cell counting should be done in duplicate to improve accuracy. For optimizing cell number please see section 3.5. Once the 20 min incubation period is complete add 20 μl. of the cell suspension per well using the Wellmate. Spin the filled plates as above and incubate at 37°C and 5% CO2 for 72 h. If siRNAs are present in either of the outermost two rows or columns, then the day after transfection add 5 μl of DMEM with 15% FBS to those wells to decrease toxic edge effects.
2.6 Viral Infection
Prepare a viral inoculum at a multiplicity of infection (moi) that produces 30 to 40% infection by IF staining of HA protein. Remove media using the 16 channel aspiration wand and add influenza A/Puerto Rico/8/34 (PR8, ATCC VR-1469) in DMEM 10% FBS. Spin plates for five minutes at 1000 rpm and return to the tissue culture incubator for 12h. For this method we have used either PR8 or influenza A/WSN/33 (WSN/33, kind gift of Dr. Peter Palese) grown in MDCK cells (ATCC) using DMEM with 2%FBS. For propagation of PR8 we use 2 μg/ml. of L-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK)-Trypsin (Worthington LS003744). The WSN/33 strain undergoes proteolytic processing of the HA protein in the absence of exogenous trypsin making it useful to screen for host factors required for budding or infectious particle maturation [6].
2.7 Immunoflourescence Assay and Processing
After 12h of viral infection the media is aspirated. Alternatively, if using WSN/33 virus, the media can be replica plated onto a fresh recipient plate of MDCK cells in a well by well manner, to assess for the levels of infectious particles. Using the Wellmate and a separate dispensing cartridge dedicated to these processing steps, add 4% v/v paraformaldehyde (PFA) solution (Sigma F1635) in D-PBS and incubate for 30 min. Wash (all wash steps are done twice with D-PBS) and then add primary antibody solution. We have used both purified antibody or conditioned hybridoma media, both from the same anti-HA secreting hybridoma cell line (anti-HA Hybridoma HA36-4-5.2, Wistar Institute, section 2.9). After a one hour incubation at room temperature, wash and then incubate with the secondary antibody, Alexa Fluor 488 goat anti-mouse, at 1:1,000 dilution (Invitrogen A11001) for 30 min. Wash and add (and let remain), D-PBS with Hoechst 33342 (DNA stain, 1:5-10,000 dilution of a 10 mg/ml stock from Invitrogen H3570). Stained cells are imaged on an automated Image Express Micro (IXM) microscope (Molecular Devices), using a 4X magnification of a single site per well acquired using both the FITC (HA-signal) and DAPI (cell nuclei) wavelengths. Images were analyzed using the Metamorph software program (Molecular Devices Inc.) using the cell scoring module (Fig. 2).
Fig. 2.
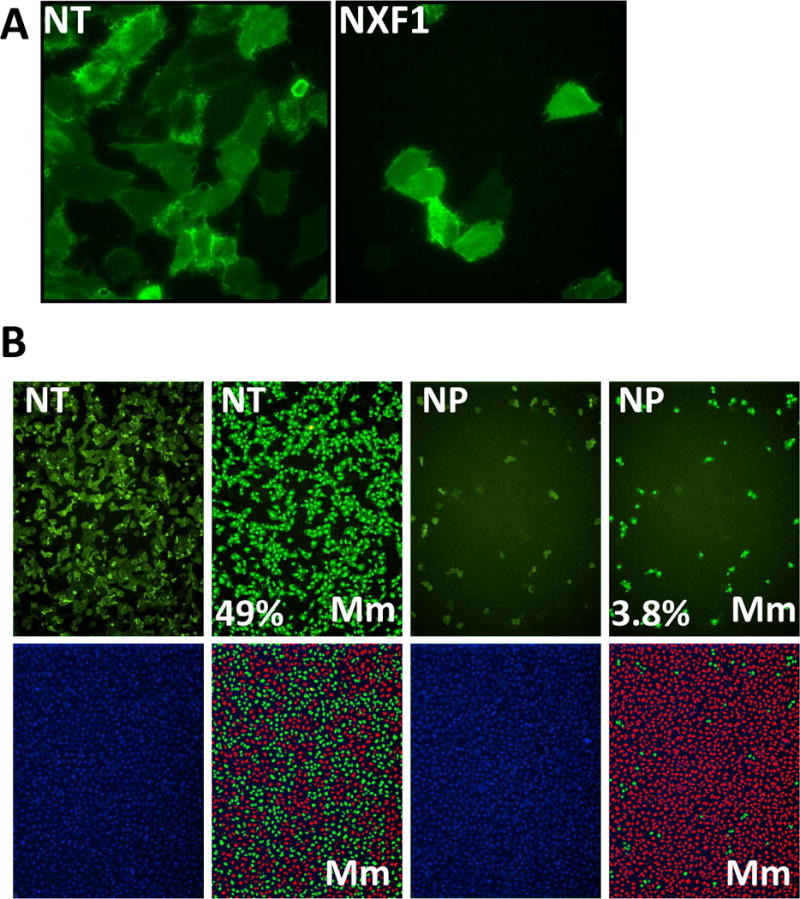
Image-based screen for host factors that modulate influenza A virus replication. A) U2OS osteosarcoma cells transfected with the indicated siRNAs for 72h, then infected with PR8 for 12 hr and stained for HA protein expression (green). Non-targeting negative control siRNA (NT), host mRNA exporter required for influenza A virus replication, NFX1. Nuclei are not depicted but were equivalent in each condition. 20× magnification. B) U2OS cells (left panels) were treated as in A) and the images analyzed with the Metamorph (Mm) cell scoring software module (right panels, denoted by Mm). Cells and their corresponding nuclei determined to be infected based on HA-staining intensity and cell size parameters are masked in green, while uninfected nuclei are masked in red. Numbers indicate percent infected cells and are provided in the lower right. 4× magnification. siRNA directed against the viral nucleoprotein (NP) [8].
2.8 Hit Selection and Validation Round Analysis
In the primary screen, siRNA pools are selected as hits if the mean of triplicate wells demonstrate that the percentage of HA-positive cells is less than 55% of the average plate mean without having less cells than 40% of the mean of the plate. Pools which increase infection by greater than 200% of the plate mean are also of interest as potential anti-viral factors i.e. IFITM3 [9]. While this is a somewhat arbitrary cutoff it is based on the ability to score a phenotype of sufficient magnitude to promote validation and further evaluation. However, if the number of genes able to be followed up on is significantly less than meet these criteria it is suggested that the researcher select high interest (novel) candidates and pathways based on the existing literature and bioinformatics, including functional class (druggable: enzymes, channels, receptors), functional clustering and pathway enrichment. In the validation round screen the individual siRNAs from each SMARTpool are rescreened individually using the identical transfection protocol as the primary screen and 50 nM final siRNA concentration. siRNAs are classified as hits using the same criteria as in the primary screen, with the exception that comparison is made to the values returned by four or more siRNA negative control wells present on each validation round plate. In the validation round, genes which have two or more individual siRNAs (out of four total) which score as positive are considered higher confidence candidates because it is unlikely that the observed phenotype was not due to depletion of the intended target. Additional validation studies are required to remove remaining false positives (section 3.10). In arraying the individual siRNAs for the validation round we leave the outside two wells around the perimeter of each plate empty to decrease edge-effects. It is also advisable to evaluate by eye the image files of the wells that score in both the primary and validation rounds to confirm the quality and accuracy of the automated imaging and cell scoring systems, as well as the library plate identity. Obviously, a prime concern is not losing the correct registry of the plates being screened.
2.9 Hybridoma Supernatant Production
Grow hybridoma cells in 250 ml.s of RPMI-1640 Medium (Sigma R0883) with 10% fetal bovine serum (Invitrogen 10437028) and 2mM L-Glutamine (Cellgro 25-005-Cl) in T225 flasks (Corning 431081) until 95% of the cells are dead as determined by trypan blue (Sigma T8154) exclusion. Pool the media from all the flasks to improve homogeneity, and then spin at 2500 rpm followed by passage through a 0.45μm vacuum filter (Corning 430514). The filtered product is then aliquoted and stored at −20°C. We titer hybridoma supernatant on WSN/33 infected U2OS cells to determine the dilution that would produce an adequate intensity signal (greater than 50 units above background signal) in the FITC channel of the IXM using a 250 millisecond exposure and a 1:1000 dilution of goat anti-mouse AF488 antibody. Usual dilutions of hybridoma supernatant vary from 1:8 to 1:10. Working stocks are diluted in 1% bovine serum albumin (BSA, BioPharm 71-010) in D-PBS and treated with Sodium Azide (Sigma S8032) at 0.02% w/v.
3. Results and Discussion
3.1 Overview
In this report we discuss an image-based siRNA screening platform to find host proteins that modify the replication of influenza A virus. Using this method, we enriched for multiple host cell pathways needed for viral replication (HA surface expression) including endosomal acidification, vesicular trafficking, mitochondrial metabolism, nucleocytoplasmic shuttling/mRNA export nuclear transport, and RNA processing. Several other RNAi screens for influenza A virus host factors using various strategies were also published and a comparisons of the screens revealed that they enriched for many of the same pathways and complexes [10–11], however, there was low overlap in exact gene identities shared among the orthologous approaches. Low inter-assay overlap has been seen previously using other functional genomic methodologies (i.e. HIV-dependency factor (HDF) siRNA screens, microarray gene expression profiling, and yeast two hybrid genetic interaction screening), and likely reflects a combination of factors, including poor siRNA knockdown, off-target-effects [12–13], stochastic cellular phenotypes [14], and differences in screening and analysis techniques [15]. Although these caveats must be recognized and appreciated, a comparison of the influenza A virus host factor screens nonetheless reveals statistically significant enrichment for multiple biological pathways and complexes, some novel and others expected based on the known viral life cycle. For example, in our efforts, the depletion of any of four components of the host’s vacuolar-ATPase (vATPase, e.g., ATP6AP1, ATP6V0B, ATP6V1G1, ATP6V0E2) limited viral replication, consistent with HA requiring an acidic environment to mediate fusion. Similar enrichments of vATPase subunits were also seen in several of the other influenza virus host factor screens [10–11].
3.2 Cell Lines
Cell lines are chosen based on viral tropism and ease of use in high throughput applications. Cell lines must be hardy enough to handle automated plating, siRNA transfection and the sample processing associated with IF staining. We have found that multiple strains of HeLa cells, and the osteosarcoma cell line, U2OS, meet these criteria and are readily infectable with the common lab strains of influenza A virus, WSN/33 or PR8. We routinely test for mycoplasma contamination (Mycoalert, Lonza LT07-318) and keep cell stocks in a suppressive concentration of the antibiotic plasmocyn (Invivogen ant-mpp) when not being used for screening. Cell lines are also frozen down in sufficient aliquots so that several tubes of cells can be thawed every 4–6 weeks during the screen. These freshly thawed cells are used for screening after they have recovered in culture for 1–2 weeks. In this way we wish to minimize changes that occur in the cell line with long term culture, such as selection for accelerated growth.
3.3 siRNA library
We have used the whole genome libraries from Dharmacon, including the updated siRNA set, the Silencer Select library from Ambion, and the esiRNA library (15,300 genes targeted) from Sigma. We have screened the Ambion library in pools of three siRNAs. Due to the significant false positives and negatives present in RNAi screening we favor screening more than one library using the primary screen platform. While we have chosen to use a pooled strategy with all of the siRNAs screened together in the primary round to save time and reagents, we concede that this method is vulnerable to a single toxic or off-target-inducing siRNA confounding interpretation of the pooled data. For example, if two or more siRNAs in a pool display discordant phenotypes, they may, due to a combination of on- and off-target events produce a negative result. Alternatively, a pool can be advantageous if two or more functionally redundant splice variants, or differentially folded mRNA structures, require depletion by specific siRNAs in order to reveal a positive phenotype.
3.4 Critical Factors in Optimization
A key factor is optimizing transfection conditions, and we do this by testing a gradation of both cell number and lipid concentration. The duration of knockdown and the length of the assay can be varied. For the most part, we have maintained a 72h period of siRNA-mediated knockdown prior to determination of phenotype or challenge with virus. Regarding transfection reagents, we have found Oligofectamine Reagent (Invitrogen 12252-011) to have low toxicity and high efficiency of knockdown. Moreover, many of our colleagues use RNAi max (Invitrogen 13778150) with excellent results. We purchase as many tubes as needed for each screen, and do this from the same lot number. We then pool the aliquots, and realiquot to improve heterogeneity. We have also found the siRNA-Oligofectamine transfection complexes to be stable for up to an hour once assembled, and we have found that this flexibility can be useful at times.
3.5 Cell Density
Arriving at the correct cell number is a critical parameter and needs to balance being too sparse and succumbing to siRNA transfection induced toxicity with being too dense leading to a decrease in transfection efficiency as well as potentially the phenotypic properties of the cells due to density [14]. Therefore starting with 650 cells per well and increasing that concentration 150 cells per well across a 384 well plate (650, 800, 950, 1100, etc.) is a usual starting point. For these experiments it is important to make the culturing conditions as close to those of the final screen. For example, if possible the cells should be plated with the automated plate filler and cultured in the same incubator as will be used for the screening cells. For removing media we use a hand held 16 channel aspiration wand with equivalent amounts of tape rolled around each end of the barrel to space the tips up from the bottom of the well, thereby minimizing cell loss from aspiration.
3.6 Phenotypic readout
This protocol uses a readout involving an automated screening microscope’s acquisition of 4X magnified images in two wave lengths, FITC and DAPI. This permits the use of an unaltered viral lab strain and a direct evaluation of viral protein replication, an easily monitored surrogate for infection. The cell surface expression of the viral HA protein was used as a readout for viral replication levels. Although measuring viral protein levels is an indirect measurement, we have measured influenza A virus replication using the IF readout side by side with plaque assays, and found that the viral protein levels as determined by IF correlated strongly with viral titers. This method is designed to detect if any host protein plays a role in the viral lifecycle from virus-host receptor binding through to the trafficking of HA to the cell surface. We have also used similar approaches to monitor the expression of intra-cellular viral proteins using a Triton-X100 (FisherBiotech BP151-500) permeabilization step prior to primary antibody staining.
3.7 Approaching the Short-comings of siRNA Screening
siRNA-based genetic screens are useful discovery tools, however due to the nature of current siRNA screening technology and methods they are not approaching saturation. This shortcoming may arise from several factors (see 3.1), including long protein half-life, catalytic activity of a protein so that the post-targeting molecules can still perform their overall function, and also technical issues such as siRNA pooling effects or edge effects. Therefore the candidate genes that are found to impact viral infection in the primary screen may be considered as functionally-defined “footholds” that may be representatives of larger pathways and complexes that are important for viral replication or host cell resistance to infection, but were not fully defined due to low assay sensitivity. For example, as noted above, the screen we performed detected multiple sub-units of the vATPase complex as did the other influenza A virus host factor siRNA screens. However, among the multiple vATPase subunits detected across multiple screens, only one component, ATP6AP1, was found in three out of four screens, with several others being unique, or found in only two of three screens i.e. ATP6AP2, ATP6VOC, ATPVOD1, ATP6V1B2, ATP6VOB. Thus, the bioinformatic analyses was instrumental in providing the information that the vATPase components were physically and functionally linked, thereby validating the discovery that vATPase was essential for influenza A virus replication. The case of the vATPase’s role in the influenza A virus lifecycle also highlights the level of false negatives occurring using this approach because it is unlikely that the subunits scoring in only one screen are not in fact required for viral replication as might be suggested by the negative data produced by the similar efforts [16]. Indeed, one of the host factors identified by just two of the screens, IFITM3, represents a novel class of anti-influenza A virus restriction factor [9, 17]. In addition, candidate genes that validate with a single siRNA are more likely to represent true-positives (on-target events) if they physically or functionally interact (perhaps as pathway components) with candidate genes that are members of enriched clusters, or that validated with two or more individual oligos. Bioinformatics can detect and assess the significance of these associations and thereby “rescue” a candidate from a low confidence designation to one significantly higher.
3.8 Statistical Methods
Statistical methodologies for siRNA screening have also been extensively discussed [18]. We have used a straightforward approach as described above to identify genes of interest. However, as the screen is being optimized, and periodically throughout the screening process, we have found it valuable to use the data-analysis programs, Spotfire (visual data mining and information visualization software license) and BioConductor/R (freely available statistical and data analysis platforms), to help detect any trends in the data that are produced by edge effects, cell number, liquid handling, and/or staining etc.
3.9 Bioinformatic Analysis
A bioinformatic analysis of both the primary screen and validated gene lists is an extremely valuable component of this approach [3, 17, 19–20]. In brief, a suggested initial strategy for bioinformatics analyses would include assessing gene ontology enrichment, along with pathway/network analysis integrated with protein-protein interaction networks. An additional contribution can come from indentifying cross-species homologs to determine if additional functions or interactions are known in these systems. An excellent resource exists for identifying homologs is available online at the Drosophila RNAi Screening Center webpage (http://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl). This database permits gene mapping across multiple organisms to corresponding candidates found in siRNA screens. This is a great way to take advantage of the tremendous amount of data that has been created in the model genetic systems, i.e. worm, fly, and frog. We have also used a number of commercially or freely available pathway tools and resources listed below for some of the suggested pathway efforts (Ingenuity – licensed pathway and enrichment application; NIH David – free gene ontology enrichment; MSigDb – free molecular signatures database; PathwayCommons – free source of protein-protein interactions Protein-protein interaction database (human protein reference database, HPRF, www.hprd.org); HomoloGene – another free source of homolog information; Cytoscape – free network visualization software. Similarly, the PathwayCommons database is very valuable for making proteomic connections, which in our experience have produced critical insights that can connect a “lone” factor to a known complex or pathway based on a physical association. The use of homologs and protein-protein interaction-generated candidate gene-mapping can then be merged with the more established pathway and complex interactions based to generate a global or systems interaction matrix map.
3.10. Validation of candidates found in the screen
This siRNA screening method will functionally define a set of candidates that modulate viral infection. We have found it to be valuable to focus on further analysis and validation of high confidence candidates as follows:
Select high confidence and high interest (novel) candidates and pathways based on a) Number of siRNAs that score for a candidate in the secondary screen (reagent redundancy [21]) and b) Bioinformatics: functional class (druggable: enzymes, channels, receptors), functional clustering and pathway enrichment.
Validate these candidates by determining if the level of siRNA-mediated knockdown correlates with the strength of the phenotype using quantitative PCR (qPCR).
Test qPCR-validated candidates using viral lifecycle assays.
Identify small molecules which modulate the actions of host factors or pathways and test their effect on viral replication.
Perform cDNA rescues of the top candidates. These cDNAs can also be used for gain-of-function studies and for affinity purification-mass spectrometry experiments to find host protein-interactors.
Test the role of the candidates in the replication of additional influenza A strains, and in primary cells.
3.11 Comparison of influenza A virus host factor siRNA screening methods
Five large scale siRNA screens were conducted for host factors required for influenza A viral replication. A comparison of these approaches is provided in the following table:
| Variable | Hao 2008 | Konig 2010 | Shapira 2010 | Karlas 2010 | Brass 2010 |
|---|---|---|---|---|---|
| Functional assay(s) | Plate reader, luciferase levels, recombinant VSV-G pseudotyped virus expressing green fluorescence protein and luciferase protein. | Plate reader, Luciferase levels, WSN/Renilla virus | Plate reader, Luciferase levels for viral replication, IFN-β production after ΔNS1 virus infection or viral RNA transfection, PR8 and ΔNS1 viruses. | Imaging of immunostaining for NP levels (part one), Luciferase levels (part two). WSN/33 virus. | Imaging of immunostaining of HA levels, PR8 virus. |
| Bioinformatic Analyses of screen data | NA | Hynet, Reactome, BIND, MINT, MCODE | BioGRID, BIND, Ingenuity, MSigDB, and INTACT | Gene Ontology, Reactome, String | GOhyperGAll module of Bioconductor for gene ontology UniProt, KEGG, Reactome, Gene Ontology, and NCBI GeneRIF OMIM Human orthologs were mapped to other species using NCBI HomoloGene |
| siRNA Hit selection | Inhibition exceeding 2.5 standard deviations from the mean in at least One of two replicates | RSA hit criterion of an activity score <0.4 | Two fold increase or decrease from the mean for any of the three above noted assays. | Z-scores below -2 | Percentage of HA-positive cells was less than 55% or greater than 200% of the plate mean, and cell numbers were not less than 40% of the plate mean. |
| siRNA transfection Controls | siRNAs against viral NP, and non-targeting negative control | siRNAs against Firefly Renilla and RPS27a, negative controls: scramble 177, scramble5701 | NA | siRNAs against viral NP, host PLK1 and the Allstars non-targeting control | siRNAs against viral NP, host NXF1, non-targeting negative control. |
| Cell types | Drosophila DL1 cells | Human A549 lung carcinoma cells | Human Bronchial Epethelial cells (part one), 293T cells part two. | Human A549 lung carcinoma cells part one, MDCK cells part two. | Human U2OS osteosarcoma cells |
| siRNA Transfection lipid | None required | 0.166% RNAi max (Invitrogen) | Hiperfect (Qiagen), concentration not available | 0.109% Hiperfect (Qiagen) | 0.32% Oligofectamine (Invitrogen). |
| siRNA library and siRNA concentration | Ambion Drosophila siRNA library, 200 ng/reaction, 384 well plate | Qiagen Human Genome 1.0 and Human Druggable Genome siRNA set versions 1 and 2, NM Set version 1, XM Set version, a kinome library from Invitrogen and the kinome library from IDT, 1 picomole/reaction, 384 well plate | Dharmacon siARRAY siRNA Library; Human Genome, G-005000-05 candidate set (1745 genes) based on expression and protein interaction experiments, 25 nM | Qiagen Human Genome 1.0 and Human Druggable Genome siRNA Set V2.0, 20 nM, 384 well plate | Dharmacon siARRAY siRNA Library; Human Genome, G- 005000-05, 50 nM. 384 well plate |
4. Conclusions
4.1
This report describes a high throughput image-based method for investigating host-viral interactions (Fig. 3). We have used this strategy and similar approaches to both support and extend previous efforts to define host-viral interactions. As discussed there was strong overlap between the mammalian screens involving multiple functional complexes and pathways, including significant overlap with the Reactome’s influenza A virus infection database (http://www.reactome.org/ReactomeGWT/entrypoint.html). Therefore, large-scale mammalian and fly genetic studies have functionally identified a number of required influenza A virus dependencies that may lead to the development of new host directed anti-viral (HDAV) targets.
Fig. 3.
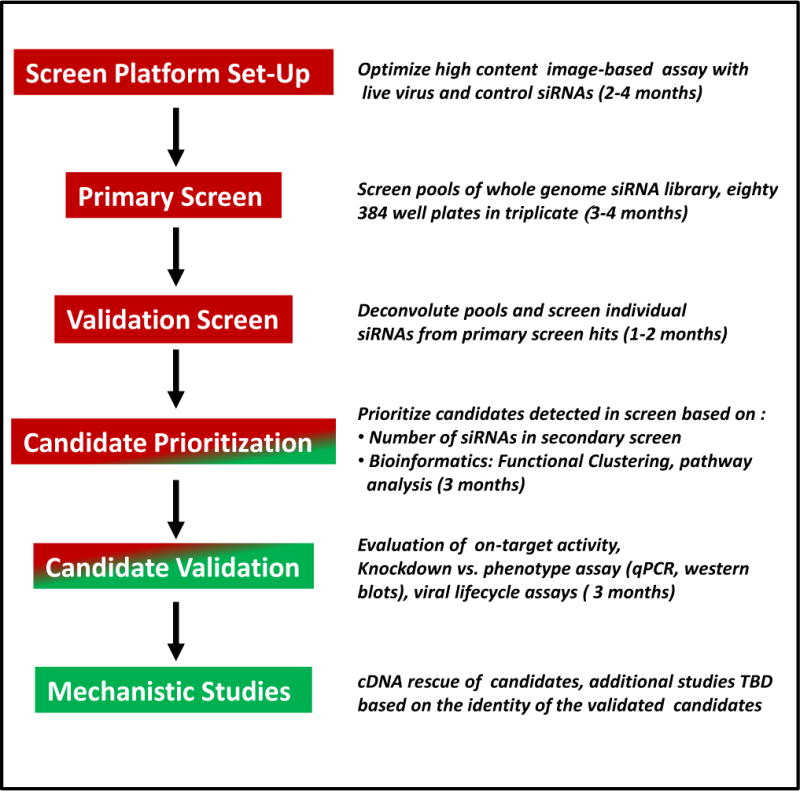
Schematic of the influenza A virus host factor siRNA screen. Time lines and milestones are included with year one activities in red and year two actions in green. To be determined (TBD), quantitative PCR (qPCR).
Although RNAi screening efforts are informative, they also suffer from limitations as evidenced by the few exact genes scoring across the collective influenza A virus screens. As discussed these caveats involve the RNAi technology employed to interfere with gene expression, as well as issues arising with the execution of cell-based high throughput screens (section 3.1). RNAi design is now in the ascendant, and newly available reagents have incorporated major advances; these include improvements in target sequence selection, analysis of target accessibility via prediction of mRNA secondary structure, and chemical modifications of the siRNA oligonucleotide that improve stability while decreasing OTEs and preventing toxicity. Moreover, attention to the rigorous validation of candidates (section 3.10) combined with improved methods for integrating data sets and analyzing primary high throughput siRNA screen data promise to boost yields. In notable comparison to other approaches [22], and when viewed collectively with short hairpin RNA screening technologies [23–24], RNAi methods are uniquely versatile, permitting innovative genetic experiments to be carried out in a wide range of mammalian cells and systems. Future directions will likely see the expanded use of genetically engineered cell lines, as well as an increasing focus on using the most physiologically relevant primary cells in siRNA screens.
Supplementary Material
S2.1 The availability of the following equipment is suggested to perform this method: sterile tissue culture hood, automated plate filler with small bore dispensing cassette for volumes of 5 μl to 30 μl (i.e. Wellmate or Multidrop, Thermo Electron), a 12-channel pipette (Rainin), a liquid handling robot with 96 and/or 384 well plate pipetting capabilities (i.e. Bravo, Velocity 11), a screening microscope, (i.e. Image Xpress Micro, Molecular Devices, or In Cell 2000 General Electric), a benchtop centrifuge with 96 or 384 well plate holders, a tissue culture incubator, and an aspiration wand (i.e. 12 channel VP-185L-1 or 16 channel VP-186L-1, V & P Scientific)
S2.2 siRNAs: To optimize conditions for siRNA transfection the depletion of polo kinase 1 (PLK1, Dharmacon SMARTpool M-003290-01) is useful because its reduction produces an easily monitored “rounded-up” cellular phenotype apparent at 18–24h post-transfection due to a G2/M cell cycle arrest. Positive control siRNAs for the influenza A virus experiments included a custom siRNA against the viral nucleoprotein (NP, GGAUCUUAUUUCCUUCGGAGUU produced by Dharmacon, [8]) and a SMARTpool directed against NFX1, a host mRNA exporter, previously found be required by influenza A virus in an elegant screen using drosophila cells (SMARTpool M-013680-01, [7]). In subsequent work based on the candidates found in the initial screens, a SMARTpool against the host vacuolar ATPase subunit, ATP6V0B (SMARTpool M-010907-02), reduced endosomal and lysosomal acidification in transfected cells with minimal toxicity, and strongly inhibited pH-dependent events such as HA’s acid-induced conformational change and viral infection. A non-targeting negative control from Dharmacon (NT, siCONTROL Non-Targeting siRNA #2, Dharmacon D-001210-02) has worked well in these assays.
S2.3 To find host factors that modulate influenza A virus replication, we performed a whole genome siRNA screen using the Dharmacon siARRAY siRNA Library (Human Genome, G-005000-05, [3], Fig. 1). This library contains SMARTpools comprised of four sequence-unique siRNAs targeting each of 17,877 genes. The remaining 3,244 pools of the original 21,121 gene library were not screened because the majority were replaced by a new sub-library of 4,506 SMARTpools (Dharmacon Human 5: RefSeq27 Reversion Pools). U2OS Cells were grown in DMEM (Invitrogen 11965) with 10% FBS (Invitrogen). Cells were kept at 50–80% confluency and were split the day before transfection. After 4 to 6 weeks in culture, the cells being used for screening were replaced with fresh cells that had been thawed and cultured for two weeks prior to use.
S2.4 A reverse transfection method using 384 well clear bottom and black wall plates (Corning 3712) was used for the genome wide screen. All procedures are carried out in sterile manner in a tissue culture hood. A master mix of Oligofectamine transfection lipid and Opti-MEM serum free media (both allowed to equilibrate to room temperature prior to use) is prepared first using the ratio of 0.1 μl lipid to 9.9 μl media for each well to be transfected.
S2.5 A Wellmate is employed to dispense 9 μl of the lipid mixture per well. The filled plates are then centrifuged at 1000 rpm for one minute to drive all of the lipid mixture to the bottom of the well. The plates are now transfected using the Bravo Velocity 11 and dispensing a desired volume of 1.5 μl of a 1 μM siRNA stock into three Corning 3712 plates containing 9 μl matured lipid-Opti-MEM mix as follows: Tips (Fluotics P30-V11.NS) on; then aspirate 5.2 μl volume from the siRNA library plate. In series, dispense 1.6 μl into the first plate, 1.55 μl into the second plate, and the remaining solution into the third plate. Then beginning with the last plate receiving the siRNA solution, perform seven sequential mixing steps of 6 μl each.
While the transfection reactions are incubating for 20 min, trypsinize and resuspend the U2OS cells in DMEM with 15% FBS. Once the 20 min incubation period is complete add 20 μl. of the cell suspension per well using the Wellmate. Spin the filled plates as above and incubate at 37°C and 5% CO2 for 72 h. If siRNAs are present in either of the outermost two rows or columns, then the day after transfection add 5 μl of DMEM with 15% FBS to those wells to decrease toxic edge effects.
S2.6 Prepare a viral inoculum at a multiplicity of infection (moi) that produces 30 to 40% infection by IF staining of HA protein. Remove media using the 16 channel aspiration wand and add influenza A/Puerto Rico/8/34 (PR8, ATCC VR-1469) in DMEM 10% FBS. Spin plates for five minutes at 1000 rpm and return to the tissue culture incubator for 12h. For this method we have used either PR8 or influenza A/WSN/33 (WSN/33, kind gift of Dr. Peter Palese) grown in MDCK cells (ATCC) using DMEM with 2%FBS.
S2.7 After 12h of viral infection the media is aspirated. Using the Wellmate and a separate dispensing cartridge dedicated to these processing steps, add 4% v/v paraformaldehyde (PFA) solution (Sigma F1635) in D-PBS and incubate for 30 min. Wash (all wash steps are done twice with D-PBS) and then add primary antibody solution. After a one hour incubation at room temperature, wash and then incubate with the secondary antibody, Alexa Fluor 488 goat anti-mouse, at 1:1,000 dilution (Invitrogen A11001) for 30 min. Wash and add (and let remain), D-PBS with Hoechst 33342 (DNA stain, 1:5-10,000 dilution of a 10 mg/ml stock from Invitrogen H3570). Stained cells are imaged on an automated Image Express Micro (IXM) microscope (Molecular Devices), using a 4X magnification of a single site per well acquired using both the FITC (HA-signal) and DAPI (cell nuclei) wavelengths. Images were analyzed using the Metamorph software program (Molecular Devices Inc.) using the cell scoring module (Fig. 2).
S2.8 In the primary screen, siRNA pools are selected as hits if the mean of triplicate wells demonstrate that the percentage of HA-positive cells is less than 55% of the average plate mean without having less cells than 40% of the mean of the plate. Pools which increase infection by greater than 200% of the plate mean are also of interest as potential anti-viral factors [9]. In the validation round screen the individual siRNAs from each SMARTpool are rescreened individually using the identical transfection protocol as the primary screen and 50 nM final siRNA concentration. siRNAs are classified as hits using the same criteria as in the primary screen, with the exception that comparison is made to the values returned by four or more siRNA negative control wells present on each validation round plate. Additional validation studies are required to remove remaining false positives (section 3.10). In arraying the individual siRNAs for the validation round we leave the outside two wells around the perimeter of each plate empty to decrease edge-effects.
S2.9 Hybridoma Supernatant Production: Grow hybridoma cells in 250 ml.s of RPMI-1640 Medium (Sigma R0883) with 10% fetal bovine serum (Invitrogen 10437028) and 2mM L-Glutamine (Cellgro 25-005-Cl) in T225 flasks (Corning 431081) until 95% of the cells are dead as determined by trypan blue (Sigma T8154) exclusion. Pool the media from all the flasks to improve homogeneity, and then spin at 2500 rpm followed by passage through a 0.45μm vacuum filter (Corning 430514). The filtered product is then aliquoted and stored at −20°C. Working stocks are diluted in 1% bovine serum albumin (BSA, BioPharm 71-010) in D-PBS and treated with Sodium Azide (Sigma S8032) at 0.02% w/v.
Highlights.
High throughput genetic screening method for investigating virus host interactions
Image-based readout for high throughput screening
Discussion of siRNA screen optimization and analysis
Acknowledgments
We thank the current and former members of the Lieberman (Derek Dykxhoorn, Nan Yan), Xavier (Yair Benita, Alwyn Ng), Fikrig (Manoj Krishnan), Brass and Elledge labs, additional invaluable colleagues that worked together with us on the influenza A virus, HIV-1, and HCV siRNA screen studies, the ICCB-Longwood Harvard Medical School Screening Facility; C. Shamu, S. Chang, J. Smith, S. Rudnicki, S. Johnston, K. Rudnicki, D. Wrobel, D. Flood, and T. Xie ; the Drosophila RNAi Screening Center; S. Mohr, and the Ragon Institute; K. Donnelly, M. Boyarina, and P. Richtmyer. A.L.B. is the recipient of a Charles H. Hood Foundation Child Health Research Award, and is supported by grants from the Phillip T. and Susan M. Ragon Institute Foundation, the Bill and Melinda Gates Foundation’s Global Health Program and the National Institute of Allergy and Infectious Diseases (R01AI091786). The influenza A virus siRNA screen study was supported by the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NIH grant U54 AI057159 to D. Kasper).
Footnotes
Supplementary Protocol: The following represents the protocol in Section 2 above but with the descriptive commentary removed to make it easier to follow.
References
- 1.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 2.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 3.Brass AL, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139(7):1243–54. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shapira SD, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139(7):1255–67. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konig R, et al. Human host factors required for influenza virus replication. Nature. 2010;463(7282):813–7. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlas A, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463(7282):818–22. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 7.Hao L, et al. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454(7206):890–3. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ge Q, et al. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci U S A. 2003;100(5):2718–23. doi: 10.1073/pnas.0437841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feeley EM, et al. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 2011;7(10):e1002337. doi: 10.1371/journal.ppat.1002337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stertz S, Shaw ML. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011;13(5):516–25. doi: 10.1016/j.micinf.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehle A, Doudna JA. A host of factors regulating influenza virus replication. Viruses. 2010;2(2):566–73. doi: 10.3390/v2020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coyne CB, Cherry S. RNAi screening in mammalian cells to identify novel host cell molecules involved in the regulation of viral infections. Methods Mol Biol. 2011;721:397–405. doi: 10.1007/978-1-61779-037-9_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigoillot FD, et al. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012 doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snijder B, Pelkmans L. Origins of regulated cell-to-cell variability. Nat Rev Mol Cell Biol. 2011;12(2):119–25. doi: 10.1038/nrm3044. [DOI] [PubMed] [Google Scholar]
- 15.Adamson B, et al. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol. 2012;14(3):318–28. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bushman FD, et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009;5(5):e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319(5865):921–6. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 18.Birmingham A, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6(8):569–75. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnan MN, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455(7210):242–5. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A. 2009;106(38):16410–5. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Echeverri CJ, et al. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat Methods. 2006;3(10):777–9. doi: 10.1038/nmeth1006-777. [DOI] [PubMed] [Google Scholar]
- 22.Carette JE, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326(5957):1231–5. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 23.Schlabach MR, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319(5863):620–4. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smogorzewska A, et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell. 2010;39(1):36–47. doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S2.1 The availability of the following equipment is suggested to perform this method: sterile tissue culture hood, automated plate filler with small bore dispensing cassette for volumes of 5 μl to 30 μl (i.e. Wellmate or Multidrop, Thermo Electron), a 12-channel pipette (Rainin), a liquid handling robot with 96 and/or 384 well plate pipetting capabilities (i.e. Bravo, Velocity 11), a screening microscope, (i.e. Image Xpress Micro, Molecular Devices, or In Cell 2000 General Electric), a benchtop centrifuge with 96 or 384 well plate holders, a tissue culture incubator, and an aspiration wand (i.e. 12 channel VP-185L-1 or 16 channel VP-186L-1, V & P Scientific)
S2.2 siRNAs: To optimize conditions for siRNA transfection the depletion of polo kinase 1 (PLK1, Dharmacon SMARTpool M-003290-01) is useful because its reduction produces an easily monitored “rounded-up” cellular phenotype apparent at 18–24h post-transfection due to a G2/M cell cycle arrest. Positive control siRNAs for the influenza A virus experiments included a custom siRNA against the viral nucleoprotein (NP, GGAUCUUAUUUCCUUCGGAGUU produced by Dharmacon, [8]) and a SMARTpool directed against NFX1, a host mRNA exporter, previously found be required by influenza A virus in an elegant screen using drosophila cells (SMARTpool M-013680-01, [7]). In subsequent work based on the candidates found in the initial screens, a SMARTpool against the host vacuolar ATPase subunit, ATP6V0B (SMARTpool M-010907-02), reduced endosomal and lysosomal acidification in transfected cells with minimal toxicity, and strongly inhibited pH-dependent events such as HA’s acid-induced conformational change and viral infection. A non-targeting negative control from Dharmacon (NT, siCONTROL Non-Targeting siRNA #2, Dharmacon D-001210-02) has worked well in these assays.
S2.3 To find host factors that modulate influenza A virus replication, we performed a whole genome siRNA screen using the Dharmacon siARRAY siRNA Library (Human Genome, G-005000-05, [3], Fig. 1). This library contains SMARTpools comprised of four sequence-unique siRNAs targeting each of 17,877 genes. The remaining 3,244 pools of the original 21,121 gene library were not screened because the majority were replaced by a new sub-library of 4,506 SMARTpools (Dharmacon Human 5: RefSeq27 Reversion Pools). U2OS Cells were grown in DMEM (Invitrogen 11965) with 10% FBS (Invitrogen). Cells were kept at 50–80% confluency and were split the day before transfection. After 4 to 6 weeks in culture, the cells being used for screening were replaced with fresh cells that had been thawed and cultured for two weeks prior to use.
S2.4 A reverse transfection method using 384 well clear bottom and black wall plates (Corning 3712) was used for the genome wide screen. All procedures are carried out in sterile manner in a tissue culture hood. A master mix of Oligofectamine transfection lipid and Opti-MEM serum free media (both allowed to equilibrate to room temperature prior to use) is prepared first using the ratio of 0.1 μl lipid to 9.9 μl media for each well to be transfected.
S2.5 A Wellmate is employed to dispense 9 μl of the lipid mixture per well. The filled plates are then centrifuged at 1000 rpm for one minute to drive all of the lipid mixture to the bottom of the well. The plates are now transfected using the Bravo Velocity 11 and dispensing a desired volume of 1.5 μl of a 1 μM siRNA stock into three Corning 3712 plates containing 9 μl matured lipid-Opti-MEM mix as follows: Tips (Fluotics P30-V11.NS) on; then aspirate 5.2 μl volume from the siRNA library plate. In series, dispense 1.6 μl into the first plate, 1.55 μl into the second plate, and the remaining solution into the third plate. Then beginning with the last plate receiving the siRNA solution, perform seven sequential mixing steps of 6 μl each.
While the transfection reactions are incubating for 20 min, trypsinize and resuspend the U2OS cells in DMEM with 15% FBS. Once the 20 min incubation period is complete add 20 μl. of the cell suspension per well using the Wellmate. Spin the filled plates as above and incubate at 37°C and 5% CO2 for 72 h. If siRNAs are present in either of the outermost two rows or columns, then the day after transfection add 5 μl of DMEM with 15% FBS to those wells to decrease toxic edge effects.
S2.6 Prepare a viral inoculum at a multiplicity of infection (moi) that produces 30 to 40% infection by IF staining of HA protein. Remove media using the 16 channel aspiration wand and add influenza A/Puerto Rico/8/34 (PR8, ATCC VR-1469) in DMEM 10% FBS. Spin plates for five minutes at 1000 rpm and return to the tissue culture incubator for 12h. For this method we have used either PR8 or influenza A/WSN/33 (WSN/33, kind gift of Dr. Peter Palese) grown in MDCK cells (ATCC) using DMEM with 2%FBS.
S2.7 After 12h of viral infection the media is aspirated. Using the Wellmate and a separate dispensing cartridge dedicated to these processing steps, add 4% v/v paraformaldehyde (PFA) solution (Sigma F1635) in D-PBS and incubate for 30 min. Wash (all wash steps are done twice with D-PBS) and then add primary antibody solution. After a one hour incubation at room temperature, wash and then incubate with the secondary antibody, Alexa Fluor 488 goat anti-mouse, at 1:1,000 dilution (Invitrogen A11001) for 30 min. Wash and add (and let remain), D-PBS with Hoechst 33342 (DNA stain, 1:5-10,000 dilution of a 10 mg/ml stock from Invitrogen H3570). Stained cells are imaged on an automated Image Express Micro (IXM) microscope (Molecular Devices), using a 4X magnification of a single site per well acquired using both the FITC (HA-signal) and DAPI (cell nuclei) wavelengths. Images were analyzed using the Metamorph software program (Molecular Devices Inc.) using the cell scoring module (Fig. 2).
S2.8 In the primary screen, siRNA pools are selected as hits if the mean of triplicate wells demonstrate that the percentage of HA-positive cells is less than 55% of the average plate mean without having less cells than 40% of the mean of the plate. Pools which increase infection by greater than 200% of the plate mean are also of interest as potential anti-viral factors [9]. In the validation round screen the individual siRNAs from each SMARTpool are rescreened individually using the identical transfection protocol as the primary screen and 50 nM final siRNA concentration. siRNAs are classified as hits using the same criteria as in the primary screen, with the exception that comparison is made to the values returned by four or more siRNA negative control wells present on each validation round plate. Additional validation studies are required to remove remaining false positives (section 3.10). In arraying the individual siRNAs for the validation round we leave the outside two wells around the perimeter of each plate empty to decrease edge-effects.
S2.9 Hybridoma Supernatant Production: Grow hybridoma cells in 250 ml.s of RPMI-1640 Medium (Sigma R0883) with 10% fetal bovine serum (Invitrogen 10437028) and 2mM L-Glutamine (Cellgro 25-005-Cl) in T225 flasks (Corning 431081) until 95% of the cells are dead as determined by trypan blue (Sigma T8154) exclusion. Pool the media from all the flasks to improve homogeneity, and then spin at 2500 rpm followed by passage through a 0.45μm vacuum filter (Corning 430514). The filtered product is then aliquoted and stored at −20°C. Working stocks are diluted in 1% bovine serum albumin (BSA, BioPharm 71-010) in D-PBS and treated with Sodium Azide (Sigma S8032) at 0.02% w/v.