

ABSTRACT
The purpose of this study was to take advantage of the nematode Caenorhabditis elegans to perform a whole-animal chemical screen to identify potential immune activators that may confer protection against bacterial infections. We identified 45 marketed drugs, out of 1,120 studied compounds, that are capable of activating a conserved p38/PMK-1 mitogen-activated protein kinase pathway required for innate immunity. One of these drugs, the last-resort antibiotic colistin, protected against infections by the Gram-negative pathogens Yersinia pestis and Pseudomonas aeruginosa but not by the Gram-positive pathogens Enterococcus faecalis and Staphylococcus aureus. Protection was independent of the antibacterial activity of colistin, since the drug was administered prophylactically prior to the infections and it was also effective against antibiotic-resistant bacteria. Immune activation by colistin is mediated not only by the p38/PMK-1 pathway but also by the conserved FOXO transcription factor DAF-16 and the transcription factor SKN-1. Furthermore, p38/PMK-1 was found to be required in the intestine for immune activation by colistin. Enhanced p38/PMK-1-mediated immune responses by colistin did not reduce the bacterial burden, indicating that the pathway plays a role in the development of host tolerance to infections by Gram-negative bacteria.
IMPORTANCE
The innate immune system represents the front line of our defenses against invading microorganisms. Given the ever-increasing resistance to antibiotics developed by bacterial pathogens, the possibility of boosting immune defenses represents an interesting, complementary approach to conventional antibiotic treatments. Here we report that the antibiotic colistin can protect against infections by a mechanism that is independent of its microbicidal activity. Prophylactic treatment with colistin activates a conserved p38/PMK-1 pathway in the intestine that helps the host better tolerate a bacterial infection. Since p38/PMK-1-mediated immune responses appear to be conserved from plants to mammals, colistin may also activate immunity in higher organisms, including humans. Antibiotics with immunomodulatory properties have the potential of improving the long-term outcome of patients with chronic infectious diseases.
INTRODUCTION
Over the past decade, the pharmaceutical industry has experienced increasing challenges in drug development since the process has become more expensive, lengthier, and riskier. This is highlighted by the fact that even though there has been a more than 10-fold increase in drug development spending during the last few years, there has been no growth in new drug approvals (1). The most important reason for this lack of growth is the difficulty of crossing the so-called “valley of death,” the gulf between finding a promising new agent and demonstrating its safety and efficacy in humans (2). Most of the hits generated by traditional screening tools turn out to be invalid once tested in mammals, resulting in a waste of funds and efforts. In recent years, due to their genetic amenability, low cost, and culture conditions compatible with large-scale screening, several small whole-animal models, such as Caenorhabditis elegans, have gained momentum as screening tools for drug discovery (3).
The innate immune system is at the front line of our defense system against invading microorganisms (4). Considering the growing demand for more efficient agents with improved pharmacological properties in the facet of antibiotic resistance, strengthening immune defenses against pathogens by boosting host innate immunity may represent an interesting, complementary approach for pharmaceutical interventions to treat infectious diseases. Therefore, we took advantage of the nematode Caenorhabditis elegans to perform a whole-animal chemical screen of marketed drugs to identify potential activators of innate immunity that may confer protection against bacterial infections.
In this study, we screened the Prestwick Library, which comprises a number of marketed drugs selected for their high chemical and pharmacological diversity and for their known bioavailability and safety in humans. This greatly reduces the risk of low-quality hits and increases the possibility of leads that can be more easily used to develop new therapeutic approaches to treat infections. Out of 1,120 studied compounds, 45 candidates that were capable of activating a conserved p38/PMK-1 mitogen-activated protein kinase (MAPK) pathway were identified. Among them, we unexpectedly found colistin, which is a last-resort antibiotic against multidrug-resistant Gram-negative bacterial strains (5). The results showed that colistin confers resistance to infections by the Gram-negative pathogens Yersinia pestis and Pseudomonas aeruginosa but not by the Gram-positive pathogens Enterococcus faecalis and Staphylococcus aureus. Further studies indicated that the immune activation by colistin is mediated by the p38 MAPK pathway in the intestine and that it requires the downstream transcription factor SKN-1. Immune activation induced by colistin also requires the conserved FOXO transcription factor DAF-16. Resistance did not correlate with a reduced bacterial burden, indicating that the immune activation mediated by colistin makes the animals more tolerant to infections.
RESULTS
C. elegans-based screen for identification of drugs capable of activating innate immunity.
We have used C. elegans as a whole-animal high-throughput system for a chemical screen for novel immunomodulators capable of controlling the conserved p38/PMK-1 pathway that is required for innate immunity (6–8). We focused on the p38/PMK-1 pathway because it comprises an NSY-1 (a MAPK kinase kinase [MAPKKK])/SEK-1 (a MAPKK)/PMK-1 (a p38 MAPK) cassette that is an evolutionarily conserved module used by mammals and nematodes in immune response against bacterial infections. In addition, it has been demonstrated that it is controlled not only at the cell-autonomous level but also at the organismal level by the nervous system (9, 10), increasing the number of putative drug targets. As a first step to identify compounds capable of modulating this conserved pathway, we screened the Prestwick chemical library. The Prestwick library has been designed to ensure maximal chemical and optimal therapeutic diversity. It contains 1,120 small molecules, 90% of which are marketed drugs, enhancing the possibility of identifying compounds that can be readily used in the clinical setting.
We used the C. elegans strain AY101, which is a p38/PMK-1 reporter strain we engineered to express green fluorescent protein (GFP) under the regulation of the promoter of a gene that is a marker of the p38/PMK-1 pathway, F35E12.5. In these transgenic animals (8), only minimal expression of GFP is observed in the presence of the food source Escherichia coli, but high levels of expression are seen in the intestine of animals exposed to bacterial pathogens or drugs capable of activating p38/PMK-1. Larval state 4 (L4) AY101 larval animals were exposed for 24 h to 1,120 compounds from the Prestwick Library (20 µg/ml, final concentration) and screened for enhanced GFP fluorescence. Animals exposed to only E. coli or Y. pestis were used as reference to determine two levels of GFP fluorescence: fluorescence strength comparable to or stronger than that induced by Y. pestis and fluorescence strength comparable to or lower than that observed in animals exposed to E. coli. The screen was repeated three times, and only those chemicals capable of inducing fluorescence strengths comparable to or stronger than that induced by Y. pestis three times were deemed candidates. As shown in Fig. 1 and Table S1 in the supplemental material, we identified 45 possible candidates, of which 16 correspond to antimicrobial agents and 22 correspond to drugs that target the nervous system. Since the p38/PMK-1 MAPK pathway appears to be controlled at the cell-nonautonomous level by the nervous system (9), it is not surprising that drugs known to target the nervous system can control it. However, the identification of 16 drugs with known antimicrobial properties as putative activators of the p38/PMK-1 pathway was unexpected.
FIG 1 .
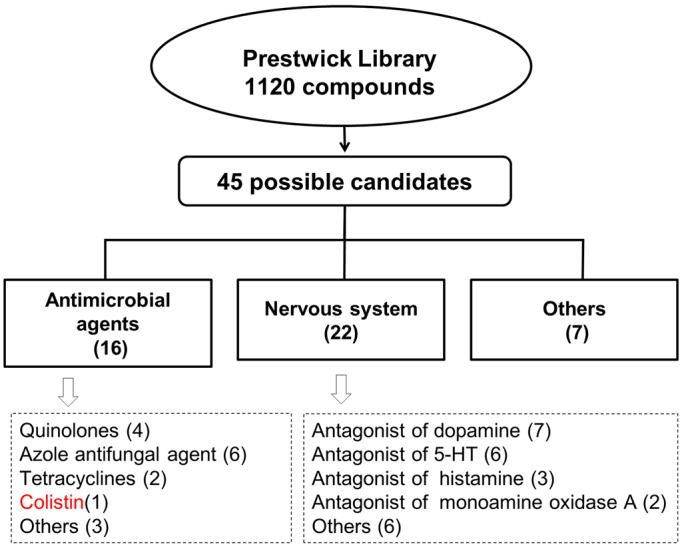
Work flow for the screening of 1,120 drugs capable of activating the p38/PMK-1 pathway. C. elegans strain AY101, which expresses GFP under the control of the promoter of a reporter gene of p38/PMK-1 activity, F35E12.5, was used to screen 1,120 drugs. Out of the 45 drugs found to enhance GFP fluorescence on AY101 animals, 16 correspond to antimicrobial agents and 22 target the nervous system.
The antibiotic colistin can activate the p38/PMK-1 MAPK pathway.
We decided to further study colistin because the identification of a drug with known antimicrobial properties as a putative activator of the p38/PMK-1 pathway was unexpected and because even though it has been rediscovered as the last-line therapy for infections caused by Gram-negative “superbugs,” it has never been reported to target the host immune system. However, our studies indicate that in addition to its antimicrobial activity, it can activate the p38/PMK-1 pathway. Consistent with the initial findings described in Fig. 1, AY101 adult animals treated with colistin exhibited higher levels of pF35E12.5::gfp expression than untreated animals (Fig. 2A). As shown in Fig. 2A, the level of pF35E12.5::gfp expression induced by colistin seems comparable to that induced by Y. pestis infection, which is known to activate p38/PMK-1 and the expression of F35E12.5 (8). Consistent with visual observation, large-particle flow cytometry (Copas Biosort instrument) demonstrated that colistin-treated animals exhibit higher levels of pF35E12.5::gfp expression than the control and that the induction of pF35E12.5::gfp expression by colistin treatment was comparable to that elicited by Y. pestis infection (Fig. 2B).
FIG 2 .

Colistin activates pF35E12.5::gfp through the p38/PMK-1 pathway. (A) AY101 acIs101[pDB09.1 (pF35E12.5::gfp), pRF4(rol-6(su1006))] animals carrying a transcriptional reporter for F35E12.5 were exposed to E. coli, E. coli plus colistin, or Y. pestis for 24 h and imaged using fluorescence microscopy. (B) GFP fluorescence intensity in AY101 animals was analyzed using the Copas Biosort instrument (Union Biometrica, Holliston, MA). The fluorescence of AY101 animals exposed to colistin or Y. pestis was compared to that of animals exposed to E. coli. GFP fluorescence intensity (FLU1) was plotted against adult animal size, measured as time of flight (TOF). Each dot represents an individual nematode. (C) AY101 animals were treated with a control vector or pmk-1-specific RNAi and exposed to colistin for 24 h. (D) Western blot analysis of active p38 expression level of N2 treated with (N2+Col) or without colistin for 24 h. Strain KU25, which carries a pmk-1 deletion allele, was used as a control. The level of active p38 in the “N2+Col” group is 1.53-fold higher than that of the N2 group (n = 3; P < 0.05). Image quantification was performed using the software program ImageJ.
We previously demonstrated that RNA interference (RNAi) inhibition of pmk-1 considerably reduced pF35E12.5::gfp expression in AY101 animals (8), indicating that inducible expression of F35E12.5 in response to Y. pestis infection was largely dependent upon PMK-1. Colistin may be able to induce F35E12.5 gene expression by targeting the PMK-1 pathway, similarly to Y. pestis infection, or it may activate a PMK-1-independent mechanism also capable of eliciting F35E12.5 expression. To distinguish between these two possibilities, we analyzed the fluorescence emitted by AY101 animals where RNAi was used to inhibit PMK-1. As shown in Fig. 2C, RNAi inhibition of pmk-1 considerably reduced pF35E12.5::gfp expression in AY101 animals (Fig. 2C), demonstrating that inducible expression of F35E12.5 in response to colistin is largely dependent upon the p38/PMK-1 pathway. We also studied whether the enhanced PMK-1 transcriptional activity with colistin treatment correlated with higher levels of active PMK-1. Western blot analysis showed that colistin-treated animals exhibited higher levels of active PMK-1 than control-treated animals (Fig. 2D). Taken together, these results show that colistin is capable of activating PMK-1, which results in the elicitation of its transcriptional activity.
Colistin acts as an overall activator of immune pathways.
To investigate other possible effects of colistin on the activation of immune pathways, we utilized Agilent’s C. elegans gene expression microarray to find genes upregulated or down-regulated in response to the drug. Animals were synchronized by hypochlorite treatment and treated with control or 20 µg/ml colistin in liquid medium supplemented with heat-killed E. coli OP 50. After 24 h, the animals were collected and RNA extraction was performed. Overall, the microarray study revealed a change in the expression of 4,402 transcripts. Of these, 2,538 genes were upregulated and 1,864 genes were down-regulated more than 2-fold, with P value less than 0.05 (Fig. 3A; see also Tables S2 and S3 in the supplemental material). Interestingly, the cluster of genes upregulated by colistin is significantly enriched in genes upregulated in response to Y. pestis infection (Fig. 3B; see also Table S4, representation factor 4.8, P < 1.05e−25), indicating that the drug activates a transcriptional profile similar to that activated in response to Y. pestis infection.
FIG 3 .

Colistin acts as a general immune activator that elicits an expression profile similar to that elicited by Y. pestis infection. (A) Microarray results shows 2,538 up- and 1,864 downregulated genes in response to colistin treatment (2-fold; P < 0.05). F35E12.5 showed a >5-fold increase in the colistin-treated group compared to expression in the untreated group (P < 0.001). (B) Venn diagrams of genes that are upregulated by colistin and positively induced by Y. pestis and PMK-1. (C) Representation factors (RFs) of gene sets linked to immune or stress responses. RF is the number of overlapping genes divided by the expected number of overlapping genes drawn from the corresponding gene sets. A representation factor below 1.0 indicates underrepresentation, whereas a value above 1.0 indicates overrepresentation.
Since the chemical screening was performed following the fluorescence emitted by GFP controlled by the promoter of gene F35E12.5, we expected to find this gene in the cluster of genes upregulated by colistin treatment. As shown in Fig. 3A (see also Table S2 in the supplemental material), F35E12.5 showed a more than 5-fold increase in the colistin-treated group (P < 0.001). To further confirm the role of colistin in the transcriptional activation of the PMK-1 pathway, we compared the 2,538 genes upregulated with colistin treatment with the transcripts positively regulated by PMK-1. As shown in Fig. 3B (see also Tables S5 and S6), there is a significant overlap between colistin-upregulated genes and genes that are positively induced by PMK-1 (representation factor 2.7; P < 7.07e−7).
In order to gain insights into the mechanisms of colistin-induced immune activation, we performed a biased gene enrichment analysis using several gene sets linked to immune or stress responses (Fig. 3C; see also Table S6 in the supplemental material). As expected, PMK-1-regulated genes showed the strongest overlap with colistin-induced genes among the selected gene sets. We found that SKN-1- and DAF-16-regulated genes are also among the most highly enriched genes (Fig. 3C). SKN-1 is phosphorylated by PMK-1 and is required to protect C. elegans from reactive oxygen species (ROS) induced by bacterial pathogens (11). DAF-16 is a FOXO transcription factor that controls both longevity and innate immunity in C. elegans (12, 13). These results suggest that colistin not only induces a PMK-1-mediated immune response but also targets additional pathways that may control the expression of additional genes required for immunity against bacterial infections.
To identify related gene groups that are transcriptionally controlled by pathways targeted by colistin, we performed an unbiased gene enrichment analysis using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/). We separately analyzed 2,538 upregulated and 1,864 downregulated genes. The top 10 enriched annotation clusters of upregulated genes with P values of <0.05 correspond to genes encoding epidermal growth factor (EGF)-like domains, Apple-like domains, C-type lectins, Kunitz metazoan domains, homeobox, CUB-like domains, laminin G, neurotransmitter-gated ion-channel, Hedgehog/intein hint domains, and EGF-like calcium-binding domains (Fig. 4A). Among these gene groups found to be upregulated by colistin treatment, C-type lectins and CUB-like domain-containing proteins have been linked to antibacterial inducible defenses (7, 8, 14). The top 10 enriched annotation clusters of downregulated genes with P values of <0.05 correspond to genes encoding protein-tyrosine phosphatase, protein kinase, serine/threonine-specific protein phosphatase, protein kinase-ATP binding site, BRCT domains, histone core domains, transcription factor-fork head domains, SCP-like extracellular protein, glutamine synthetase, and zinc finger domains (Fig. 4B).
FIG 4 .

DAVID functional annotation cluster analysis of genes up- and downregulated by colistin. (A) Top 10 clusters of genes upregulated by colistin. Gray bars show 2 clusters that are also positively induced in response to Y. pestis infection. (B) Top 10 clusters of genes downregulated by colistin.
The major overlap between genes upregulated by Y. pestis infection and colistin treatment (Fig. 3B; see also Table S4 in the supplemental material) suggests that similar immune mechanisms are activated by the two stimuli and that similar gene groups should be upregulated. To test this hypothesis, we performed DAVID analysis on the clusters of genes misregulated in response to Y. pestis infection (see Fig. S1) and compared the results with those obtained by analyzing genes misregulated in response to colistin treatment. As shown in Fig. 4A, two of the three upregulated functional annotation clusters exhibiting the highest enrichment scores, EGF-like domains and C-type lectins, are induced by both Y. pestis infection and colistin treatment.
Prophylactic administration of colistin protects from infection by Gram-negative bacteria by inducing tolerance.
Since colistin treatment induces the expression of classes of transcripts that are also induced in response to Y. pestis (Fig. 3B and 4A), we hypothesized that colistin treatment may protect from bacterial infection. An advantage of using Y. pestis as a model pathogen is that it is naturally resistant to colistin (15). Our results also suggest that colistin does not affect the growth of Y. pestis at a 160-µg/ml concentration, which is much higher than the dosage used for treating the animals (see Fig. S2 in the supplemental material). Thus, the use of Y. pestis should allow us to distinguish the antimicrobial properties of colistin from its immunomodulatory properties. To further rule out the possibility that colistin may protect from bacterial infections due to any antimicrobial property, we decided to pretreat the animals prior to pathogen exposure. Thus, we treated C. elegans with colistin for 24 h prior to exposing it to Y. pestis. As shown in Fig. 5A, pretreatment with colistin enhances the resistance of the animals to Y. pestis-mediated killing. However, the resistance to Y. pestis may be due to an advantage in overall fitness conferred by colistin pretreatment. To rule out this possibility, we pretreated the wild-type animals with colistin and exposed them to nonpathogenic E. coli. As shown in Fig. S3A, a colistin treatment capable of inducing resistance to bacterial infections does not affect the life span of the animal. Similar results were also found using chronic exposure to different concentrations of colistin (see Fig. S3B).
FIG 5 .

Colistin protects C. elegans from infection by Gram-negative bacteria Y. pestis and P. aeruginosa. (A and B) L4 N2 animals treated with or without colistin for 24 h were transferred to plates containing Y. pestis or P. aeruginosa and scored for survival (Y. pestis, P < 0.01; P. aeruginosa, P < 0.001). Shown are representative assays of more than three independent experiments (n = 60 animals per treatment). WT, wild type. (C and D) Wild-type N2 animals treated with or without colistin (Col) for 24 h were exposed to P. aeruginosa expressing GFP for 48 h and then visualized using a fluorescence microscope. (E and F) Wild-type N2 animals treated with or without colistin for 24 h were exposed to Y. pestis expressing GFP for 96 h. These merged images show bacterial fluorescence (green channel) and the gut autofluorescence (red channel). Multiple animals are shown; arrows point at bacteria expressing GFP in the intestinal lumen of an animal. (G and H) Wild-type N2 animals treated with or without colistin for 24 h were exposed to P. aeruginosa/GFP for 48 h and Y. pestis/GFP for 96 h, and the CFU were quantified. Ten animals were used for each condition. The graph represents combined results of three independent experiments. Bars represent means ± SEM.
Y. pestis KIM5 infection in C. elegans is characterized as causing a persistent colonization of the gut that initiates in the posterior part of the intestinal track (16), which is the region of the animal where most upregulated F35E12.5 with Y. pestis infection and colistin treatment is observed (Fig. 2A). In contrast, pathogens such as P. aeruginosa kill C. elegans by a mechanism that does not involve persistent colonization of the intestine (17). Thus, we studied whether colistin could protect from a pathogen that kills C. elegans using a different mode of infection. Pretreatment with colistin also enhanced the resistance of the animals to P. aeruginosa-mediated killing (Fig. 5B). To analyze the prophylactic effect of colistin in the context of infections by Gram-positive bacteria, we used E. faecalis and S. aureus, which are capable of killing C. elegans by causing persistent and nonpersistent infections, respectively (18). Interestingly, pretreatment with colistin does not have an impact on the resistance to either E. faecalis- or S. aureus-mediated killing of C. elegans (see Fig. S4 in the supplemental material).
To provide further insights into the mechanisms by which colistin confers protection against Y. pestis and P. aeruginosa infection, we analyzed the bacterial burden following the profile of accumulation of bacteria expressing GFP. Live bacteria in infected animals were quantified by grinding up the nematodes to recover live bacteria and counting the CFU after plating on the appropriate medium. As shown in Fig. 5C to H, colistin pretreatment did not reduce the bacterial burden. Taken together, these results indicate that while the animals cannot clear the infection, they are better equipped to tolerate it.
Colistin protects from infection by Gram-negative bacteria in a PMK-1-dependent manner.
We studied whether the prophylactic effect of colistin requires PMK-1 by treating KU25 animals, which carry the pmk-1(km25) deletion allele, with colistin prior to Y. pestis or P. aeruginosa infection. Figure 6A and B shows that the survival of pmk-1(km25)-treated animals was comparable to that of untreated animals, indicating that the beneficial effects of colistin require a functional PMK-1 pathway.
FIG 6 .

PMK-1 is required for colistin-induced resistance to the Gram-negative bacteria Y. pestis and P. aeruginosa. (A and B) Wild-type N2 and pmk-1(km25) L4 animals treated with or without colistin for 24 h were transferred to plates containing Y. pestis or P. aeruginosa and scored for survival. (C and D) MGH171 sid-1(qt9); Is[vha-6::sid-1::SL2::gfp] L4 animals fed with E. coli strain HT115 carrying a vector control or expressing double-stranded RNA (dsRNA) targeting pmk-1 were treated with or without colistin for 24 h, transferred to plates containing Y. pestis or P. aeruginosa, and scored for survival (n = 60 animals per treatment). Shown are representative results of at least two independent experiments.
Since PMK-1 functions in the intestine to confer resistance to both Y. pestis and P. aeruginosa infection (19, 20), we hypothesized that colistin may require intestinal PMK-1 for protection against infections. To evaluate the intestinal contribution of PMK-1 in response to colistin, we utilized a C. elegans strain capable of RNAi activity only in the intestine (strain MGH171). Enriched RNAi knockdown of pmk-1 in the intestine of MGH171 animals completely abolished the protection conferred by colistin (Fig. 6C and D). This result indicates that intestine is the major site where colistin functions to modulate the immune response in C. elegans.
Colistin-induced resistance to Gram-negative bacterium infections depends on the transcription factors SKN-1 and DAF-16.
It is known that ATF-7, an ortholog of mammalian ATF2/ATF7, is phosphorylated by PMK-1 and is responsible for the induction of PMK-1-regulated genes during pathogen infection (21). To study whether ATF-7 plays a role in the protective effect of colistin, we analyzed the effect of colistin on the survival of pathogen infection of animals where RNAi was used to inhibit ATF-7. Consistent with previous results, atf-7 RNAi animals were susceptible to both Y. pestis and P. aeruginosa infections compared to results for control animals (Fig. 7A and B, dotted black line versus black line; P < 0.01). atf-7 RNAi also suppressed the enhanced resistance to pathogen infection induced by colistin (Fig. 7A and B, dotted red line versus red line; P < 0.001). However, colistin was capable of enhancing resistance to pathogen infection in atf-7 RNAi animals (Fig. 7A and B, dotted black line versus dotted red line; P < 0.01), indicating that the enhanced resistance to pathogen infection induced by colistin is not dependent on ATF-7 activity.
FIG 7 .

Colistin-induced resistance to the Gram-negative bacteria Y. pestis and P. aeruginosa depends on the transcription factors SKN-1 and DAF-16. (A and B) Wild-type N2 L4 animals fed with E. coli strain HT115 carrying a vector control or expressing dsRNA targeting atf-7 were treated with or without colistin for 24 h, transferred to plates containing Y. pestis or P. aeruginosa, and scored for survival. (C and D) Wild-type N2 L4 animals fed with E. coli strain HT115 carrying a vector control or expressing dsRNA targeting skn-1 were treated with or without colistin for 24 h, transferred to plates containing Y. pestis or P. aeruginosa, and scored for survival. (E and F) Wild-type N2 and daf-16(mu86) L4 animals treated with or without colistin for 24 h were transferred to plates containing Y. pestis or P. aeruginosa and scored for survival (n = 60 animals per treatment). Shown are representative results of at least two independent experiments.
The gene enrichment analysis indicates that there is a significant overlap between colistin-upregulated genes and genes positively regulated by SKN-1 and DAF-16 (Fig. 3C). Thus, we sought to investigate whether SKN-1 and DAF-16 are needed for colistin-induced immune activation. skn-1 RNAi completely abolished the beneficial effect of colistin (Fig. 7C and D). Colistin-induced resistance to infections was also found to be dependent on DAF-16, since daf-16(mu86) mutation completely suppressed the enhanced resistance to both Y. pestis and P. aeruginosa upon colistin treatment (Fig. 7E and F). Taken together, these results indicate that colistin protection for the Gram-negative bacteria Y. pestis and P. aeruginosa depends on both SKN-1 and DAF-16.
DISCUSSION
We have shown here that prophylactic administration of colistin protects C. elegans from infection by Y. pestis and P. aeruginosa. This effect appears to involve conserved mechanisms that control immune responses in C. elegans, including those regulated by the p38/PMK-1 MAPK and DAF-16/SKN-1 pathways. Furthermore, our studies suggest that the enhancement of immune responses by colistin requires intestinal PMK-1 and that it does not protect the animals from the infection by reducing the bacterial burden but rather by making the animals more tolerant to the infection. Together, these findings identify an alternative mechanism by which the antibiotic colistin may protect from bacterial infections.
Colistin is a rapid bactericide against Gram-negative bacteria, interacting with the lipid A moiety of lipopolysaccharide to cause disorganization of the outer membrane. Colistin was first introduced to the clinic in 1952, but due to reports of nephrotoxicity and neurotoxicity in the 1970s, it was largely replaced by other antibiotics. However, the increase in widespread multidrug resistance of Gram-negative pathogens during recent decades has led to the reconsideration of colistin as a therapeutic option. Although a large number of studies concerning colistin investigate its clinical use, antibacterial activity, and mechanism of microbial resistance, there is no information regarding specific immune pathways that may be targeted by the drug.
The results presented herein clearly show that colistin acts as an activator of conserved immune pathways. C. elegans has a number of physical and enzymatic xenobiotic defenses, including various detoxification enzymes that can act as xenobiotic efflux pumps (22). However, the direct exposure of the intestinal cells of the animals to 20 µg/ml of colistin for 24 h appears to circumvent the aforementioned xenobiotic detoxification mechanisms, allowing colistin to reach intracellular concentrations appropriate to activate immune pathways and confer resistance to pathogen infections. Indeed, the expression profile study showed that colistin can induce a transcriptional response in C. elegans that is very similar to that induced by pathogen infection. In addition, colistin treatment results in the upregulation of a number of genes that are markers of immune activation, including those that encode EGF-like domains, C-type lectins, and CUB-like domains. The biased gene enrichment analyses highlighted pathways that are controlled by PMK-1, DAF-16, and SKN-1 (Fig. 3C). Both DAF-16 and SKN-1 are independently inhibited by the insulin receptor DAF-2 (23). DAF-16 is the sole ortholog of the FOXO family of transcription factors and responsible for being the primary transcription factor required for the profound life span extension and resistance to pathogen infection upon mutation of the insulin-like receptor DAF-2 (12, 24, 25). Consistent with previous findings demonstrating that PMK-1 plays a crucial role in immunity since it is critical for immunity even when the DAF-16 pathway is hyperactivated by daf-2 mutation (7), we found that protection against bacterial infections induced by colistin is completely abolished by PMK-1 inhibition.
For decades, it has been noted that certain antibiotics have immunomodulatory properties, which can improve the long-term outcome of patients with some chronic diseases (26). The best-investigated family of antibiotics is macrolides. Several clinical trials have confirmed the beneficial effects of long-term treatment with macrolides such as azithromycin in patients with chronic inflammatory pulmonary diseases, such as mainly diffuse panbronchiolitis (DPB) and cystic fibrosis (CF) (27, 28). The concept of taking advantage of the immunomodulatory properties of antibiotics to reduce the severity of pulmonary diseases has been so convincing that investigations have been expanded to other classes of antibiotics, such as tetracyclines and fluoroquinolones. However, a role for the polymycin antibiotic colistin in the control of immunity has not been reported. Here we showed that a short, nontoxic treatment with colistin can activate the innate immunity of C. elegans in a p38/PMK-1-dependent manner. Since p38/PMK-1-mediated immune responses appear to be conserved from plants to mammals, it is conceivable that colistin may also activate immunity in higher organisms, including humans.
MATERIALS AND METHODS
Nematode and bacterial strains.
C. elegans strains used in this study were N2 Bristol, AY101 acIs101[pDB09.1(pF35E12.5::gfp), pRF4(rol-6(su1006))] (8), KU25 pmk-1(km25), CF1038 daf-16(mu86), and MGH171 sid-1(qt9); Is[Pvha-6::sid-1::SL2::gfp] (29). All strains were maintained at 20°C on nematode growth medium (NGM) and fed with E. coli OP 50. Bacterial strains used were Escherichia coli OP 50 (30), Pseudomonas aeruginosa PA14 (31), Yersinia pestis KIM5 (32), Enterococcus faecalis OG1RF (33), and Staphylococcus aureus MSSA476 (34). E. coli, P. aeruginosa, and S. aureus cultures were grown in Luria-Bertani (LB) broth overnight at 37°C. E. faecalis was grown in brain heart infusion (BHI) broth overnight at 37°C. Y. pestis culture was grown in LB broth at 25°C.
Chemical library screen.
The Prestwick Library that comprises 1,120 small molecules was used (see Table S1 in the supplemental material). More than 95% of its compounds are marketed drugs (all off patent). Eggs from C. elegans strain AY101 were extracted from gravid adults and cultured in S-basal medium (5.8 g NaCl, 50 ml 1 M KPO4 [pH 6.0], and 1 ml 5-mg/ml cholesterol per liter) for 22 h. L1 larval animals were transferred to regular SB plates seeded with E. coli OP 50 and grown at 20°C until they reached the L4 larval stage. L4 animals were transferred into 96-well plates containing different drugs in S-basal medium and screened for enhanced GFP fluorescence after 24 h at 20°C. The chemicals were diluted in S-basal to 1:10. Each well contained 87 µl of S-basal (about 20 animals), 3 µl of E. coli OP 50, and 10 µl of each compound (final concentration, 20 µg/ml). E. coli OP 50 was replaced by Y. pestis KIM5 as a positive control.
Prophylactic treatment with colistin.
Animals were synchronized by hypochlorite treatment. Arrested L1 animals were placed on 10-cm NGM plates seeded with E. coli OP 50 and grown at 20°C until late L4 larval stage. Animals were collected after a wash with M9 buffer (3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl, and 1 ml 1 M MgSO4 [per liter]) and subsequently kept at 20°C in 9 ml of liquid S-basal medium with or without 20 µg/ml colistin and supplemented with 300 µl of heat-killed E. coli OP 50. Twenty-four hours later, the animals were harvested for RNA extractions, killing assays, or Western blotting. Colistin was dissolved in water to a 20-mg/ml stock concentration. Subsequent dilutions were performed using S-basal.
RNA interference.
RNA interference was used to generate loss-of-function RNAi phenotypes by feeding nematodes with E. coli strain HT115(DE3) expressing double-stranded RNA that is homologous to a target gene. E. coli HT115(DE3) was grown in LB broth containing ampicillin (100 µg/ml) at 37°C overnight and plated onto NGM containing 100 µg/ml ampicillin and 3 mM isopropyl 1-thio-β-d-galactopyranoside. RNAi-expressing bacteria were allowed to grow overnight at 37°C. L2 or L3 larval animals were placed on RNAi or vector control plates for 2 days at 20°C until nematodes became gravid. Gravid adults were then transferred to fresh RNAi-expressing bacterial lawns and allowed to lay eggs for 2 h to synchronize a second-generation RNAi population. For nematodes that were subjected to colistin pretreatment, L4 animals were harvested and treated with colistin as mentioned above. skn-1 RNAi started at the L1 stage and was performed by placing synchronized L1 animals on RNAi plates. unc-22 RNAi was included as a positive control in all experiments to account for RNAi efficiency.
Copas biosorter GFP analysis.
Expression levels of the pF35E12.5::gfp reporter in AY101 transgenic animals were analyzed using the Copas Biosort instrument for large-particle flow cytometry (Union Biometrica, Holliston, MA). Synchronized animals treated with control vector or pmk-1 RNAi were exposed to E. coli or Y. pestis for 24 h and washed in M9 buffer prior to analysis. Fluorescence data were acquired from a minimum of 400 adult animals for each experimental sample. Plots were constructed using FlowJo flow cytometry analysis software (Tree Star, Inc., Ashland, OR).
Microarray analysis.
RNA was extracted from three biological replicates using the TRIzol reagent (Invitrogen). Residual genomic DNA was removed by DNase treatment (Ambion, Austin, TX). Samples were hybridized to the C. elegans Gene Expression Microarray (Agilent Technologies) by the Duke Microarray Facility. Data were analyzed using Partek Genomics Suite software. Raw data were preprocessed, including background correction, normalization, and summarization using robust multiarray average analysis, and expression data were log2 transformed. Principal-component analysis (PCA) was performed to identify outliers and evaluate whether batch effects, ethnicity, or intrinsic subtype significantly affected the data. Gene lists were created using a cutoff P value of <0.05, 2-fold change (see Tables S2 and S3 in the supplemental material). For subsequent analyses, the list of up- and downregulated genes was curated by removing duplicates and using the WormBase Converter (http://wormbasemanager.sourceforge.net), WB220.
Western blot analysis.
Whole-worm lysates were prepared in the presence of protease and phosphorylase inhibitors. Active PMK-1 was detected using an Anti-Active p38 polyclonal antibody (pAb), rabbit (pTGpY), from Promega, Inc. β-Actin was detected using a monoclonal anti-β-actin antibody produced in mouse from Sigma. Blots were developed using SuperSignal chemiluminescence substrate (Pierce).
Functional gene enrichment analysis for all differentially expressed genes.
DAVID (Database for Annotation Visualization and Integrated Discovery; http://david.abcc.ncifcrf.gov/) was used for functional annotation bioinformatics microarray analysis to determine the functional enrichment and Gene Ontology annotation; clusters with P values of <0.05 were selected as significant. Probability calculations of enrichment were performed using an online hypergeometric probability tool to calculate the statistical significance of the overlap of gene groups (http://www.geneprof.org/GeneProf/tools/hypergeometric.jsp). The representation factor is the number of overlapping genes divided by the expected number of overlapping genes drawn from two independent groups. A representation factor below 1.0 indicates underrepresentation, whereas a value above 1.0 indicates overrepresentation.
C. elegans killing assays.
The bacterial lawns used for C. elegans killing assays were prepared by placing 20 µl of an overnight culture of the bacterial strains on modified NGM agar medium (0.35% instead of 0.25% peptone) in 3.5-cm diameter plates. Plates were incubated overnight at 37°C (25°C for Y. pestis) before animals were added. Synchronized L4 C. elegans hermaphrodites treated with or without colistin for 24 h at 20°C were then transferred to modified NGM plates containing bacterial lawns and incubated at 25°C. Animals were scored every 4 to 24 h for survival and transferred to fresh pathogen lawns each day. Animals were scored at the times indicated and were considered dead when they failed to respond to touch. Animal survival was plotted as a nonlinear regression curve using the PRISM (version 4.00) computer program. Survival curves were considered different when P values were <0.05. PRISM uses the product limit or Kaplan-Meier method to calculate survival fractions and the log rank test, which is equivalent to the Mantel-Haenszel test, to compare survival curves.
Profile of bacterial accumulation in the nematode intestine.
After pretreatment with colistin for 24 h at 20°C, N2 animals were then transferred to plates seeded with either P. aeruginosa/GFP for 48 h or Y. pestis/GFP for 96 h at 25°C. Animals were transferred to fresh pathogen lawns each day. At 48 h or 96 h, animals were transferred to an NGM plate seeded with E. coli for 15 min and transferred again to a new NGM plate seeded with E. coli for 30 min to eliminate P. aeruginosa/GFP or Y. pestis/GFP adhered to the body of the nematodes. Animals were then visualized using a fluorescence stereomicroscope.
Quantification of intestinal bacterial loads.
After exposure to P. aeruginosa/GFP for 48 h or Y. pestis/GFP for 96 h, animals were transferred to an NGM plate seeded with E. coli for 15 min to eliminate P. aeruginosa/GFP or Y. pestis/GFP adhered to the body of the worms. Animals were transferred to a new NGM plate seeded with E. coli for 30 min to further eliminate external P. aeruginosa/GFP or Y. pestis/GFP. Ten nematodes per condition were transferred into 50 μl of phosphate-buffered saline (PBS) containing 0.1% Triton and grounded. Serial dilutions of the lysates (10−1, 10−2, 10−3, and 10−4) were plated onto LB-kanamycin plates and grown for 24 h at 37°C and 48 h at 25°C to select for P. aeruginosa/GFP or Y. pestis/GFP cells, respectively.
SUPPLEMENTAL MATERIAL
DAVID functional annotation cluster analysis of genes up- and downregulated by Y. pestis infection. (A) Top 6 clusters of genes upregulated by Y. pestis infection. (B) Top 3 clusters of genes downregulated by Y. pestis infection. Download
Colistin does not affect the growth of Y. pestis. Y. pestis KIM5 was cultured on modified NGM agar plates used for C. elegans killing assays supplemented with 160 µg/ml of colistin. At the indicated times, serial dilutions were prepared and plated to quantify CFU. Download
Prophylactic or chronic treatment with colistin does not prolong life span on exposure to E. coli. (A) Wild-type N2 L4 animals treated with or without 20 µg/ml colistin for 24 h were transferred to modified NGM plates containing E. coli and scored for survival. (B) Wild-type N2 animals were fed with E. coli grown on modified NGM plates supplemented with different concentrations of colistin. Shown are representative assays of two independent experiments (n = 60 adult animals per treatment). Download
Prophylactic treatment of colistin does not prolong life span on exposure to the Gram-positive pathogens E. faecalis and S. aureus. Wild-type N2 L4 animals treated with or without 20 µg/ml colistin for 24 h were transferred to modified NGM plates containing E. faecalis (A) or S. aureus (B) and scored for survival. Shown are representative assays of two independent experiments (n = 60 animals per treatment). Download
Forty-five market drugs from the Prestwick Library capable of inducing pF35E12.5::gfp expression
Genes upregulated by colistin
Genes downregulated by colistin
Fifty-four overlap genes upregulated by colistin and genes positively induced by Y. pestis
Twenty-seven overlap genes upregulated by colistin and genes positively induced by p38/PMK-1
Overlap of pathways linked to immune or stress responses with genes upregulated by colistin
ACKNOWLEDGMENTS
We thank the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN) for strains used in this study, Justine Melo from the Ruvkun Lab (Massachusetts General Hospital, Harvard Medical School, Boston, MA) for providing the intestine-specific RNAi strain MGH171, and Dennis Kim (Massachusetts Institute of Technology, Cambridge, MA) for providing the atf-7 RNAi bacterial clone.
This work was supported by National Institutes of Health grant GM0709077 (to A.A.) and Beijing Natural Science Foundation of China grant 7132168 (to Y.C.).
Footnotes
Citation Cai Y, Cao X, Aballay A. 2014. Whole-animal chemical screen identifies colistin as a new immunomodulator that targets conserved pathways. mBio 5(4):e01235-14. doi:10.1128/mBio.01235-14.
REFERENCES
- 1. Scannell JW, Blanckley A, Boldon H, Warrington B. 2012. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 11:191–200. 10.1038/nrd3681 [DOI] [PubMed] [Google Scholar]
- 2. Coller BS, Califf RM. 2009. Traversing the valley of death: a guide to assessing prospects for translational success. Sci. Transl. Med. 1:10cm19. 10.1126/scitranslmed.3000265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giacomotto J, Ségalat L. 2010. High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 160:204–216. 10.1111/j.1476-5381.2010.00725.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Janeway CA, Jr, Medzhitov R. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. 10.1146/annurev.immunol.20.083001.084359 [DOI] [PubMed] [Google Scholar]
- 5. Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, Paterson DL. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 6:589–601. 10.1016/S1473-3099(06)70580-1 [DOI] [PubMed] [Google Scholar]
- 6. Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, Ausubel FM. 2002. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297:623–626. 10.1126/science.1073759 [DOI] [PubMed] [Google Scholar]
- 7. Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH. 2006. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet. 2:e183. 10.1371/journal.pgen.0020183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bolz DD, Tenor JL, Aballay A. 2010. A conserved PMK-1/p38 MAPK is required in Caenorhabditis elegans tissue-specific immune response to Yersinia pestis infection. J. Biol. Chem. 285:10832–10840. 10.1074/jbc.M109.091629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun J, Singh V, Kajino-Sakamoto R, Aballay A. 2011. Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332:729–732. 10.1126/science.1203411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aballay A. 2013. Role of the nervous system in the control of proteostasis during innate immune activation: insights from C. elegans. PLoS Pathog. 9:e1003433. 10.1371/journal.ppat.1003433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van der Hoeven R, McCallum KC, Cruz MR, Garsin DA. 2011. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog. 7: e1002453. 10.1371/journal.ppat.1002453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM. 2003. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300:1921. 10.1126/science.1080147 [DOI] [PubMed] [Google Scholar]
- 13. Irazoqui JE, Urbach JM, Ausubel FM. 2010. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat. Rev. Immunol. 10:47–58. 10.1038/nri2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kerry S, TeKippe M, Gaddis NC, Aballay A. 2006. GATA transcription factor required for immunity to bacterial and fungal pathogens. PLoS One 1:e77. 10.1371/journal.pone.0000077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hernandez E, Girardet M, Ramisse F, Vidal D, Cavallo JD. 2003. Antibiotic susceptibilities of 94 isolates of Yersinia pestis to 24 antimicrobial agents. J. Antimicrob. Chemother. 52:1029–1031. 10.1093/jac/dkg484 [DOI] [PubMed] [Google Scholar]
- 16. Styer KL, Hopkins GW, Bartra SS, Plano GV, Frothingham R, Aballay A. 2005. Yersinia pestis kills Caenorhabditis elegans by a biofilm-independent process that involves novel virulence factors. EMBO Rep. 6:992–997. 10.1038/sj.embor.7400516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aballay A, Ausubel FM. 2002. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr. Opin. Microbiol. 5:97–101. 10.1016/S1369-5274(02)00293-X [DOI] [PubMed] [Google Scholar]
- 18. Garsin DA, Sifri CD, Mylonakis E, Qin X, Singh KV, Murray BE, Calderwood SB, Ausubel FM. 2001. A simple model host for identifying Gram-positive virulence factors. Proc. Natl. Acad. Sci. U. S. A. 98:10892–10897. 10.1073/pnas.191378698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bolz DD, Tenor JL, Aballay A. 2010. A conserved PMK-1/p38 MAPK is required in Caenorhabditis elegans tissue-specific immune response to Yersinia pestis infection. J. Biol. Chem. 285:10832–10840. 10.1074/jbc.M109.091629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shivers RP, Kooistra T, Chu SW, Pagano DJ, Kim DH. 2009. Tissue-specific activities of an immune signaling module regulate physiological responses to pathogenic and nutritional bacteria in C. elegans. Cell Host Microbe 6:321–330. 10.1016/j.chom.2009.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shivers RP, Pagano DJ, Kooistra T, Richardson CE, Reddy KC, Whitney JK, Kamanzi O, Matsumoto K, Hisamoto N, Kim DH. 2010. Phosphorylation of the conserved transcription factor ATF-7 by PMK-1 p38 MAPK regulates innate immunity in Caenorhabditis elegans. PLoS Genet. 6:e1000892. 10.1371/journal.pgen.1000892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lindblom TH, Dodd AK. 2006. Xenobiotic detoxification in the nematode Caenorhabditis elegans. J. Exp. Zool. A Comp. Exp. Biol. 305:720–730. 10.1002/jez.a.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. 2008. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132:1025–1038. 10.1016/j.cell.2008.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. 1997. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389:994–999. 10.1038/40194 [DOI] [PubMed] [Google Scholar]
- 25. Singh V, Aballay A. 2006. Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc. Natl. Acad. Sci. U. S. A. 103:13092–13097. 10.1073/pnas.0604050103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tauber SC, Nau R. 2008. Immunomodulatory properties of antibiotics. Curr. Mol. Pharmacol. 1:68–79. 10.2174/1874467210801010068 [DOI] [PubMed] [Google Scholar]
- 27. Rubin BK, Henke MO. 2004. Immunomodulatory activity and effectiveness of macrolides in chronic airway disease. Chest 125(2 Suppl):70S–78S. 10.1378/chest.125.2_suppl.70S [DOI] [PubMed] [Google Scholar]
- 28. Saiman L, Marshall BC, Mayer-Hamblett N, Burns JL, Quittner AL, Cibene DA, Coquillette S, Fieberg AY, Accurso FJ, Campbell PW, Study Macrolide Group 2003. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 290:1749–1756. 10.1001/jama.290.13.1749 [DOI] [PubMed] [Google Scholar]
- 29. Melo JA, Ruvkun G. 2012. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149:452–466. 10.1016/j.cell.2012.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77:71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan MW, Rahme LG, Sternberg JA, Tompkins RG, Ausubel FM. 1999. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc. Natl. Acad. Sci. U. S. A. 96:2408–2413. 10.1073/pnas.96.5.2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Une T, Brubaker RR. 1984. In vivo comparison of avirulent Vwa- and Pgm- or Pstr phenotypes of yersiniae. Infect. Immun. 43:895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray BE, Singh KV, Ross RP, Heath JD, Dunny GM, Weinstock GM. 1993. Generation of restriction map of Enterococcus faecalis OG1 and investigation of growth requirements and regions encoding biosynthetic function. J. Bacteriol. 175:5216–5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38:1008–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DAVID functional annotation cluster analysis of genes up- and downregulated by Y. pestis infection. (A) Top 6 clusters of genes upregulated by Y. pestis infection. (B) Top 3 clusters of genes downregulated by Y. pestis infection. Download
Colistin does not affect the growth of Y. pestis. Y. pestis KIM5 was cultured on modified NGM agar plates used for C. elegans killing assays supplemented with 160 µg/ml of colistin. At the indicated times, serial dilutions were prepared and plated to quantify CFU. Download
Prophylactic or chronic treatment with colistin does not prolong life span on exposure to E. coli. (A) Wild-type N2 L4 animals treated with or without 20 µg/ml colistin for 24 h were transferred to modified NGM plates containing E. coli and scored for survival. (B) Wild-type N2 animals were fed with E. coli grown on modified NGM plates supplemented with different concentrations of colistin. Shown are representative assays of two independent experiments (n = 60 adult animals per treatment). Download
Prophylactic treatment of colistin does not prolong life span on exposure to the Gram-positive pathogens E. faecalis and S. aureus. Wild-type N2 L4 animals treated with or without 20 µg/ml colistin for 24 h were transferred to modified NGM plates containing E. faecalis (A) or S. aureus (B) and scored for survival. Shown are representative assays of two independent experiments (n = 60 animals per treatment). Download
Forty-five market drugs from the Prestwick Library capable of inducing pF35E12.5::gfp expression
Genes upregulated by colistin
Genes downregulated by colistin
Fifty-four overlap genes upregulated by colistin and genes positively induced by Y. pestis
Twenty-seven overlap genes upregulated by colistin and genes positively induced by p38/PMK-1
Overlap of pathways linked to immune or stress responses with genes upregulated by colistin