

Abstract
The immune system plays a complex role in the development and progression of pancreatic cancer. Inflammation can promote the formation of premalignant lesions and accelerate pancreatic cancer development. Conversely, pancreatic cancer is characterized by an immunosuppressive environment, which is thought to promote tumor progression and invasion. Here we review the current literature describing the role of the immune response in the progressive development of pancreatic cancer, with a focus on the mechanisms that drive recruitment and activation of immune cells at the tumor site, and our current understanding of the function of the immune cell types at the tumor. Recent clinical and preclinical data are reviewed, detailing the involvement of the immune response in pancreatitis and pancreatic cancer, including the role of specific cytokines and implications for disease outcome. Acute pancreatitis is characterized by a predominantly innate immune response, while chronic pancreatitis elicits an immune response that involves both innate and adaptive immune cells, and often results in profound systemic immune-suppression. Pancreatic adenocarcinoma is characterized by marked immune dysfunction driven by immunosuppressive cell types, tumor-promoting immune cells, and defective or absent inflammatory cells. Recent studies reveal that immune cells interact with cancer stem cells and tumor stromal cells, and these interactions have an impact on development and progression of pancreatic ductal adenocarcinoma (PDAC). Finally, current PDAC therapies are reviewed and the potential for harnessing the actions of the immune response to assist in targeting pancreatic cancer using immunotherapy is discussed.
Keywords: Immune system, Pancreatitis, Pancreatic ductal adenocarcinoma, Immunosuppression, Immunotherapy, Inflammation
Core tip: The development and progression of pancreatic cancer is heavily influenced by the immune response. Inflammation of the pancreas (pancreatitis) is a significant risk factor for pancreatic cancer. Immune cells recruited to the inflamed pancreas release additional cytokines and potentiate damage to the tissue. Pancreatic cancer is characterized by profound immune suppression thought to be caused by signals originating from the tumor cells. Additionally, a subset of immune cells has been shown to support the growth of pancreatic cancer cells. Novel therapies for pancreatic cancer aim to utilize this unique immune environment to target this deadly disease.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer, and with a 5-year survival rate of 6%, it is one of the deadliest cancers worldwide[1]. The development and progression of PDAC is strongly influenced by the presence of inflammation[2]. Inflammation of the pancreas (pancreatitis) is a strong risk factor for PDAC development[3,4], and has been described as a critical factor in the initiation[5] and maintenance[6,7] of pancreatic disease. Additionally, PDAC is characterized by marked immunosuppression[6], which is thought to promote tumor progression and invasion.
This review highlights the role(s) of the immune response in the development of PDAC, focusing primarily on inflammation. Inflammatory conditions of the pancreas that can lead to increased risk for PDAC (pancreatitis), as well as the role of the immune response in the progressive stages of pancreas disease development, are discussed. Data from clinical studies and rodent models are integrated to present an up-to-date consensus of the role of inflammation in the initiation and progression of pancreatic cancer.
IMMUNE SYSTEM: A BRIEF OVERVIEW
The immune system is characteristically activated in response to infection by a foreign pathogen. In the case of cancer, tumor-specific antigens are recognized by the immune system, turning the cancer cell, in essence, into a foreign pathogen[8]. This allows the immune system to act as an extrinsic tumor-suppressor system. However, over time the chronic immune response to the tumor drives immunoselection of tumor cells that are able to thrive in an immunocompetent environment[9], much like the resistant bacterial strains that emerge as a result of exposure to antibiotics.
The mechanism by which the immune system can initially protect a host from tumor growth, but in some cases subsequently promotes cancer progression, is termed cancer immunoediting[10]. Cancer immunoediting is a dynamic process consisting of three phases, (1) elimination, when the immune system overcomes and eliminates the tumor before it can progress to a clinically relevant disease; (2) equilibrium, when the immune system does not eliminate the tumor, but controls tumor growth; and (3) escape, which occurs when the tumor has overcome the immune system and progresses to a clinically apparent disease. This third stage is generally seen as a failure in the adaptive immune system to provide long-term protection from tumor development due to selection of less immunogenic tumor cell variants during the equilibrium stage. Additionally, tumor escape can be facilitated by active immunosuppression induced by the tumor itself or some form of immune compromise or immune deficiency[11]. It is through this process that the cells of the immune system can act as both friend and foe to the body in the face of cancer.
Innate immunity
The innate immune system is composed of those immune cells that are already present in the body and can be immediately recruited to a site of infection during the process of “inflammation”. Innate immune cells include granulocytes, macrophages, mast cells, natural killer cells (NKCs) and dendritic cells (DCs). Table 1 reviews the mechanism(s) of action attributed to the innate immune cells in both the normal immune response and during the process of cancer development. Neutrophils are by far the most abundant granulocytes in the body and typically one of the first cell types to respond; they secrete cytokines and chemokines, modulating other cells in the immune response[12]. Macrophages, another major cell type of the innate immune response, remove dead or dying cells and associated debris via phagocytosis, as well as play a role in the adaptive immune response. Macrophage maturation in response to various signals produces one of two cell types, M1-polarized cells, which initiate the inflammatory response, and M2-polarized cells, which restrain the inflammatory response[13]. M2-polarized macrophages are immunosuppressive and can limit adaptive immunity by inhibiting T-lymphocyte proliferation, thus impeding the T-lymphocyte response[14].
Table 1.
Major functions of the innate immune cells in both inflammation and cancer
| Inflammatory cell | Immune function | Role in cancer |
| Neutrophil | Secretion of cytokines/chemokines to modulate other cells in the immune response | Maintenance of pro-angiogenic phenotype[242] |
| Release of cytotoxic granules | Suppression of anti-tumor immunity[243] | |
| Phagocytosis | Promotion of metastasis[244] | |
| Mast cell | Release of cytotoxic granules | Suppression of anti-tumor immunity[245] |
| Enhancement of immune cell recruitment | Stimulation of angiogenesis[152] | |
| Permeabilization of blood vessels | Direct stimulation of cancer cell growth[246] | |
| Secretion of mitogenic factors[157] | ||
| Macrophage | Phagocytosis | |
| Promotion of T-lymphocyte activation | ||
| M1 | Initiation of immune cell response | |
| Antigen presentation | ||
| M2 | Wound repair | |
| Immunosuppression | ||
| Tissue remodeling | ||
| Resolution of immune response | ||
| TAM | Secretion of Arginase-1[19] | |
| Support of Treg activation[247,248] | ||
| Promotion of angiogenesis[16] | ||
| Enhancement of tumor metastasis[17] | ||
| MDSC | Suppression of NKC and T-lymphocyte activation | Production of ROS[22] |
| Secretion of peroxynitrite[23] | ||
| Secretion of Arginase-1[24,25] | ||
| Induction of Treg[26] | ||
| Depletion of cysteine[27] | ||
| NKC | Release of cytotoxic granules | Tumor cytotoxicity[28] |
| DC | Antigen presentation |
TAM: Tumor-associated macrophage; MDSC: Myeloid-derived suppressor cell; NKC: Natural killer cell; DC: Dendritic cells.
In the context of cancer, two types of macrophages emerge, tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSC). Both cell types are similar to M2 macrophages and are recruited by tumor cells to thwart the anti-tumor immune activity. TAMs inhibit T-lymphocyte responses[15] and secrete cytokines that promote the tumor phenotype and metastasis[16,17]. In addition to their ability to directly induce T-lymphocyte apoptosis[18], TAMs produce arginase-1[19], a metalloenzyme that metabolizes and depletes the environment of arginine, an essential compound for T-lymphocyte proliferation[20,21].
The second type of immunosuppressive macrophage often found in the tumor microenvironment is the MDSC. Similar to TAMs in function, they differ mainly by their cell surface markers. MDSCs have been shown to exhibit powerful immunosuppressive properties, in part through production of reactive oxygen species and peroxynitrite[22,23], expression of arginase-1[24,25], induction of regulatory T-lymphocytes (Tregs)[26], and depletion of available cysteine, another amino acid required by T-lymphocytes[27].
NKCs are circulating immune cells that are able to kill target cells via induction of programmed cell death. NKCs have been shown to eliminate tumor cells, and treated cancer patients with high circulating NKCs have significantly longer metastasis-free survival[28]. The gap between the innate and adaptive immune systems is bridged by DCs[29]. Activated, antigen-presenting-DCs travel to the lymphoid organs where they interact with, and activate, B- and T-lymphocytes. In this way, activation of DCs by foreign pathogens can lead to the activation of both the innate and adaptive immune responses, allowing the body to fully respond to the perceived threat[30].
Adaptive immunity
Innate immunity is a powerful first defense against invading pathogens; however, it is most effective against cells bearing antigens that are common to many pathogens, and can be subverted by quickly evolving cell types, as in the case with cancer cells. The adaptive arm of the immune system is responsible for controlling those pathogens that have overcome the innate response and can provide long-lasting immunity against specific infectious agents.
T- and B-lymphocytes comprise the adaptive arm of the immune response. T-lymphocytes are grouped into classes based on their cell-surface proteins which mediate distinct effector functions. Cytotoxic T-lymphocytes express CD8 and are responsible for killing cells expressing foreign antigen by activating the target cell’s apoptosis program, leading to subsequent cell death[30]. Helper T-lymphocytes express CD4 on their surface and assist in the activation of CD8+ T-lymphocytes, B-lymphocytes and macrophages via secretion of specific cytokines, thus extending their function across both the innate and adaptive immune responses[30]. A subset of CD4+ lymphocytes, Tregs, protects the body from autoimmune responses. Tregs suppress T-lymphocyte activation in a cytokine independent, cell-contact-dependent manner[31].
B-lymphocytes are one of the main cell types responsible for the body’s ability to mount a long-term pathogen-specific response. Each B-lymphocyte produces a single species of antibody, and once activated, proliferates into an antibody-secreting effector cell. It is largely through the action of B-lymphocytes that the body is able to maintain an immunological memory and initiate and immediate response to foreign pathogens it has already encountered[32].
Although inflammation serves to protect the body from harm, it also plays a major role the development of disease, including cancer[33]. Pancreatic cancer, in particular, is heavily influenced by the inflammatory response associated with pancreatic injury and disease. Pancreatitis, or inflammation of the pancreas, is a relatively common condition that often leads to irreversible pancreatic damage and leaves the pancreas vulnerable to the development of neoplastic disease.
PANCREATITIS
The pancreas is comprised of both exocrine (acinar and ductal cells) and endocrine (islets of Langerhans) cells. The exocrine pancreas functions to produce and secrete digestive enzymes into the small intestine whereas the endocrine pancreas is primarily responsible for producing hormones crucial for glucose homeostasis. Dysfunction of the pancreas due to disease or injury can lead to impaired digestion, hypoglycemia and diabetes[34]. Acute pancreatitis (AP) is one of the most commonly diagnosed gastrointestinal diseases, with over 200000 patients admitted to the hospital each year[35]. Patients typically present with acute abdominal pain which may be accompanied by nausea and vomiting, and display increased serum concentrations of amylase and lipase[36]. Pathologically, AP presents as acinar degranulation, increased occurrences of autophagosomes, formation of dilated acinar lumina and autodigestive fat necrosis[37]. Risk factors for AP include alcohol consumption, gallstones, and smoking, however many cases are idiopathic[38-41]. Although the clinical symptoms of AP often resolve completely, more severe cases can lead to serious complications and even death in a small percentage of patients[42]. Complications associated with AP include pancreatic necrosis[43], infection leading to sepsis[44,45], and systemic inflammatory response syndrome (SIRS) leading to distant organ damage and failure (multiple organ dysfunction syndrome, MODS)[46]. These complications significantly increase the risk of mortality from AP. Additionally, recurrent bouts of AP lead to fibrosis/damage and chronic pancreatitis (CP), a risk factor for the development of pancreatic cancer[4,47].
CP is characterized by acinar loss, extensive fibrosis and immune cell infiltrate, and is a strong risk factor for pancreatic cancer[47-49], which may develop 10-20 years following CP diagnosis[3]. Although CP shares many of the same risk factors, causes, and symptoms as AP, the clinical presentation differs dramatically. Serum levels of pancreatic enzymes amylase and lipase are elevated in AP due to the acute damage caused to the pancreas. In contrast, these enzymes are either normal or only mildly elevated in CP[50]. The chronic inflammation that is the hallmark of CP leads to permanent damage and loss of pancreatic function, leading to diabetes and pancreatic insufficiency[51]. Comprehensive reviews of the diagnosis and etiology of these diseases, as well as factors that distinguish CP from AP, are available[52-54].
Studies from both clinical cases and rodent models of pancreatitis have contributed to the understanding of the inflammatory response to AP and associated syndromes. AP is thought to originate with uncontrolled activation of pancreatic acinar cells and release of digestive enzyme stores leading to autodigestion of the pancreatic cells. This autodigestion releases cellular contents, triggering the recruitment of inflammatory cells. Those inflammatory cells release cytokines and other modulating factors that can amplify the inflammatory response, causing systemic inflammation that can progress to SIRS and MODS. The events responsible for initiating the premature activation of digestive enzymes are not fully understood, but include trypsin auto-digestion[55], generation of reactive oxygen species, disturbances in microcirculation[56], calcium overload, and leukocyte overstimulation[57], and are reviewed in detail elsewhere[58-61].
There is a close relationship between the inflammatory response to AP and clinical severity of the disease[62]. Indeed, overstimulation of leukocytes, specifically neutrophils, results in systemic activation and is thought to be a major cause for severe AP-associated death[57,63]. Many animal models for AP exist in which pancreatitis is induced by pancreatic injury or surgical blockage. While insights into the possible mechanisms of the immune response in the pancreatic disease process can be gained from these models, they do not exactly replicate the clinical disease. Additionally, it should be noted that whereas rodents are the model of choice to study the immune system and its interaction with organ systems, significant differences exist between the human and rodent immune systems in the balance of leukocyte subsets, and in the expression of inflammatory mediators, cytokines and cytokine receptors, as well as the significantly different immunological environments occupied by either species[64]. For these reasons, animal models of pancreatitis will be addressed separately in this section of the review.
Inflammatory response to AP: Clinical findings
AP is characterized by early recruitment and activation of polymorphonuclear cells, the majority of which are neutrophils[57]. Activation of neutrophils can be identified by rising levels of serum neutrophil elastase in the early course of AP, typically within the first two days of diagnosis[62,65,66]. This is accompanied by the detection of metabolically hyperactive granulocytes in the pancreatic tissue of AP patients within 48 h of admission to the hospital, suggesting that granulocyte activation is an early AP event[67]. Neutrophil recruitment is followed by recruitment of the macrophage-monocyte system in the subsequent 2-3 d, as determined by rising levels of C-reactive protein (CRP) in the serum[62,68]. As elastase and CRP are released by neutrophils and macrophages, respectively, the presence of these proteins in the blood of pancreatitis patients is used as an indirect indicator of the recruitment and activation of these inflammatory cells in the pancreas.
Complications associated with AP can be grouped into two phases: immune overactivation and immune suppression. In the first phase, control is lost over the local inflammatory response, leading to excessive and uncontrolled systemic activation of inflammatory cells and mediators[69]. This often leads to the development of SIRS and MODS, and is associated with death within one week of disease[70]. In a subset of patients, the body responds to systemic inflammation with compensatory anti-inflammatory response syndrome (CARS)[71]. CARS initiates the second phase of complications associated with AP and can lead to immune deficiency or suppression, rendering the body susceptible to infection. These patients go on to develop AP-associated infections[44] that are associated with excessive CARS.
Systemically, a decrease in circulating lymphocytes, including B- and T-lymphocytes (both CD4+ and CD8+), as well as NKCs, is often seen in AP[67,72,73]. A decrease in circulating lymphocytes is associated with more severe disease and is often predictive of AP-associated systemic infection[74-76]. Kylänpää-Bäck et al[77] demonstrated a significant decrease in monocyte surface expression of human leukocyte antigen-DR (HLA-DR), a hallmark for systemic immunosuppression, in the first 24-48 h following AP diagnosis. Many independent studies have confirmed that a decrease in serum lymphocytes, as well as monocyte HLA-DR, correlates with more severe disease and increased mortality[73-75,77,78].
It is important to understand that the clinical timeline of inflammatory response to AP is a relative concept, as data is dependent on the timing of the patient’s admission to the hospital. Additionally, as biopsies are not routinely performed on AP patients, a detailed histological analysis of the pancreatic tissue is often not available. It is these limitations of clinical analysis that led to the use of rodent models in an attempt to more fully understand the mechanism of disease initiation and progression.
Animal models of AP
Several animal models have been developed to study the complex interactions that occur between the pancreatic epithelium and the many inflammatory cell types recruited to the pancreas. These models can be used to more precisely map the time course of the inflammatory response in AP, as well as to determine the mechanism(s) of damage and recovery in order to identify potential therapeutic targets. Although many species have been used to evaluate AP, this review will focus on data generated using rodents. Rodent models are used most often because of their cost-effectiveness and ease of characterization and genetic manipulation; however, no one model completely recapitulates all components of human disease.
The most common method of modeling pancreatitis in rodents is secretagogue hyperstimulation leading to premature intrapancreatic activation of digestive proteases. In this model, administration of high concentrations of the intestinal hormone cholecystokinin, or its molecular ortholog, caerulein, leads to autodigestion of the pancreas[79] and pancreatitis-like pathology including vacuolization, edema, acinar degranulation, dilated acinar lumina, necrosis, lung injury, and cytoplasmic destruction of pancreatic acini[80,81]. Figure 1 shows the histological similarities between human AP and caerulein-induced rodent AP. Another model for AP is surgical ligation of the pancreatic duct. Although this method was designed to mimic clinical gallstone-induced pancreatitis, it often produces a milder form of the disease and is more technically demanding and invasive[82]. Other models of AP include administration of high concentration of L-arginine leading to acinar necrosis[83], feeding of a choline-deficient diet, leading to severe necrotizing pancreatitis[84,85], and overstimulation of the immune system using bacteria or toxins[86].
Figure 1.
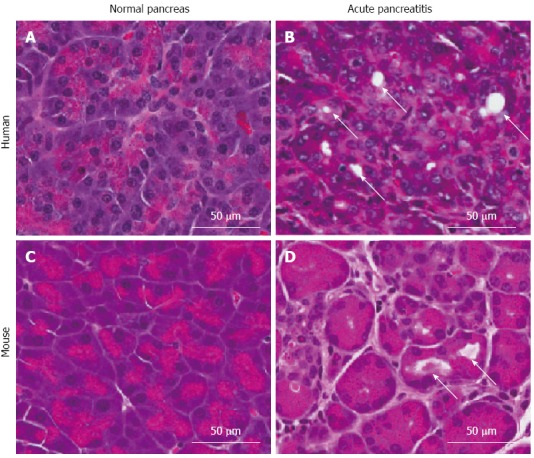
Comparison of histology of human and mouse pancreatic tissue. A: Normal human pancreas; B: Human acute pancreatitis (AP); C: Normal mouse pancreas; D: Mouse AP. Arrows indicate acini with dilated lumina, as commonly seen in AP.
Inflammatory response to AP: Animal models
Animal models of AP allow histological analysis of all stages of the disease, providing much information regarding the pathogenesis of AP. Although induced AP can present differently depending on animal model utilized, nearly all models result in a recruitment of neutrophils within hours of treatment (Figure 2), thus confirming neutrophils as one of the first responders to pancreatic damage[81,87-91]. Neutrophils have been shown to mediate systemic remote organ injury and death in a murine model of hemorrhagic pancreatitis[92]. Significant macrophage infiltration to the pancreas is observed shortly after caerulein-induced AP (Figure 2), and macrophage-derived macrophage-inflammatory protein-2 (MIP-2) is known to play a role in progression of AP through attraction of leukocytes, promoting tissue injury[93]. Therefore, results from caerulein-treated mice are consistent with clinical data that macrophages also contribute to the early inflammatory response to AP[93].
Figure 2.
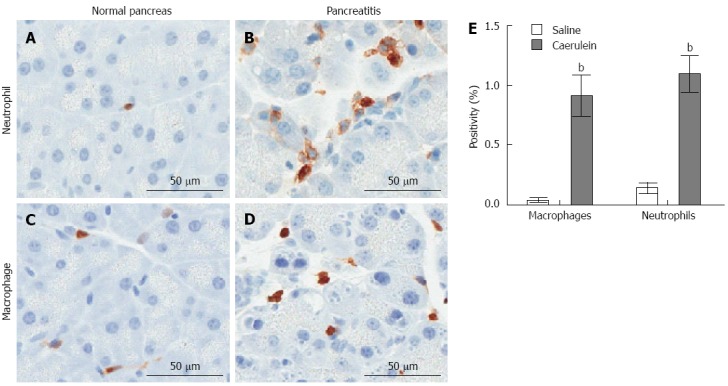
Induction of innate immune response in a mouse model of acute pancreatitis. Mice were injected intraperitoneally with 50 μg/kg caerulein or saline hourly for 7 h (Mayo Clinic IACUC protocol A48510). The pancreas was isolated one hour following the last injection and analyzed immunohistochemically for the presence of neutrophils (Ly6B.2: AbSerotec) or macrophages (Mac-2; Cedarlane Diagnostics). A: Neutrophil infiltration in saline-treated mice; B: Neutrophil infiltration in caerulein-treated mice; C: Macrophage infiltration in saline-treated mice; D: Macrophage infiltration in cearulein-treated mice; E: Stained slides were imaged using ScanScope XT (Aperio, Vista, California) and immunohistochemical staining was quantified using the Aperio ImageScope reader. Mean ± SD is plotted for each group of n ≥ 4 mice, bP < 0.01 vs saline group.
Mast cells are present in the normal pancreas and undergo degranulation upon induction of AP[87,94]. Inhibition of mast cell degranulation decreases pancreatic inflammation without affecting pancreatic damage[94], suggesting that mast cells primarily play a role in releasing or activating additional inflammatory mediators. DCs are rare in the normal pancreas, but pancreata of caerulein-treated mice exhibit a significant increase in mature DCs. These DCs are crucial for pancreatic viability during injury, as their depletion during caerulein- and L-arginine-induced AP leads to massive pancreatic cell death[95].
A distinct decrease in B- and T-lymphocytes is seen in serum from patients with AP, suggesting a systemic inhibition of the immune system. In support of impaired cell-mediated immunity, interleukin (IL)-2, a product of T-lymphocytes, is decreased in mononuclear splenic cells in a murine model of AP[96]. However, it has been theorized that decreases in circulating lymphocytes are not due to immune suppression but instead to a redistribution of lymphocytes from the blood pool to the pancreas[67]. This concept is difficult to test clinically, as most AP patients will not undergo pancreatic biopsy. In support of this theory, Demols et al[97] demonstrated that T-lymphocytes, specifically CD4+ cells, are increased in the murine pancreas following induced AP. This study showed that T-lymphocytes have a role in mediating tissue injury, as depletion of CD4+ T-lymphocytes, or elimination of T-lymphocytes using genetic models, reduced the severity of AP, and this injury was reversible by a T-lymphocyte transfer[97].
Role of cytokines in AP
AP is characterized by excessive recruitment and activation of leukocytes within the pancreas, and in many cases, other organs. Recently, a theory has emerged that the damaged pancreatic epithelium itself is responsible for expression of the first wave of inflammatory mediators, effectively launching the inflammatory cascade that leads to recruitment and activation of immune cells. This is supported by the fact that acinar cells are capable of producing a number of inflammatory mediators in response to damage or noxious stimuli[98-100]. When activated or stressed, isolated acinar cells have been shown to express cytokines[101-104] and chemokines[105] (Table 2 and Figure 3). Detailed reviews regarding the major roles of these mediators in AP and associated conditions are available[106-108]. In addition, activated immune cells secrete numerous chemotactic factors which are capable of perpetuating the immune cell activation cascade.
Table 2.
Cytokines released during pancreatic injury
| Cytokine | Expressed by | Acts on |
| GM-CSF | Acini[194] | Neutrophils[231], MDSCs[194] |
| IL-1β | Acini[104,238], activated macrophages[237], neutrophils[236] | Macrohpage (inhibition)[227] |
| IL-6 | Acini[239], monocytes[30], macrophages[30], endothelial cells[30] fibroblasts[30], smooth muscle cells[30], IL-1b[30], TNFα[30] | CRP[228,229], T-lymphocyte[30], DCs (inhibition)[174] |
| IL-8 | Acini[105], IL-1[241], TNFα[241], macrophages[241], neutrophils[235] | Neutrophils[223] |
| IL-10 | Acini[240], Treg[233] | T-lymphocytes (inhibition)[233], DCs (inhibition)[174], macrophages (inhibition)[232,234] |
| IL-12 | Activated macrophages[231], dendritic cells[30] | NKCs[231] |
| IL-33 | Acini[105] | Mast cells[103] |
| KC | Acini[105], monocytes and neutrophils[250] | Neutrophils[225] |
| MCP-1 | Acini[105] | Macrophages[230] |
| MIP-2 | Acini[105], activated macrophages and neutrophils[93] | Neutrophils[224] |
| TNFα | Acini[105], activated macrophages[99], neutrophils[236], mast cells[226] | Neutrophils[226], NKC[231] |
IL: Interleukin; MCP-1: Monocyte chemotactic protein-1; MIP-2: Macrophage inflammatory protein 2; TNFα: Tumor necrosis factor-α; MDSC: Myeloid-derived suppressor cell; NKC: Natural killer cell; DC: Dendritic cells.
Figure 3.
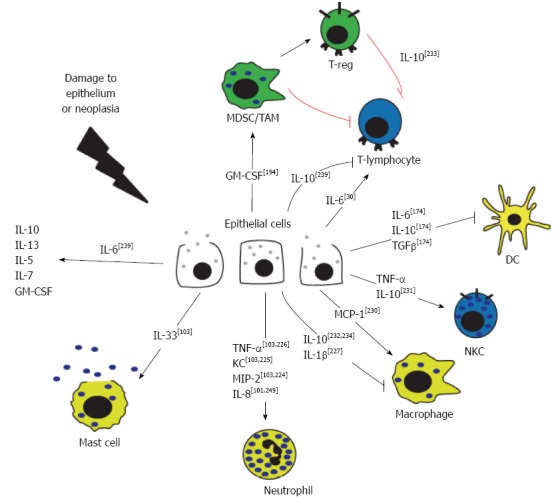
Cytokine signaling in the pancreatic epithelium. In response to damage or disease, the pancreatic epithelial cells release cytokines and chemokines. Activated immune cells secrete numerous chemotactic factors in response to, and in addition to, those secreted by the epithelial cells. TNF: Tumor necrosis factor; IL: Interleukin; MDSC: Myeloid-derived suppressor cells; TAM: Tumor-associated macrophage.
IL-1β and tumor necrosis factor (TNF) α production by pancreatic cells is thought to be a relatively early event in the activation of the cytokine cascade in AP, with IL-6 secretion occurring later in the disease process[109]. Serum IL-6 correlates with disease severity in humans[110,111] and is useful as an early diagnostic marker of AP[112,113]. In a rat model of pancreatitis, TNFα levels were shown to rise over time following induction of pancreatitis[114], and neutralization of TNFα via antibody improved all aspects of pancreatitis (elevated serum amylase, hematocrit, ascites) supporting an important role for TNFα in the pathogenesis of AP[114]. Blockade of IL-1 signaling using IL-1 receptor antagonist (IL-1ra) attenuates the rise in both IL-6 and TNFα, as well as lessens pancreatic damage in the context of AP, supporting early expression of IL-1 and confirming its role as an important mediator for subsequent cytokines[99].
IL-8 is another cytokine implicated in the early stages of the disease process. IL-8 is an established secretory product of activated macrophages[115], but has also been detected in the pancreatic epithelial cells of clinical and pre-clinical pancreatitis tissue[101,102], suggesting a damaged pancreatic epithelium as a possible source of IL-8. Gross et al[116] determined that IL-8 in the serum of pancreatitis patients correlates with disease severity, and showed a significant positive correlation between serum IL-8 and neutrophil elastase, a marker of neutrophils found upregulated in the early stages of AP. As IL-8 is a potent neutrophil activator[117], infiltration of neutrophils may be initiated by secretion of IL-8 from the pancreatic parenchyma. Resident and infiltrating macrophages can also secrete IL-8, recruiting more neutrophils and effectively reinforcing the cycle of inflammation.
Based on clinical studies and analyses of murine models of AP, our knowledge of the inflammatory events in AP can be summarized as: Damage to the pancreas, either caused by, and/or resulting in pancreatic autodigestion, leads to the release of inflammatory mediators from acinar cells. Many of these mediators are potent neutrophil attractants and are likely responsible for neutrophil recruitment to the pancreas as the first wave of inflammatory response. Once in the pancreas, activated neutrophils secrete additional cytokines, thus amplifying the inflammatory response by recruiting additional neutrophils as well as macrophages and other cells of the innate immune response. These cell types are often able to resolve the damage to the pancreas, with limited activation of the adaptive immune response. However, recurrent bouts of AP can lead to a much more serious condition, CP, which presents as a significant risk factor for the development of pancreatic cancer.
Whereas AP is often a self-limiting condition, CP is defined as longstanding inflammation of the pancreas that leads to progressive and irreversible changes. Clinically, CP is characterized by macrophage and T- and B-lymphocyte infiltration into the pancreas[118-122], although peripheral T-lymphocytes appear to decrease[123,124]. Infiltrating mast cells have also been described in the pancreas of CP patients[120,125,126], positively associating with pancreatic fibrosis[126] and pain[120,125]. Finally, immunosuppressive Tregs have been identified in the bone marrow, blood and lesions of CP patients[127].
In general, animal models of CP are generated by repeated induction of AP[128]. In a rat model of CP, both macrophages and CD8+ T-lymphocytes were prevalent in the connective tissue and parenchyma, and CD8+ T cells infiltrated the pancreatic lobules[118]. In a mouse model of CP, a significant increase in both macrophages and neutrophils were observed in response to caerulein (Figure 4), and depletion of these inflammatory cell types significantly reduced pancreatic injury as determined by serum amylase release, and pancreatic lesion formation and fibrosis[129,130]. Similar to clinical findings, mast cells were found to play a significant role in the pathogenesis of pain in a mouse model of CP[125]. Although a serious disease on its own, CP is also a significant risk factor for PDAC and may represent a condition that promotes PDAC development[131].
Figure 4.
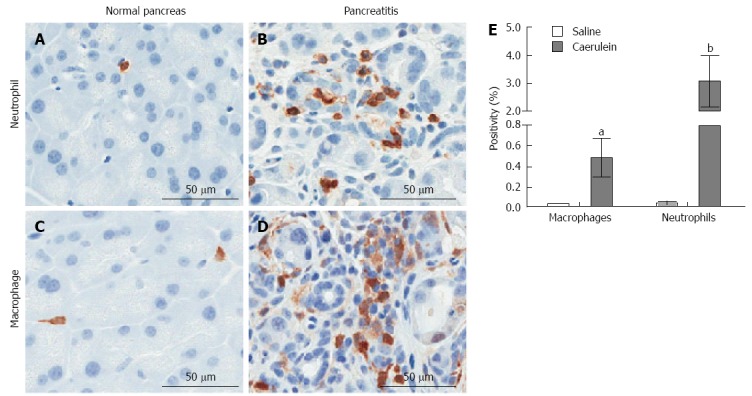
Induction of innate immune response in a mouse model of chronic pancreatitis. Mice were injected intraperatoneally with 250 μg/kg caerulein or saline twice daily, six days per week for two weeks (Mayo Clinic IACUC protocol A48510). The pancreas was isolated 24 h following the last injection and analyzed immunohistochemically for the presence of neutrophils (Ly6B.2: AbSerotec) or macrophages (Mac-2; Cedarlane Diagnostics). A: Neutrophil infiltration in saline-treated mice; B: Neutrophil infiltration in caerulein-treated mice; C: Macrophage infiltration in saline-treated mice; D: Macrophage infiltration in cearulein-treated mice; E: Stained slides were imaged using ScanScope XT (Aperio, Vista, California) and staining was quantified using the Aperio ImageScope reader. aP < 0.05, bP < 0.01 vs corresponding saline treated control. Mean ± SD is plotted for each group of n ≥ 5 mice.
PANCREATIC CANCER
PDAC is the most common form of pancreatic cancer. PDAC is thought to develop by progression through a distinct series of pre-cancerous stages, pancreatic intraepithelial neoplasias (PanINs), before advancing to adenocarcinoma[132]. Greater than 90% of all pancreatic adenocarcinomas contain an activating mutation in codon 12 of the Kirsten rat sarcoma viral oncogene homolog (Kras)[133]. This mutation is thought to occur early in the disease process and drive initiation and progression of PDAC. Indeed, Kras mutations are prevalent even in early stage PanINs and their presence correlates with disease progression[134]. In a classic PDAC mouse model, tissue specific expression of oncogenic KrasG12D from the endogenous Kras promoter (Lox-Stop-Lox- KrasG12D; Pdx-1-cre, KC mouse model) in the mouse pancreatic epithelium recapitulates the full spectrum of human PDAC, including development of PanINs, with progression to adenocarcinoma at approximately 1-2 years of age[135]. For this reason, KrasG12D-expressing mouse models are commonly used to study the initiation and development of PDAC.
It is well established that inflammation plays a major role in the development and progression of pancreatic cancer. Chronic inflammation of the pancreas (pancreatitis) is a significant risk factor for development of PDAC, and PDAC itself is characterized by marked leukocyte infiltration[4,47,48]. Notably, multiple immunosuppressive cell types are observed in pancreatic cancer tissue, suggesting a dysfunction of the immune response, likely mediated by the cancer itself (as described below). Immune dysfunction in PDAC is typified by (1) the recruitment and activation of immunosuppressive cell types; (2) the presence of tumor-supportive immune cells; and (3) a lack of immunity due to defective or absent immune cells. Clinical data and experimental rodent models contribute to the current understanding of the inflammatory response to PDAC as well as the impairment of that response that is a common evasion tool of most cancers.
Immunosuppression in PDAC
Clinical and murine evidence support the existence of profound immunosuppression in PDAC tissues. Clark et al[6] characterized the immune cell influx in various stages of PDAC ranging from normal pancreas to PanINs to invasive carcinoma using the KC mouse model of pancreatic cancer. The study describes an early immunosuppressive phenotype in pancreatic cancer, challenging the classic immunoediting “elimination” phase and suggesting that tumor cells “escape” from immune control almost immediately. At the early PanIN stages, Tregs and MDSCs dominate the immune infiltrate. As the disease progresses to PDAC, CD4+ and CD8+ cells are inconsistently found associated with the tumor, and those CD8+ cells associated with the tumor lack evidence of activation, suggesting a suppressed immune environment[6]. In all stages of disease, there is a strong inverse correlation between MDSCs and CD8+ T-lymphocytes, suggesting that MDSCs are a mediator of tumor immunosuppression[6].
Clinically, lymphocytes are prevalent in pancreatic cancer. CD8+ cells are elevated in the circulation of PDAC patients[136], and leukocytes, the majority of which are T-lymphocytes, surround the pancreatic lesion[137]. T-lymphocytes are found more frequently in the fibrotic interstitial tissue than in the intraepithelial area of the pancreatic cancer[138], and distribute heterogeneously within the tissue, presenting as both scattered cells and focal areas of high accumulation[139]. An example of T-lymphocyte accumulation in progressive stages of the KC model of PDAC is shown in Figure 5.
Figure 5.
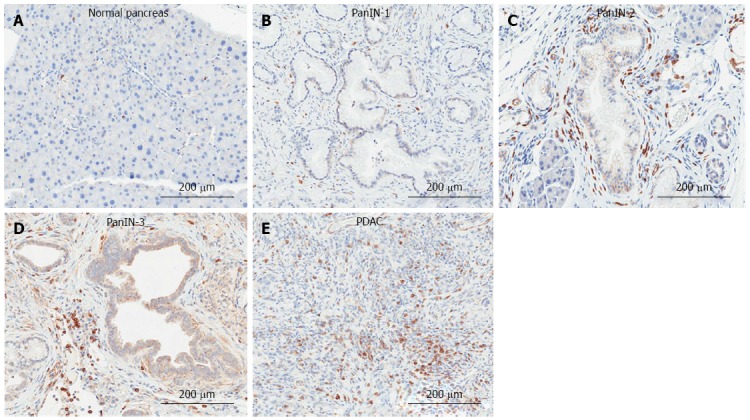
Immunohistochemical detection of T-lymphocytes in the KrasG12D-induced mouse model of pancreatic ductal adenocarcinoma. A: Normal pancreas; B: Pancreatic intraepithelial neoplasia (PanIN)-1; C: PanIN-2; D: PanIN-3; E: Pancreatic ductal adenocarcinoma (PDAC) tissue was analyzed by immunohistochemistry for the presence of T-lymphocytes (CD3: abcam).
The majority of the T-lymphocytes in PDAC are CD4+ Tregs, supporting an immunosuppressive phenotype. Tregs are significantly increased in the blood of PDAC patients as well as in the pancreatic tissue[140]. They are found typically in the stromal areas of the tumor, and only occasionally in association with tumor epithelial cells[141]. Hiraoka et al[141] examined clinical samples of pre-malignant lesions and found that Treg accumulation correlates with the progression of both of the major preneoplastic lesions in pancreatic cancer, PanINs and intraductal papillary mucinous neoplasms (IPMN). The association of Tregs with IPMN progression has been independently confirmed[142]. Additionally, Tregs correlate with metastasis[143] and tumor grade, and negatively correlate with patient survival[141].
Treg infiltration in the development of pancreatic cancer is confirmed in murine models of PDAC. A significant accumulation of Tregs is found in the KC model of PDAC[6]. In another murine tumor model, subcutaneous injection of mouse pancreatic tumor cells into syngeneic mice results in a significant increase in Tregs in the spleen and tumor-draining lymph nodes of these mice[144]. Tan et al[145] have shown in both human PDAC and a murine model of pancreatic cancer that tumor cells produce elevated levels of ligands for the CCR5 receptor, a receptor preferentially expressed by Tregs. Interruption of this receptor-ligand signaling reduces tumor size, as well as Treg recruitment to that tumor, suggesting Tregs are likely recruited in response to direct signaling from the tumor cells.
MDSCs are another immunosuppressive cell type prevalent in PDAC that contributes to immune dysfunction. Whereas MDSCs are absent in the healthy human pancreas, they are readily detected in the stroma of PDAC, comprising approximately 67% of the infiltrating leukocytes[146]. MDSCs are also found in the blood and bone marrow of PDAC patients, and are significantly higher in cases of metastatic disease compared to patients with local tumor[146-148], supporting a previous report that MDSC count correlates with disease stage[149]. Additionally, circulating MDSC numbers are found to be an independent prognostic factor for survival[147].
The functional role of MDSCs in tumor promotion has been characterized in murine models of PDAC[146,147,150]. Following subcutaneous injection of the non-metastatic pancreatic cancer cell line, Pan02, MDSC infiltration is detected in bone marrow, spleen, and tumor[146]. This increase is associated with decreased levels of CD4 and CD8 and increased levels of Tregs in circulation. These MDSCs are able to suppress CD8+ T-lymphocytes in vitro and promote initial tumor growth when co-injected with Pan02[146]. Similarly, in a spontaneous murine model of tumor formation [driven by pancreas-specific overexpression of transforming growth factor (TGF) α and loss of Trp53], MDSC numbers increase in the lymph nodes, blood and pancreas as early as the pre-malignant lesion stage, and increase further upon tumor development. In vitro, these tumor-associated MDSCs were shown to possess arginase activity and suppress T-lymphocyte responses[151]. In the KPC model of metastatic pancreatic cancer (driven by pancreas-specific expression of oncogenic KrasG12D and mutant Trp53R172H), MDSCs accumulate in tumor and spleen and comprise 20%-30% of all leukocytes. MDSCs are closely associated with tumor cells and metastases, suppress proliferation of T-lymphocytes, and express high levels of arginase and nitrite upon stimulation[150]. Collectively, these animal models strongly support a role for MDSCs in tumor promotion through T-lymphocyte inhibition and resulting immunosuppression.
To summarize, both clinical and animal models provide strong evidence for accumulation of immunosuppressive cells types, and subsequent inhibition of immune function in PDAC. The consequence of a suppressed immune system is not direct tumor promotion, but rather alleviation of an important barrier to tumor growth. As described by the cancer immunoediting model, a functional immune system can act as an extrinsic tumor suppressor, identifying and eliminating tumor cells[10]. Alterations in tumor immunogenicity or suppression of the immune response are crucial mechanisms by which this barrier is surmounted, allowing the cancer to progress.
Tumor-supportive immune cells
In addition to the immunosuppressive cell types that inhibit effector T-lymphocyte immunity, other immune cells adopt a tumor-supportive role in the context of PDAC. Once a tumor is established, factors secreted by effector immune cells can often promote, rather than prevent, tumor progression. Mast cells accumulate in PDAC tissue and, along with macrophages, express tumor-promoting factors including vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF)[152]. VEGF and FGF have been shown to stimulate the growth of pancreatic tumors and maintain blood vessels[153,154]. Mast cell accumulation correlates with higher tumor grade, worse survival[155], lymphatic and microvascular invasion[156], and lymph node metastasis[152,156]. In support of the idea that mast cells can support tumor growth, in vitro analyses demonstrate that mast cell-derived factors can promote PDAC cell migration, proliferation, and invasion[155,157]. Interestingly, the importance of mast cells in PDAC appears to be zone specific, with the presence of mast cells in the intratumoral border zone, and not in the intratumoral center or peritumoral zones, correlating with microvessel proliferation, lymph node metastases, and lymphatic and microvascular invasion[156]. Additionally, mast cell accumulation within the intratumoral border is an independent prognostic factor for survival[156]. The significance of mast cell localization likely stems from the ability to establish a pro-tumor microenvironment by degrading tissue surrounding the tumor to promote invasion and by remodeling blood vessels[156]. Taken together, preclinical PDAC models support a role for mast cells in tumor growth and for mast cell accumulation and function at the tumor border[158].
Inflammation can drive the early stages of pancreatic lesion formation in mouse models[159]. A recent study demonstrated that two macrophage secreted inflammatory cytokines could mediate the effects of infiltrating macrophage on acinar cell metaplasia[5]. Matrix metalloproteinase (MMP)-9 and RANTES (regulated on activation, normal T cell expressed and secreted), secreted by pancreas-infiltrating macrophages, are implicated in driving the very first stages of pancreatic lesion formation in a mouse model of cearulein-induced pancreatitis[5].
Immune factors that are responsible for pathogen killing can also promote tumor cell migration and metastasis[160]. Additionally, tumor-promoting immune factors can promote tumor cells to adopt a more fibroblast-like morphology[155,157,160-162] and remodel the tumor environment[156,163] to facilitate migration. In these ways, neutrophil- and B-lymphocyte-derived proteins are implicated in PDAC invasion[160-163]. Neutrophil-derived elastase has been shown to mediate epithelial-mesenchymal transition (EMT) of PDAC cells in vitro[161,162]. Neutrophil-derived MMP-9 promotes PDAC tumor growth and angiogenesis[163], and MMP-9-producing neutrophils have been identified at the leading edge of PDAC in a murine model of pancreatic cancer as well at the invasive front of PDAC metastases[164]. Neutrophils, typically one of the body’s first lines of defense, are not prevalent in PDAC epithelia[165]. However, they are associated with the predominant stromal component of PDAC, and several reports describe a significant increase in neutrophils when measuring levels in both PDAC tissue and stroma together[162,166]. In PDAC, neutrophils are associated with micropapillary carcinoma of the pancreas and correlate with poor prognosis[165,167]. Although density of neutrophil infiltrate does not correlate with tumor clinical stage[162], a patient’s neutrophil: lymphocyte ratio is an independent prognostic indicator of survival following resection of PDAC tumor[168,169].
B-lymphocytes are also able to promote the tumor phenotype by secretion of B-lymphocyte activating factor (BAFF). Koizumi et al[160] described in vitro data demonstrating that BAFF mediates EMT, increases invasion and promotes motility of PDAC cells, supporting a role for B-lymphocyte-secreted BAFF tumor progression and metastasis. Additionally, BAFF-expressing B-lymphocytes surround and infiltrate tumor cells in clinical samples of PDAC, correlating with increased serum levels of BAFF[160].
Defective immune cells in PDAC
DCs are the link between innate and adaptive immunity, recognizing the presence of foreign pathogens and alerting the adaptive immune cells via antigen presentation. However, in a tumor environment, DCs often display an immature phenotype and are defective in their antigen-presenting abilities[170-173]. Tumors express various molecules which are thought to repress DC maturation[174] including IL-10[175,176], IL-4[177], VEGF[178], TGF-β[179], IL-6[180,181], and macrophage-colony stimulating factor[181]. Muc-1, a protein highly expressed on PanIN cells profoundly affects the maturation of DCs. Monti et al[182] demonstrated that DCs exposed to tumor mucins do not fully mature and are characterized by a tolerogenic/regulatory cytokine profile, expressing high levels of IL-10 and low levels of IL-12. Peripherally, circulating DCs are significantly decreased in patients with PDAC[183-186], and demonstrate an impaired ability to stimulate T-lymphocyte proliferation[184].
Absent immune cells in PDAC
Similar to chronic pancreatitis, few NKCs are found in PDAC tissue[138]. The basal systemic NKC activity is decreased in PDAC patients, as well as the NKC response to interferon-α, a potent enhancer of NKC activity[187,188]. Decreased NKC activity has been linked to poor patient prognosis[188] and may be indicative of a suppressed innate immune response.
A role for epithelial cells in dysfunction of the immune response
Several lines of evidence point to a strong role for the neoplastic pancreatic epithelial cells in establishing a dysfunctional immune environment. As previously mentioned, PDAC cells can recruit Tregs[145], promote mast cell migration and activation[157], and repress DC maturation[182]. In vitro, human PDAC cells inhibit T-lymphocyte proliferation and migration, and this is accompanied by an increase in immunosuppressive cytokines including IL-8 and TGFβ[189]. Immunosuppressive compounds TGFβ and IL-10 are upregulated in PDAC and pancreatitis patient sera[190]. Finally, Muc-1, a mucin highly expressed in PanIN-1 early lesions, can suppress T-lymphocyte proliferation[191,192] and it has been suggested that Muc-1 is responsible for tumor escape from recognition and destruction by immune cells[193].
Murine models of PDAC provide additional support for the role of pancreatic epithelial cells in maintaining an immunosuppressive tumor environment. In the KC mouse model, KrasG12D-dependent upregulation of granulocyte macrophage-colony stimulating factor (GM-CSF) is detected in PanINs, and results in recruitment of MDSCs and concomitant inhibition of CD8+ T-lymphocytes[194]. This data is supported by the work of Bayne et al[150] who describe a model in which tumor-derived GM-CSF recruits myeloid inflammatory cells resulting in the negative regulation of CD8+ T cells.
Interaction(s) between immune cells and stroma in PDAC
The development of PDAC is marked by increasing desmoplasia, resulting in the development of a vast stroma that often equals or exceeds the epithelial component of the tumor. The stroma is composed of extracellular matrix proteins and contains various non-epithelial cell types including stellate cells, endothelial cells, and immune cells, but the majority of the stromal cells are activated pancreatic stellate cells (PSCs) and fibroblasts[195]. Activated PSCs promote cancer cell growth and immune cell dysfunction in PDAC[196,197]. PSCs have been implicated in promoting transformed growth, cellular invasion and EMT of PDAC cells, as well as in promoting PDAC tumor incidence, size, and metastasis in an orthotopic mouse model[198-200].
PSCs can have profound effects on the immune cell milieu of PDAC, and have been shown to express a number of growth factors and cytokines[201]. Inflammatory cells recruited to the tumor site are most often found in the stroma rather than infiltrating the epithelial cells[202]. Indeed, activated PSCs have been shown to attract and adhere to CD8+ T-lymphocytes, sequestering them in a juxtatumoral compartment (< 100 μm from tumor) and preventing their access to PDAC tumor cells[197]. One report showed that 94% of tumor-associated T-lymphocytes were either inactivated or did not make it to tumor because they were trapped in the tumor stroma[190]. A potential mechanism by which PSCs may regulate T-lymphocyte trafficking in PDAC is via CXCL12 expression, as PDAC T-lymphocytes express elevated levels of the CXCL12 receptor[197]. Additionally, activated PSCs express a number of cytokines and adhesion-mediating molecules[203], and have been shown to produce MDSC-promoting cytokines, including IL-6, M-CSF, and VEGF, and to promote differentiation of MDSCs from peripheral blood mononuclear cells[204]. Finally, PSCs stimulate mast cell activation, and, conversely, mast cell-derived factors can stimulate PSC proliferation[157]. These data suggest that not only can activated stellate cells directly promote the cancer cell phenotype, but they also contribute to the immunosuppressive phenotype that characterizes PDAC and hampers immunotherapy efforts.
Cancer stem cells
In addition to the immune dysfunction that is prevalent in PDAC, new evidence is emerging to support a role for immune-cytokines in promoting cancer metastasis via interaction with cancer stem cells (CSCs). CSCs are characterized by their ability to self-renew, capability to develop into multiple lineages, enhanced tumor-initiating ability, and resistance to typical cancer therapies[205]. CSCs are thought to be responsible for cancer progression, resistance to standard therapies and tumor relapse. Specifically, pancreatic CSCs have been shown to play a crucial role in the aggressive nature of pancreatic cancer and the resistance to therapy that is a hallmark of this cancer (reviewed in Dorado et al[206]). They share an intimate relationship with the tumor microenvironment, regulating, and being regulated by, cells present therein[207,208]. Specifically, TAMs regulate CSC tumorigenicity and anticancer drug resistance through production of specific growth factors[208]. Additionally, inflammatory cytokines play a role in mediating CSC self-renewal[209].
Recent work suggests that inflammatory cells can promote dissemination of pancreatic CSCs. Rhim et al[210] reported that pancreatic cancer cells exhibiting cancer stem cell properties, including enhanced tumor-initiating capacity, survival, and self-renewal, left the pancreas and were detected in the circulation at the immediate early stages of pancreatic cancer development (PanIN formation), prior to overt tumor formation. The presence of CSCs in the circulation was significantly elevated following induction of pancreatitis, and conversely, treatment with an anti-inflammatory drug, dexamethasone, resulted in a significant decrease in the circulating CSCs. These data support a role for inflammation in promoting dissemination of pancreatic CSCs and potentially in PDAC metastasis.
THERAPY FOR PDAC
Immunotherapy, therapeutic modulation of the immune response, has emerged as a promising line of treatment for many cancers including PDAC[211,212]. Types of immunotherapy that are currently being tested in clinical trials for pancreatic cancer include whole cell, peptide/DNA, antigen pulsed DC, and monoclonal antibody vaccines. Whole cell vaccines typically use irradiated pancreatic cancer cells as the immunogen. These cells have the potential to elicit a robust immune response because they express the full complement of tumor-associated antigens. There have been significant survival advantages reported in resected pancreatic cancer patients using whole cell vaccines such as Algenpantucel-L, an irradiated, live combination of two allogenic pancreatic cell lines[213]. Vaccines comprised of peptides or DNA corresponding to tumor antigens are designed to enhance the cytotoxic T-lymphocyte response. Peptides corresponding to oncogenic Ras, telomerase, VEG-F receptor, carcinoembryonic antigen (CEA), survivin, and Muc-1 have all been successful at prolonging life in pancreatic cancer patients in clinical trials[211,213].
Antigen-pulsed DC vaccines take advantage of the antigen-presenting abilities of DCs to elicit a robust adaptive immune response specific to a tumor antigen of choice[214]. DCs pulsed with Muc-1 and CEA antigens have both been used in clinical trials for pancreatic cancer[215]. Finally, monoclonal antibodies against cell surface tumor antigens are used to induce antibody-dependent cell cytotoxicity[211]. Clinical trials have evaluated antibodies against mesothelin, CEA, and epidermal growth factor receptor for pancreatic cancer treatment[211,215]. Of importance, the success of an anti-cancer vaccine relies on its ability to elicit an immune response in the host, and thus may not exhibit uniform effectiveness in all patients depending on the ability of their immune system to generate a response to treatment.
The stromal compartment has drawn recent interest as a target for PDAC therapy. The stroma creates an inflammatory environment and promotes tumor progression; and the extent of activated stroma has been identified as a novel independent prognostic marker in PDAC[216]. Several potential PDAC therapies have targeted the stroma[217-219]. One method in particular aims at overcoming the immunosuppression often found in, and potentially caused by, cells of the stroma. CD40 agonists take advantage of the inflammatory cells found within the stroma[218]. CD40 is the co-stimulatory factor needed for activation of the T-lymphocyte-dependent anti-tumor response of the immune system. It is thought that activating stromal T-lymphocytes may overcome the immunosuppressive environment that is a hallmark of PDAC. Co-treatment with a CD40 agonist and gemcitabine showed therapeutic efficacy in patients with metastatic PDA[218].
Whereas cancer immunotherapy has typically involved treatment with cancer antigens to stimulate or boost the anti-cancer immune response, the immunosuppressive environment found in PDAC presents obvious challenges. Vaccination with self-antigens has been associated with induction of immunosuppressive cell types, thus potentiating, rather than inhibiting, tumor growth[220]. Successfully overcoming the immunosuppressive environment that characterizes PDAC will likely require a multifaceted approach due to the multiple mechanisms by which tumor-associated immune dysfunction seems to occur. However, new evidence suggests that immunotherapy can be successful for pancreatic cancer, if stimulation of the immune system is combined with control over the immunosuppressive environment[221,222]. Developing new methods to overcome immunosuppression or exploit the immune response to target PDAC may be utilized in combination with conventional or novel chemotherapy to enhance the survival of this currently deadly disease.
CONCLUSION
The immune system protects the host from invading pathogens and foreign materials. The aberrant expression profile and uncontrollable proliferation that characterizes tumor cells should allow for recognition as non-self by the immune system. However, we now know that from a very early stage in PDAC development, the ability of the immune system to identify and eliminate neoplastic cells is compromised. This suggests that an immunosuppressive environment is established early in tumor development to effectively thwart the immune response to neoplastic cells at the onset of tumor development. Figure 6 depicts the progressive changes in immune cell infiltrate found during various stages of pancreatic disease.
Figure 6.
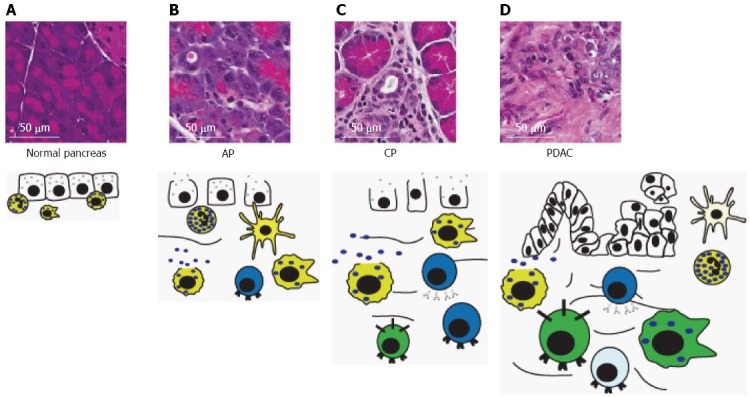
Immune cells in progressive pancreatic disease. A: The normal pancreas contains sparse, mostly innate, inflammatory cells and lacks the dense stroma typically seen in chronic pancreatitis (CP) and pancreatic ductal adenocarcinoma (PDAC); B: Acute pancreatitis (AP) is characterized by acinar degranulation, edema and recruitment of mostly innate inflammatory cells, but also some T-lymphocytes in response to acinar damage. Pancreatic mast cells begin to degranulate; C: CP is characterized by development of stroma surrounding degranulated acinar cells, acinar-to-ductal metaplasia and edema. Additionally, there is an increased presence of macrophages, and T- and B-lymphocytes, and further degranulation of mast cells; D: Development of pancreatic intraepithelial neoplasias and subsequent PDAC leads to a significant increase in immunosuppressive cell types including tumor-associated macrophages, myeloid-derived suppressor cells, and Tregs. Degranulated mast cells, neutrophils, dendritic cells and B- and T-lymphocytes are also present. However, T-lymphocytes and dendritic cells are typically inhibited and defective, respectively. Size of immune cell represents relative abundance. Cell color denotes immune cell type: yellow, innate immune cells; blue, adaptive immune cells; green, immunosuppressive cells. For description of graphical representation of cell types, see Figure 3 legend. Cells that are inactivated or defective are represented by a lighter color.
Based on data reviewed here, neoplastic cells produce compounds (such as GM-CSF) at a very early stage of pancreatic cancer development, that recruit immunosuppressive immune cells, potentially facilitating progression to later PanIN stages and PDAC[190-192,194]. Once PDAC cells are present, they actively prevent the maturation of dendritic cells, inhibiting their antigen presenting activity and effectively cutting off a major communication between the innate and adaptive immune response[182]. In addition, immunosuppressive Tregs and MDSCs accumulate in the blood, stroma and PDAC tissue and inhibit T-lymphocyte proliferation[6,140,141,144-146]. In this setting, even immune cell types considered pro-inflammatory add to the tumor-supportive environment: neutrophils accumulate in the stroma and secrete proteases that aid in EMT, tumor motility and invasion[161,163] and mast cells accumulate in the tumor tissue and express factors used by the tumor to sustain its growth[152]. It is clear that a complex relationship exists between the immune system and the developing pancreatic cancer, and that these interactions have important implications for disease prevention and control. Immunotherapy can potentially be a powerful component of PDAC treatment. Further study of the mechanisms by which immunosuppression is initiated in PDAC, and ways to overcome it, will facilitate the development of this treatment option[22,104,223-249].
Footnotes
Supported by The National Institute of Health (NIH/NCI R01CA140290-3 to Murray NR)
P- Reviewer: Ghiorzo P, Schuurman HJ, Tanaka S, Zhong QY S- Editor: Zhai HH L- Editor: A E- Editor: Wang CH
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Evans A, Costello E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front Physiol. 2012;3:270. doi: 10.3389/fphys.2012.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American Cancer Society. Cancer Facts and Figures 2013. Atlanta: American Cancer Society; 2013. [Google Scholar]
- 4.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Liou GY, Döppler H, Necela B, Krishna M, Crawford HC, Raimondo M, Storz P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J Cell Biol. 2013;202:563–577. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 7.Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, Kato K, Kurokawa T, Suzuoki M, Nakakubo Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–e31. doi: 10.1097/00006676-200401000-00023. [DOI] [PubMed] [Google Scholar]
- 8.Burnet FM. Immunological surveillance. Oxford: Pergamon Press; 1970. [Google Scholar]
- 9.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 10.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 11.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 12.Cassatella MA. Neutrophil-derived proteins: selling cytokines by the pound. Adv Immunol. 1999;73:369–509. doi: 10.1016/s0065-2776(08)60791-9. [DOI] [PubMed] [Google Scholar]
- 13.Hao NB, Lü MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098. doi: 10.1155/2012/948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber S, Hoffmann R, Muskens F, Voehringer D. Alternatively activated macrophages inhibit T-cell proliferation by Stat6-dependent expression of PD-L2. Blood. 2010;116:3311–3320. doi: 10.1182/blood-2010-02-271981. [DOI] [PubMed] [Google Scholar]
- 15.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 16.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 17.Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saio M, Radoja S, Marino M, Frey AB. Tumor-infiltrating macrophages induce apoptosis in activated CD8(+) T cells by a mechanism requiring cell contact and mediated by both the cell-associated form of TNF and nitric oxide. J Immunol. 2001;167:5583–5593. doi: 10.4049/jimmunol.167.10.5583. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109:1568–1573. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munder M, Choi BS, Rogers M, Kropf P. L-arginine deprivation impairs Leishmania major-specific T-cell responses. Eur J Immunol. 2009;39:2161–2172. doi: 10.1002/eji.200839041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 23.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol. 2007;601:213–223. doi: 10.1007/978-0-387-72005-0_22. [DOI] [PubMed] [Google Scholar]
- 24.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 26.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 27.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2:850–861. doi: 10.1038/nrc928. [DOI] [PubMed] [Google Scholar]
- 29.Clark GJ, Angel N, Kato M, López JA, MacDonald K, Vuckovic S, Hart DN. The role of dendritic cells in the innate immune system. Microbes Infect. 2000;2:257–272. doi: 10.1016/s1286-4579(00)00302-6. [DOI] [PubMed] [Google Scholar]
- 30.Janeway C. Immunobiology: the immune system in health and disease. 6th ed. New York: Garland Science; 2005. [Google Scholar]
- 31.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J. Molecular Biology of the Cell. 3 rd ed. New York, NY: Garland Publishing Inc; 1994. [Google Scholar]
- 33.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura T, Takeuchi T, Tando Y. Pancreatic dysfunction and treatment options. Pancreas. 1998;16:329–336. doi: 10.1097/00006676-199804000-00020. [DOI] [PubMed] [Google Scholar]
- 35.Russo MW, Wei JT, Thiny MT, Gangarosa LM, Brown A, Ringel Y, Shaheen NJ, Sandler RS. Digestive and liver diseases statistics, 2004. Gastroenterology. 2004;126:1448–1453. doi: 10.1053/j.gastro.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 36.Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet. 2008;371:143–152. doi: 10.1016/S0140-6736(08)60107-5. [DOI] [PubMed] [Google Scholar]
- 37.Klöppel G, Dreyer T, Willemer S, Kern HF, Adler G. Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch A Pathol Anat Histopathol. 1986;409:791–803. doi: 10.1007/BF00710764. [DOI] [PubMed] [Google Scholar]
- 38.Vidarsdottir H, Möller PH, Vidarsdottir H, Thorarinsdottir H, Björnsson ES. Acute pancreatitis: a prospective study on incidence, etiology, and outcome. Eur J Gastroenterol Hepatol. 2013;25:1068–1075. doi: 10.1097/MEG.0b013e3283640fc8. [DOI] [PubMed] [Google Scholar]
- 39.Takuma K, Kamisawa T, Hara S, Tabata T, Kuruma S, Chiba K, Kuwata G, Fujiwara T, Egashira H, Koizumi K, et al. Etiology of recurrent acute pancreatitis, with special emphasis on pancreaticobiliary malformation. Adv Med Sci. 2012;57:244–250. doi: 10.2478/v10039-012-0041-7. [DOI] [PubMed] [Google Scholar]
- 40.Lowenfels AB, Maisonneuve P, Sullivan T. The changing character of acute pancreatitis: epidemiology, etiology, and prognosis. Curr Gastroenterol Rep. 2009;11:97–103. doi: 10.1007/s11894-009-0016-4. [DOI] [PubMed] [Google Scholar]
- 41.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252–1261. doi: 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Appelros S, Borgström A. Incidence, aetiology and mortality rate of acute pancreatitis over 10 years in a defined urban population in Sweden. Br J Surg. 1999;86:465–470. doi: 10.1046/j.1365-2168.1999.01049.x. [DOI] [PubMed] [Google Scholar]
- 43.Renner IG, Savage WT, Pantoja JL, Renner VJ. Death due to acute pancreatitis. A retrospective analysis of 405 autopsy cases. Dig Dis Sci. 1985;30:1005–1018. doi: 10.1007/BF01308298. [DOI] [PubMed] [Google Scholar]
- 44.Beger HG, Bittner R, Block S, Büchler M. Bacterial contamination of pancreatic necrosis. A prospective clinical study. Gastroenterology. 1986;91:433–438. doi: 10.1016/0016-5085(86)90579-2. [DOI] [PubMed] [Google Scholar]
- 45.Büchler MW, Gloor B, Müller CA, Friess H, Seiler CA, Uhl W. Acute necrotizing pancreatitis: treatment strategy according to the status of infection. Ann Surg. 2000;232:619–626. doi: 10.1097/00000658-200011000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2002;9:401–410. doi: 10.1007/s005340200049. [DOI] [PubMed] [Google Scholar]
- 47.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, Domellöf L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 48.Malka D, Hammel P, Maire F, Rufat P, Madeira I, Pessione F, Lévy P, Ruszniewski P. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut. 2002;51:849–852. doi: 10.1136/gut.51.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinho AV, Chantrill L, Rooman I. Chronic pancreatitis: a path to pancreatic cancer. Cancer Lett. 2014;345:203–209. doi: 10.1016/j.canlet.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 50.Freedman SD. Clinical manifestations and diagnosis of chronic pancreatitis in adults. In: Basow DS, editor. Waltham, MA: UpToDate; 2013. [Google Scholar]
- 51.Büchler MW, Martignoni ME, Friess H, Malfertheiner P. A proposal for a new clinical classification of chronic pancreatitis. BMC Gastroenterol. 2009;9:93. doi: 10.1186/1471-230X-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Etemad B, Whitcomb DC. Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology. 2001;120:682–707. doi: 10.1053/gast.2001.22586. [DOI] [PubMed] [Google Scholar]
- 53.Mergener K, Baillie J. Chronic pancreatitis. Lancet. 1997;350:1379–1385. doi: 10.1016/S0140-6736(97)07332-7. [DOI] [PubMed] [Google Scholar]
- 54.Klöppel G, Maillet B. Pathology of acute and chronic pancreatitis. Pancreas. 1993;8:659–670. doi: 10.1097/00006676-199311000-00001. [DOI] [PubMed] [Google Scholar]
- 55.Hirota M, Kuwata K, Ohmuraya M, Ogawa M. From acute to chronic pancreatitis: the role of mutations in the pancreatic secretory trypsin inhibitor gene. JOP. 2003;4:83–88. [PubMed] [Google Scholar]
- 56.Zhou ZG, Chen YD, Sun W, Chen Z. Pancreatic microcirculatory impairment in experimental acute pancreatitis in rats. World J Gastroenterol. 2002;8:933–936. doi: 10.3748/wjg.v8.i5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rinderknecht H. Fatal pancreatitis, a consequence of excessive leukocyte stimulation? Int J Pancreatol. 1988;3:105–112. doi: 10.1007/BF02798921. [DOI] [PubMed] [Google Scholar]
- 58.Wang GJ, Gao CF, Wei D, Wang C, Ding SQ. Acute pancreatitis: etiology and common pathogenesis. World J Gastroenterol. 2009;15:1427–1430. doi: 10.3748/wjg.15.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frick TW. The role of calcium in acute pancreatitis. Surgery. 2012;152:S157–S163. doi: 10.1016/j.surg.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 60.Gerasimenko OV, Gerasimenko JV. Mitochondrial function and malfunction in the pathophysiology of pancreatitis. Pflugers Arch. 2012;464:89–99. doi: 10.1007/s00424-012-1117-8. [DOI] [PubMed] [Google Scholar]
- 61.Cuthbertson CM, Christophi C. Disturbances of the microcirculation in acute pancreatitis. Br J Surg. 2006;93:518–530. doi: 10.1002/bjs.5316. [DOI] [PubMed] [Google Scholar]
- 62.Viedma JA, Pérez-Mateo M, Agulló J, Domínguez JE, Carballo F. Inflammatory response in the early prediction of severity in human acute pancreatitis. Gut. 1994;35:822–827. doi: 10.1136/gut.35.6.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banks RE, Evans SW, Alexander D, McMahon MJ, Whicher JT. Is fatal pancreatitis a consequence of excessive leukocyte stimulation? The role of tumor necrosis factor alpha. Cytokine. 1991;3:12–16. doi: 10.1016/1043-4666(91)90004-w. [DOI] [PubMed] [Google Scholar]
- 64.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 65.Novovic S, Andersen AM, Nord M, Astrand M, Ottosson T, Jørgensen LN, Hansen MB. Activity of neutrophil elastase reflects the progression of acute pancreatitis. Scand J Clin Lab Invest. 2013;73:485–493. doi: 10.3109/00365513.2013.807935. [DOI] [PubMed] [Google Scholar]
- 66.Uhl W, Büchler M, Malfertheiner P, Martini M, Beger HG. PMN-elastase in comparison with CRP, antiproteases, and LDH as indicators of necrosis in human acute pancreatitis. Pancreas. 1991;6:253–259. doi: 10.1097/00006676-199105000-00001. [DOI] [PubMed] [Google Scholar]
- 67.Widdison AL, Cunningham S. Immune function early in acute pancreatitis. Br J Surg. 1996;83:633–636. doi: 10.1002/bjs.1800830514. [DOI] [PubMed] [Google Scholar]
- 68.Regnér S, Manjer J, Appelros S, Hjalmarsson C, Sadic J, Borgström A. Protease activation, pancreatic leakage, and inflammation in acute pancreatitis: differences between mild and severe cases and changes over the first three days. Pancreatology. 2008;8:600–607. doi: 10.1159/000161011. [DOI] [PubMed] [Google Scholar]
- 69.Kylänpää ML, Repo H, Puolakkainen PA. Inflammation and immunosuppression in severe acute pancreatitis. World J Gastroenterol. 2010;16:2867–2872. doi: 10.3748/wjg.v16.i23.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKay CJ, Evans S, Sinclair M, Carter CR, Imrie CW. High early mortality rate from acute pancreatitis in Scotland, 1984-1995. Br J Surg. 1999;86:1302–1305. doi: 10.1046/j.1365-2168.1999.01246.x. [DOI] [PubMed] [Google Scholar]
- 71.Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–1128. doi: 10.1097/00003246-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 72.Pezzilli R, Billi P, Beltrandi E, Maldini M, Mancini R, Morselli Labate AM, Miglioli M. Circulating lymphocyte subsets in human acute pancreatitis. Pancreas. 1995;11:95–100. doi: 10.1097/00006676-199507000-00010. [DOI] [PubMed] [Google Scholar]
- 73.Dabrowski A, Osada J, Dabrowska MI, Wereszczynska-Siemiatkowska U. Monocyte subsets and natural killer cells in acute pancreatitis. Pancreatology. 2008;8:126–134. doi: 10.1159/000123605. [DOI] [PubMed] [Google Scholar]
- 74.Pezzilli R, Maldini M, Morselli-Labate AM, Barakat B, Romboli E, Beltrandi E, Migliori M, Tomassetti P, Corinaldesi R. Early activation of peripheral lymphocytes in human acute pancreatitis. J Clin Gastroenterol. 2003;36:360–363. doi: 10.1097/00004836-200304000-00016. [DOI] [PubMed] [Google Scholar]
- 75.Ueda T, Takeyama Y, Yasuda T, Shinzeki M, Sawa H, Nakajima T, Ajiki T, Fujino Y, Suzuki Y, Kuroda Y. Immunosuppression in patients with severe acute pancreatitis. J Gastroenterol. 2006;41:779–784. doi: 10.1007/s00535-006-1852-8. [DOI] [PubMed] [Google Scholar]
- 76.Takeyama Y, Takas K, Ueda T, Hori Y, Goshima M, Kuroda Y. Peripheral lymphocyte reduction in severe acute pancreatitis is caused by apoptotic cell death. J Gastrointest Surg. 2000;4:379–387. doi: 10.1016/s1091-255x(00)80016-5. [DOI] [PubMed] [Google Scholar]
- 77.Kylänpää-Bäck ML, Takala A, Kemppainen E, Puolakkainen P, Kautiainen H, Jansson SE, Haapiainen R, Repo H. Cellular markers of systemic inflammation and immune suppression in patients with organ failure due to severe acute pancreatitis. Scand J Gastroenterol. 2001;36:1100–1107. doi: 10.1080/003655201750422738. [DOI] [PubMed] [Google Scholar]
- 78.Richter A, Nebe T, Wendl K, Schuster K, Klaebisch G, Quintel M, Lorenz D, Post S, Trede M. HLA-DR expression in acute pancreatitis. Eur J Surg. 1999;165:947–951. doi: 10.1080/110241599750008053. [DOI] [PubMed] [Google Scholar]
- 79.Yamaguchi H, Kimura T, Mimura K, Nawata H. Activation of proteases in cerulein-induced pancreatitis. Pancreas. 1989;4:565–571. doi: 10.1097/00006676-198910000-00007. [DOI] [PubMed] [Google Scholar]
- 80.Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch A Pathol Anat Histol. 1977;373:97–117. doi: 10.1007/BF00432156. [DOI] [PubMed] [Google Scholar]
- 81.Song MH, Kim MH, Lee SK, Seo DW. The clinical usefulness of secretin-enhanced magnetic resonance pancreatography in patients with pancreas divisum and idiopathic acute pancreatitis. Gastrointest Endosc. 2002;55:454–455. doi: 10.1067/mge.2002.121601. [DOI] [PubMed] [Google Scholar]
- 82.Walker NI. Ultrastructure of the rat pancreas after experimental duct ligation. I. The role of apoptosis and intraepithelial macrophages in acinar cell deletion. Am J Pathol. 1987;126:439–451. [PMC free article] [PubMed] [Google Scholar]
- 83.Mizunuma T, Kawamura S, Kishino Y. Effects of injecting excess arginine on rat pancreas. J Nutr. 1984;114:467–471. doi: 10.1093/jn/114.3.467. [DOI] [PubMed] [Google Scholar]
- 84.Lombardi B, Estes LW, Longnecker DS. Acute hemorrhagic pancreatitis (massive necrosis) with fat necrosis induced in mice by DL-ethionine fed with a choline-deficient diet. Am J Pathol. 1975;79:465–480. [PMC free article] [PubMed] [Google Scholar]
- 85.Nutrition classics from The Journal of Biological Chemistry, 131: 567-577, 1937. Choline metabolism. I. The occurrence and prevention of hemorrhagic degeneration in young rats on a low choline diet. By Wendell H. Griffith and Nelson J. Wade. Nutr Rev. 1974;32:112–114. doi: 10.1111/j.1753-4887.1974.tb06292.x. [DOI] [PubMed] [Google Scholar]
- 86.THAL A, BRACKNEY E. Acute hemorrhagic pancreatic necrosis produced by local Shwartzman reaction: experimental study on pancreatitis. J Am Med Assoc. 1954;155:569–574. doi: 10.1001/jama.1954.73690240003011. [DOI] [PubMed] [Google Scholar]
- 87.Meyerholz DK, Samuel I. Morphologic characterization of early ligation-induced acute pancreatitis in rats. Am J Surg. 2007;194:652–658. doi: 10.1016/j.amjsurg.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pastor CM, Vonlaufen A, Georgi F, Hadengue A, Morel P, Frossard JL. Neutrophil depletion--but not prevention of Kupffer cell activation--decreases the severity of cerulein-induced acute pancreatitis. World J Gastroenterol. 2006;12:1219–1224. doi: 10.3748/wjg.v12.i8.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sandoval D, Gukovskaya A, Reavey P, Gukovsky S, Sisk A, Braquet P, Pandol SJ, Poucell-Hatton S. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology. 1996;111:1081–1091. doi: 10.1016/s0016-5085(96)70077-x. [DOI] [PubMed] [Google Scholar]
- 90.Folch E, Prats N, Hotter G, López S, Gelpi E, Roselló-Catafau J, Closa D. P-selectin expression and Kupffer cell activation in rat acute pancreatitis. Dig Dis Sci. 2000;45:1535–1544. doi: 10.1023/a:1005552725243. [DOI] [PubMed] [Google Scholar]
- 91.Awla D, Abdulla A, Zhang S, Roller J, Menger MD, Regnér S, Thorlacius H. Lymphocyte function antigen-1 regulates neutrophil recruitment and tissue damage in acute pancreatitis. Br J Pharmacol. 2011;163:413–423. doi: 10.1111/j.1476-5381.2011.01225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kyriakides C, Jasleen J, Wang Y, Moore FD, Ashley SW, Hechtman HB. Neutrophils, not complement, mediate the mortality of experimental hemorrhagic pancreatitis. Pancreas. 2001;22:40–46. doi: 10.1097/00006676-200101000-00007. [DOI] [PubMed] [Google Scholar]
- 93.Pastor CM, Rubbia-Brandt L, Hadengue A, Jordan M, Morel P, Frossard JL. Role of macrophage inflammatory peptide-2 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. Lab Invest. 2003;83:471–478. doi: 10.1097/01.lab.0000063928.91314.9f. [DOI] [PubMed] [Google Scholar]
- 94.Lopez-Font I, Gea-Sorlí S, de-Madaria E, Gutiérrez LM, Pérez-Mateo M, Closa D. Pancreatic and pulmonary mast cells activation during experimental acute pancreatitis. World J Gastroenterol. 2010;16:3411–3417. doi: 10.3748/wjg.v16.i27.3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bedrosian AS, Nguyen AH, Hackman M, Connolly MK, Malhotra A, Ibrahim J, Cieza-Rubio NE, Henning JR, Barilla R, Rehman A, et al. Dendritic cells promote pancreatic viability in mice with acute pancreatitis. Gastroenterology. 2011;141:1915–26.e1-14. doi: 10.1053/j.gastro.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Curley P, Nestor M, Collins K, Saporoschetz I, Mendez M, Mannick JA, Rodrick ML. Decreased interleukin-2 production in murine acute pancreatitis: potential for immunomodulation. Gastroenterology. 1996;110:583–588. doi: 10.1053/gast.1996.v110.pm8566607. [DOI] [PubMed] [Google Scholar]
- 97.Demols A, Le Moine O, Desalle F, Quertinmont E, Van Laethem JL, Devière J. CD4(+ )T cells play an important role in acute experimental pancreatitis in mice. Gastroenterology. 2000;118:582–590. doi: 10.1016/s0016-5085(00)70265-4. [DOI] [PubMed] [Google Scholar]
- 98.Dios ID. Inflammatory role of the acinar cells during acute pancreatitis. World J Gastrointest Pharmacol Ther. 2010;1:15–20. doi: 10.4292/wjgpt.v1.i1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Norman JG, Fink GW, Franz MG. Acute pancreatitis induces intrapancreatic tumor necrosis factor gene expression. Arch Surg. 1995;130:966–970. doi: 10.1001/archsurg.1995.01430090052018. [DOI] [PubMed] [Google Scholar]
- 100.Norman J, Franz M, Riker A, Fabri P, Gower W. Rapid Elevation of Systemic Cytokines During Acute Pancreatitis and Their Origination Within the Pancreas. Surgical Forum. 1994;45:148–150. [Google Scholar]
- 101.Xie MJ, Motoo Y, Su SB, Mouri H, Sawabu N. Induction of chemokines in rat pancreatic acinar cell injury. Pancreas. 2002;24:198–204. doi: 10.1097/00006676-200203000-00012. [DOI] [PubMed] [Google Scholar]
- 102.Motoo Y, Xie MJ, Mouri H, Sawabu N. Expression of interleukin-8 in human obstructive pancreatitis. JOP. 2004;5:138–144. [PubMed] [Google Scholar]
- 103.Kempuraj D, Twait EC, Williard DE, Yuan Z, Meyerholz DK, Samuel I. The novel cytokine interleukin-33 activates acinar cell proinflammatory pathways and induces acute pancreatic inflammation in mice. PLoS One. 2013;8:e56866. doi: 10.1371/journal.pone.0056866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu JH, Kim KH, Kim H. Suppression of IL-1beta expression by the Jak 2 inhibitor AG490 in cerulein-stimulated pancreatic acinar cells. Biochem Pharmacol. 2006;72:1555–1562. doi: 10.1016/j.bcp.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 105.Blinman TA, Gukovsky I, Mouria M, Zaninovic V, Livingston E, Pandol SJ, Gukovskaya AS. Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. Am J Physiol Cell Physiol. 2000;279:C1993–C2003. doi: 10.1152/ajpcell.2000.279.6.C1993. [DOI] [PubMed] [Google Scholar]
- 106.Bhatia M. Inflammatory response on the pancreatic acinar cell injury. Scand J Surg. 2005;94:97–102. doi: 10.1177/145749690509400203. [DOI] [PubMed] [Google Scholar]
- 107.Granger J, Remick D. Acute pancreatitis: models, markers, and mediators. Shock. 2005;24 Suppl 1:45–51. doi: 10.1097/01.shk.0000191413.94461.b0. [DOI] [PubMed] [Google Scholar]
- 108.Bhatia M, Brady M, Shokuhi S, Christmas S, Neoptolemos JP, Slavin J. Inflammatory mediators in acute pancreatitis. J Pathol. 2000;190:117–125. doi: 10.1002/(SICI)1096-9896(200002)190:2<117::AID-PATH494>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 109.Norman JG, Fink GW, Denham W, Yang J, Carter G, Sexton C, Falkner J, Gower WR, Franz MG. Tissue-specific cytokine production during experimental acute pancreatitis. A probable mechanism for distant organ dysfunction. Dig Dis Sci. 1997;42:1783–1788. doi: 10.1023/a:1018886120711. [DOI] [PubMed] [Google Scholar]
- 110.Viedma JA, Pérez-Mateo M, Domínguez JE, Carballo F. Role of interleukin-6 in acute pancreatitis. Comparison with C-reactive protein and phospholipase A. Gut. 1992;33:1264–1267. doi: 10.1136/gut.33.9.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heath DI, Cruickshank A, Gudgeon M, Jehanli A, Shenkin A, Imrie CW. Role of interleukin-6 in mediating the acute phase protein response and potential as an early means of severity assessment in acute pancreatitis. Gut. 1993;34:41–45. doi: 10.1136/gut.34.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pezzilli R, Miniero R, Cappelletti O, Barakat B. Behavior of serum interleukin 12 in human acute pancreatitis. Pancreas. 1999;18:247–251. doi: 10.1097/00006676-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 113.Matull WR, Pereira SP, O’Donohue JW. Biochemical markers of acute pancreatitis. J Clin Pathol. 2006;59:340–344. doi: 10.1136/jcp.2002.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Grewal HP, Kotb M, el Din AM, Ohman M, Salem A, Gaber L, Gaber AO. Induction of tumor necrosis factor in severe acute pancreatitis and its subsequent reduction after hepatic passage. Surgery. 1994;115:213–221. [PubMed] [Google Scholar]
- 115.Sorimachi K, Akimoto K, Hattori Y, Ieiri T, Niwa A. Secretion of TNF-alpha, IL-8 and nitric oxide by macrophages activated with polyanions, and involvement of interferon-gamma in the regulation of cytokine secretion. Cytokine. 1999;11:571–578. doi: 10.1006/cyto.1998.0472. [DOI] [PubMed] [Google Scholar]
- 116.Gross V, Andreesen R, Leser HG, Ceska M, Liehl E, Lausen M, Farthmann EH, Schölmerich J. Interleukin-8 and neutrophil activation in acute pancreatitis. Eur J Clin Invest. 1992;22:200–203. doi: 10.1111/j.1365-2362.1992.tb01826.x. [DOI] [PubMed] [Google Scholar]
- 117.Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84:1045–1049. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamada T, Hashimoto T, Sogawa M, Kobayashi S, Kaneda K, Nakamura S, Kuno A, Sano H, Ando T, Kobayashi S, et al. Role of T cells in development of chronic pancreatitis in male Wistar Bonn/Kobori rats: effects of tacrolimus. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1397–G1404. doi: 10.1152/ajpgi.2001.281.6.G1397. [DOI] [PubMed] [Google Scholar]
- 119.Emmrich J, Weber I, Nausch M, Sparmann G, Koch K, Seyfarth M, Löhr M, Liebe S. Immunohistochemical characterization of the pancreatic cellular infiltrate in normal pancreas, chronic pancreatitis and pancreatic carcinoma. Digestion. 1998;59:192–198. doi: 10.1159/000007488. [DOI] [PubMed] [Google Scholar]
- 120.Demir IE, Schorn S, Schremmer-Danninger E, Wang K, Kehl T, Giese NA, Algül H, Friess H, Ceyhan GO. Perineural mast cells are specifically enriched in pancreatic neuritis and neuropathic pain in pancreatic cancer and chronic pancreatitis. PLoS One. 2013;8:e60529. doi: 10.1371/journal.pone.0060529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grundsten M, Liu GZ, Permert J, Hjelmstrom P, Tsai JA. Increased central memory T cells in patients with chronic pancreatitis. Pancreatology. 2005;5:177–182. doi: 10.1159/000085269. [DOI] [PubMed] [Google Scholar]
- 122.Ebert MP, Ademmer K, Müller-Ostermeyer F, Friess H, Büchler MW, Schubert W, Malfertheiner P. CD8+CD103+ T cells analogous to intestinal intraepithelial lymphocytes infiltrate the pancreas in chronic pancreatitis. Am J Gastroenterol. 1998;93:2141–2147. doi: 10.1111/j.1572-0241.1998.00610.x. [DOI] [PubMed] [Google Scholar]
- 123.Bhatnagar A, Wig JD, Majumdar S. Immunological findings in acute and chronic pancreatitis. ANZ J Surg. 2003;73:59–64. doi: 10.1046/j.1445-2197.2003.02621.x. [DOI] [PubMed] [Google Scholar]
- 124.Ockenga J, Jacobs R, Kemper A, Benschop RJ, Schmidt RE, Manns MP. Lymphocyte subsets and cellular immunity in patients with chronic pancreatitis. Digestion. 2000;62:14–21. doi: 10.1159/000007772. [DOI] [PubMed] [Google Scholar]
- 125.Hoogerwerf WA, Gondesen K, Xiao SY, Winston JH, Willis WD, Pasricha PJ. The role of mast cells in the pathogenesis of pain in chronic pancreatitis. BMC Gastroenterol. 2005;5:8. doi: 10.1186/1471-230X-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Esposito I, Friess H, Kappeler A, Shrikhande S, Kleeff J, Ramesh H, Zimmermann A, Büchler MW. Mast cell distribution and activation in chronic pancreatitis. Hum Pathol. 2001;32:1174–1183. doi: 10.1053/hupa.2001.28947. [DOI] [PubMed] [Google Scholar]
- 127.Schmitz-Winnenthal H, Pietsch DH, Schimmack S, Bonertz A, Udonta F, Ge Y, Galindo L, Specht S, Volk C, Zgraggen K, et al. Chronic pancreatitis is associated with disease-specific regulatory T-cell responses. Gastroenterology. 2010;138:1178–1188. doi: 10.1053/j.gastro.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 128.Aghdassi AA, Mayerle J, Christochowitz S, Weiss FU, Sendler M, Lerch MM. Animal models for investigating chronic pancreatitis. Fibrogenesis Tissue Repair. 2011;4:26. doi: 10.1186/1755-1536-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bai H, Chen X, Zhang L, Dou X. The effect of sulindac, a non-steroidal anti-inflammatory drug, attenuates inflammation and fibrosis in a mouse model of chronic pancreatitis. BMC Gastroenterol. 2012;12:115. doi: 10.1186/1471-230X-12-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van Westerloo DJ, Florquin S, de Boer AM, Daalhuisen J, de Vos AF, Bruno MJ, van der Poll T. Therapeutic effects of troglitazone in experimental chronic pancreatitis in mice. Am J Pathol. 2005;166:721–728. doi: 10.1016/S0002-9440(10)62293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Farrow B, Evers BM. Inflammation and the development of pancreatic cancer. Surg Oncol. 2002;10:153–169. doi: 10.1016/s0960-7404(02)00015-4. [DOI] [PubMed] [Google Scholar]
- 132.Pérez-Mancera PA, Guerra C, Barbacid M, Tuveson DA. What we have learned about pancreatic cancer from mouse models. Gastroenterology. 2012;142:1079–1092. doi: 10.1053/j.gastro.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 133.Hruban RH, van Mansfeld AD, Offerhaus GJ, van Weering DH, Allison DC, Goodman SN, Kensler TW, Bose KK, Cameron JL, Bos JL. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am J Pathol. 1993;143:545–554. [PMC free article] [PubMed] [Google Scholar]
- 134.Löhr M, Klöppel G, Maisonneuve P, Lowenfels AB, Lüttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 136.Fogar P, Sperti C, Basso D, Sanzari MC, Greco E, Davoli C, Navaglia F, Zambon CF, Pasquali C, Venza E, et al. Decreased total lymphocyte counts in pancreatic cancer: an index of adverse outcome. Pancreas. 2006;32:22–28. doi: 10.1097/01.mpa.0000188305.90290.50. [DOI] [PubMed] [Google Scholar]
- 137.Emmrich J, Sparmann G, Hopt U, Löhr M, Liebe S. Typing of leukocytes in pancreatic tissue surrounding human pancreatic carcinoma. Ann N Y Acad Sci. 1999;880:171–174. doi: 10.1111/j.1749-6632.1999.tb09520.x. [DOI] [PubMed] [Google Scholar]
- 138.Ademmer K, Ebert M, Müller-Ostermeyer F, Friess H, Büchler MW, Schubert W, Malfertheiner P. Effector T lymphocyte subsets in human pancreatic cancer: detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin Exp Immunol. 1998;112:21–26. doi: 10.1046/j.1365-2249.1998.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ryschich E, Nötzel T, Hinz U, Autschbach F, Ferguson J, Simon I, Weitz J, Fröhlich B, Klar E, Büchler MW, et al. Control of T-cell-mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res. 2005;11:498–504. [PubMed] [Google Scholar]
- 140.Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 141.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12:5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 142.Ikemoto T, Shimada M, Komatsu M, Yamada S, Saito Y, Mori H, Morine Y, Imura S, Bando Y, Utsunomiya T. Indoleamine 2,3-dioxygenase affects the aggressiveness of intraductal papillary mucinous neoplasms through Foxp3+CD4+CD25+ T cells in peripheral blood. Pancreas. 2013;42:130–134. doi: 10.1097/MPA.0b013e3182575e4a. [DOI] [PubMed] [Google Scholar]
- 143.Ikemoto T, Yamaguchi T, Morine Y, Imura S, Soejima Y, Fujii M, Maekawa Y, Yasutomo K, Shimada M. Clinical roles of increased populations of Foxp3+CD4+ T cells in peripheral blood from advanced pancreatic cancer patients. Pancreas. 2006;33:386–390. doi: 10.1097/01.mpa.0000240275.68279.13. [DOI] [PubMed] [Google Scholar]
- 144.Linehan DC, Goedegebuure PS. CD25+ CD4+ regulatory T-cells in cancer. Immunol Res. 2005;32:155–168. doi: 10.1385/IR:32:1-3:155. [DOI] [PubMed] [Google Scholar]
- 145.Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, Linehan DC. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, Gillanders WE, Linehan DC, Goedegebuure P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother. 2012;61:1373–1385. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Basso D, Fogar P, Falconi M, Fadi E, Sperti C, Frasson C, Greco E, Tamburrino D, Teolato S, Moz S, et al. Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PLoS One. 2013;8:e54824. doi: 10.1371/journal.pone.0054824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhao F, Obermann S, von Wasielewski R, Haile L, Manns MP, Korangy F, Greten TF. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology. 2009;128:141–149. doi: 10.1111/j.1365-2567.2009.03105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57:630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tsuzuki Y, Mouta Carreira C, Bockhorn M, Xu L, Jain RK, Fukumura D. Pancreas microenvironment promotes VEGF expression and tumor growth: novel window models for pancreatic tumor angiogenesis and microcirculation. Lab Invest. 2001;81:1439–1451. doi: 10.1038/labinvest.3780357. [DOI] [PubMed] [Google Scholar]
- 154.Yamazaki K, Nagao T, Yamaguchi T, Saisho H, Kondo Y. Expression of basic fibroblast growth factor (FGF-2)-associated with tumour proliferation in human pancreatic carcinoma. Virchows Arch. 1997;431:95–101. doi: 10.1007/s004280050074. [DOI] [PubMed] [Google Scholar]
- 155.Strouch MJ, Cheon EC, Salabat MR, Krantz SB, Gounaris E, Melstrom LG, Dangi-Garimella S, Wang E, Munshi HG, Khazaie K, et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res. 2010;16:2257–2265. doi: 10.1158/1078-0432.CCR-09-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cai SW, Yang SZ, Gao J, Pan K, Chen JY, Wang YL, Wei LX, Dong JH. Prognostic significance of mast cell count following curative resection for pancreatic ductal adenocarcinoma. Surgery. 2011;149:576–584. doi: 10.1016/j.surg.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 157.Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013;73:3927–3937. doi: 10.1158/0008-5472.CAN-12-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chang DZ, Ma Y, Ji B, Wang H, Deng D, Liu Y, Abbruzzese JL, Liu YJ, Logsdon CD, Hwu P. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2011;17:7015–7023. doi: 10.1158/1078-0432.CCR-11-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 160.Koizumi M, Hiasa Y, Kumagi T, Yamanishi H, Azemoto N, Kobata T, Matsuura B, Abe M, Onji M. Increased B cell-activating factor promotes tumor invasion and metastasis in human pancreatic cancer. PLoS One. 2013;8:e71367. doi: 10.1371/journal.pone.0071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gaida MM, Steffen TG, Günther F, Tschaharganeh DF, Felix K, Bergmann F, Schirmacher P, Hänsch GM. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur J Immunol. 2012;42:3369–3380. doi: 10.1002/eji.201242628. [DOI] [PubMed] [Google Scholar]
- 162.Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Hänsch GM, Gaida MM. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. 2012;2012:720768. doi: 10.1155/2012/720768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bausch D, Pausch T, Krauss T, Hopt UT, Fernandez-del-Castillo C, Warshaw AL, Thayer SP, Keck T. Neutrophil granulocyte derived MMP-9 is a VEGF independent functional component of the angiogenic switch in pancreatic ductal adenocarcinoma. Angiogenesis. 2011;14:235–243. doi: 10.1007/s10456-011-9207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Benson DD, Meng X, Fullerton DA, Moore EE, Lee JH, Ao L, Silliman CC, Barnett CC. Activation state of stromal inflammatory cells in murine metastatic pancreatic adenocarcinoma. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1067–R1075. doi: 10.1152/ajpregu.00320.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, Altinel D, Adsay V. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol. 2011;24:1612–1619. doi: 10.1038/modpathol.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Phan VT, Wu X, Cheng JH, Sheng RX, Chung AS, Zhuang G, Tran C, Song Q, Kowanetz M, Sambrone A, et al. Oncogenic RAS pathway activation promotes resistance to anti-VEGF therapy through G-CSF-induced neutrophil recruitment. Proc Natl Acad Sci USA. 2013;110:6079–6084. doi: 10.1073/pnas.1303302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Khayyata S, Basturk O, Adsay NV. Invasive micropapillary carcinomas of the ampullo-pancreatobiliary region and their association with tumor-infiltrating neutrophils. Mod Pathol. 2005;18:1504–1511. doi: 10.1038/modpathol.3800460. [DOI] [PubMed] [Google Scholar]
- 168.Bhatti I, Peacock O, Lloyd G, Larvin M, Hall RI. Preoperative hematologic markers as independent predictors of prognosis in resected pancreatic ductal adenocarcinoma: neutrophil-lymphocyte versus platelet-lymphocyte ratio. Am J Surg. 2010;200:197–203. doi: 10.1016/j.amjsurg.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 169.Wang DS, Luo HY, Qiu MZ, Wang ZQ, Zhang DS, Wang FH, Li YH, Xu RH. Comparison of the prognostic values of various inflammation based factors in patients with pancreatic cancer. Med Oncol. 2012;29:3092–3100. doi: 10.1007/s12032-012-0226-8. [DOI] [PubMed] [Google Scholar]
- 170.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 171.Nestle FO, Burg G, Fäh J, Wrone-Smith T, Nickoloff BJ. Human sunlight-induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell co-stimulatory molecules and are impaired as antigen-presenting cells. Am J Pathol. 1997;150:641–651. [PMC free article] [PubMed] [Google Scholar]
- 172.Chaux P, Favre N, Bonnotte B, Moutet M, Martin M, Martin F. Tumor-infiltrating dendritic cells are defective in their antigen-presenting function and inducible B7 expression. A role in the immune tolerance to antigenic tumors. Adv Exp Med Biol. 1997;417:525–528. doi: 10.1007/978-1-4757-9966-8_86. [DOI] [PubMed] [Google Scholar]
- 173.Kiertscher SM, Luo J, Dubinett SM, Roth MD. Tumors promote altered maturation and early apoptosis of monocyte-derived dendritic cells. J Immunol. 2000;164:1269–1276. doi: 10.4049/jimmunol.164.3.1269. [DOI] [PubMed] [Google Scholar]
- 174.Bellone G, Carbone A, Smirne C, Scirelli T, Buffolino A, Novarino A, Stacchini A, Bertetto O, Palestro G, Sorio C, et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J Immunol. 2006;177:3448–3460. doi: 10.4049/jimmunol.177.5.3448. [DOI] [PubMed] [Google Scholar]
- 175.Allavena P, Piemonti L, Longoni D, Bernasconi S, Stoppacciaro A, Ruco L, Mantovani A. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur J Immunol. 1998;28:359–369. doi: 10.1002/(SICI)1521-4141(199801)28:01<359::AID-IMMU359>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 176.De Smedt T, Van Mechelen M, De Becker G, Urbain J, Leo O, Moser M. Effect of interleukin-10 on dendritic cell maturation and function. Eur J Immunol. 1997;27:1229–1235. doi: 10.1002/eji.1830270526. [DOI] [PubMed] [Google Scholar]
- 177.Menetrier-Caux C, Thomachot MC, Alberti L, Montmain G, Blay JY. IL-4 prevents the blockade of dendritic cell differentiation induced by tumor cells. Cancer Res. 2001;61:3096–3104. [PubMed] [Google Scholar]
- 178.Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 179.Strobl H, Knapp W. TGF-beta1 regulation of dendritic cells. Microbes Infect. 1999;1:1283–1290. doi: 10.1016/s1286-4579(99)00256-7. [DOI] [PubMed] [Google Scholar]
- 180.Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. 2000;1:510–514. doi: 10.1038/82763. [DOI] [PubMed] [Google Scholar]
- 181.Menetrier-Caux C, Montmain G, Dieu MC, Bain C, Favrot MC, Caux C, Blay JY. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood. 1998;92:4778–4791. [PubMed] [Google Scholar]
- 182.Monti P, Leone BE, Zerbi A, Balzano G, Cainarca S, Sordi V, Pontillo M, Mercalli A, Di Carlo V, Allavena P, et al. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J Immunol. 2004;172:7341–7349. doi: 10.4049/jimmunol.172.12.7341. [DOI] [PubMed] [Google Scholar]
- 183.Tjomsland V, Sandström P, Spångeus A, Messmer D, Emilsson J, Falkmer U, Falkmer S, Magnusson KE, Borch K, Larsson M. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: an indicator of disease severity? BMC Cancer. 2010;10:87. doi: 10.1186/1471-2407-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tjomsland V, Spångeus A, Sandström P, Borch K, Messmer D, Larsson M. Semi mature blood dendritic cells exist in patients with ductal pancreatic adenocarcinoma owing to inflammatory factors released from the tumor. PLoS One. 2010;5:e13441. doi: 10.1371/journal.pone.0013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Takahashi K, Toyokawa H, Takai S, Satoi S, Yanagimoto H, Terakawa N, Araki H, Kwon AH, Kamiyama Y. Surgical influence of pancreatectomy on the function and count of circulating dendritic cells in patients with pancreatic cancer. Cancer Immunol Immunother. 2006;55:775–784. doi: 10.1007/s00262-005-0079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yanagimoto H, Takai S, Satoi S, Toyokawa H, Takahashi K, Terakawa N, Kwon AH, Kamiyama Y. Impaired function of circulating dendritic cells in patients with pancreatic cancer. Clin Immunol. 2005;114:52–60. doi: 10.1016/j.clim.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 187.Funa K, Nilsson B, Jacobsson G, Alm GV. Decreased natural killer cell activity and interferon production by leucocytes in patients with adenocarcinoma of the pancreas. Br J Cancer. 1984;50:231–233. doi: 10.1038/bjc.1984.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Aparicio-Pagés MN, Verspaget HW, Peña AS, Jansen JB, Lamers CB. Natural killer cell activity in patients with neuroendocrine tumours of the gastrointestinal tract; relation with circulating gastrointestinal hormones. Neuropeptides. 1991;20:1–7. doi: 10.1016/0143-4179(91)90033-f. [DOI] [PubMed] [Google Scholar]
- 189.Fogar P, Basso D, Fadi E, Greco E, Pantano G, Padoan A, Bozzato D, Facco M, Sanzari MC, Teolato S, et al. Pancreatic cancer alters human CD4+ T lymphocyte function: a piece in the immune evasion puzzle. Pancreas. 2011;40:1131–1137. doi: 10.1097/MPA.0b013e31822077b8. [DOI] [PubMed] [Google Scholar]
- 190.von Bernstorff W, Voss M, Freichel S, Schmid A, Vogel I, Jöhnk C, Henne-Bruns D, Kremer B, Kalthoff H. Systemic and local immunosuppression in pancreatic cancer patients. Clin Cancer Res. 2001;7:925s–932s. [PubMed] [Google Scholar]
- 191.Chang JF, Zhao HL, Phillips J, Greenburg G. The epithelial mucin, MUC1, is expressed on resting T lymphocytes and can function as a negative regulator of T cell activation. Cell Immunol. 2000;201:83–88. doi: 10.1006/cimm.2000.1643. [DOI] [PubMed] [Google Scholar]
- 192.Agrawal B, Krantz MJ, Reddish MA, Longenecker BM. Cancer-associated MUC1 mucin inhibits human T-cell proliferation, which is reversible by IL-2. Nat Med. 1998;4:43–49. doi: 10.1038/nm0198-043. [DOI] [PubMed] [Google Scholar]
- 193.Plate JM, Shott S, Harris JE. Immunoregulation in pancreatic cancer patients. Cancer Immunol Immunother. 1999;48:270–279. doi: 10.1007/s002620050575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rasheed ZA, Matsui W, Maitra A. Pathology of pancreatic stroma in PDAC. In: Grippo PJ, Munshi HG, editors. Source Pancreatic Cancer and Tumor Microenvironment. 1st ed. Trivandrum (India): Transworld Research Network; 2012. [PubMed] [Google Scholar]
- 196.Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2009;7:S44–S47. doi: 10.1016/j.cgh.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121–1132. doi: 10.1053/j.gastro.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Fujita H, Ohuchida K, Mizumoto K, Egami T, Miyoshi K, Moriyama T, Cui L, Yu J, Zhao M, Manabe T, et al. Tumor-stromal interactions with direct cell contacts enhance proliferation of human pancreatic carcinoma cells. Cancer Sci. 2009;100:2309–2317. doi: 10.1111/j.1349-7006.2009.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, Satoh K, Egawa S, Unno M, Shimosegawa T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun. 2010;403:380–384. doi: 10.1016/j.bbrc.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 201.Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S, Esposito I, Friess H, Gress TM, Habisch HJ, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61:172–178. doi: 10.1136/gutjnl-2011-301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tjomsland V, Niklasson L, Sandström P, Borch K, Druid H, Bratthäll C, Messmer D, Larsson M, Spångeus A. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin Dev Immunol. 2011;2011:212810. doi: 10.1155/2011/212810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, Clevers H, Hart IR, Kocher HM. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-β-catenin signaling to slow tumor progression. Gastroenterology. 2011;141:1486–197, 1486-197. doi: 10.1053/j.gastro.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 204.Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, Fuchs JR, Eubank TD, Frankel WL, Bekaii-Saab T, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007–3018. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 206.Dorado J, Lonardo E, Miranda-Lorenzo I, Heeschen C. Pancreatic cancer stem cells: new insights and perspectives. J Gastroenterol. 2011;46:966–973. doi: 10.1007/s00535-011-0422-x. [DOI] [PubMed] [Google Scholar]
- 207.Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 208.Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, Yagita H, Takaoka A, Tahara H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci USA. 2011;108:12425–12430. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Korkaya H, Liu S, Wicha MS. Regulation of cancer stem cells by cytokine networks: attacking cancer’s inflammatory roots. Clin Cancer Res. 2011;17:6125–6129. doi: 10.1158/1078-0432.CCR-10-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dodson LF, Hawkins WG, Goedegebuure P. Potential targets for pancreatic cancer immunotherapeutics. Immunotherapy. 2011;3:517–537. doi: 10.2217/imt.11.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Koos D, Josephs SF, Alexandrescu DT, Chan RC, Ramos F, Bogin V, Gammill V, Dasanu CA, De Necochea-Campion R, Riordan NH, et al. Tumor vaccines in 2010: need for integration. Cell Immunol. 2010;263:138–147. doi: 10.1016/j.cellimm.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 213.Gunturu KS, Rossi GR, Saif MW. Immunotherapy updates in pancreatic cancer: are we there yet? Ther Adv Med Oncol. 2013;5:81–89. doi: 10.1177/1758834012462463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD. Peptide-pulsed dendritic cells induce antigen-specific CTL-mediated protective tumor immunity. J Exp Med. 1996;183:283–287. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Niccolai E, Prisco D, D’Elios MM, Amedei A. What is recent in pancreatic cancer immunotherapy? Biomed Res Int. 2013;2013:492372. doi: 10.1155/2013/492372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A, Giese NA, Esposito I, Friess H, Kleeff J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2008;6:1155–1161. doi: 10.1016/j.cgh.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 217.Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, Ouellette MM, Hollingsworth MA. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nishikawa H, Kato T, Tanida K, Hiasa A, Tawara I, Ikeda H, Ikarashi Y, Wakasugi H, Kronenberg M, Nakayama T, et al. CD4+ CD25+ T cells responding to serologically defined autoantigens suppress antitumor immune responses. Proc Natl Acad Sci USA. 2003;100:10902–10906. doi: 10.1073/pnas.1834479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bazhin AV, Shevchenko I, Umansky V, Werner J, Karakhanova S. Two immune faces of pancreatic adenocarcinoma: possible implication for immunotherapy. Cancer Immunol Immunother. 2014;63:59–65. doi: 10.1007/s00262-013-1485-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Finke JH, Rayman PA, Ko JS, Bradley JM, Gendler SJ, Cohen PA. Modification of the tumor microenvironment as a novel target of renal cell carcinoma therapeutics. Cancer J. 2013;19:353–364. doi: 10.1097/PPO.0b013e31829da0ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ribeiro RA, Flores CA, Cunha FQ, Ferreira SH. IL-8 causes in vivo neutrophil migration by a cell-dependent mechanism. Immunology. 1991;73:472–477. [PMC free article] [PubMed] [Google Scholar]
- 224.Wolpe SD, Sherry B, Juers D, Davatelis G, Yurt RW, Cerami A. Identification and characterization of macrophage inflammatory protein 2. Proc Natl Acad Sci USA. 1989;86:612–616. doi: 10.1073/pnas.86.2.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bozic CR, Kolakowski LF, Gerard NP, Garcia-Rodriguez C, von Uexkull-Guldenband C, Conklyn MJ, Breslow R, Showell HJ, Gerard C. Expression and biologic characterization of the murine chemokine KC. J Immunol. 1995;154:6048–6057. [PubMed] [Google Scholar]
- 226.Zhang Y, Ramos BF, Jakschik BA. Neutrophil recruitment by tumor necrosis factor from mast cells in immune complex peritonitis. Science. 1992;258:1957–1959. doi: 10.1126/science.1470922. [DOI] [PubMed] [Google Scholar]
- 227.Schmid MC, Avraamides CJ, Foubert P, Shaked Y, Kang SW, Kerbel RS, Varner JA. Combined blockade of integrin-α4β1 plus cytokines SDF-1α or IL-1β potently inhibits tumor inflammation and growth. Cancer Res. 2011;71:6965–6975. doi: 10.1158/0008-5472.CAN-11-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Castell JV, Gómez-Lechón MJ, David M, Hirano T, Kishimoto T, Heinrich PC. Recombinant human interleukin-6 (IL-6/BSF-2/HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Lett. 1988;232:347–350. doi: 10.1016/0014-5793(88)80766-x. [DOI] [PubMed] [Google Scholar]
- 229.Castell JV, Gómez-Lechón MJ, David M, Fabra R, Trullenque R, Heinrich PC. Acute-phase response of human hepatocytes: regulation of acute-phase protein synthesis by interleukin-6. Hepatology. 1990;12:1179–1186. doi: 10.1002/hep.1840120517. [DOI] [PubMed] [Google Scholar]
- 230.Yoshimura T, Yuhki N, Moore SK, Appella E, Lerman MI, Leonard EJ. Human monocyte chemoattractant protein-1 (MCP-1). Full-length cDNA cloning, expression in mitogen-stimulated blood mononuclear leukocytes, and sequence similarity to mouse competence gene JE. FEBS Lett. 1989;244:487–493. doi: 10.1016/0014-5793(89)80590-3. [DOI] [PubMed] [Google Scholar]
- 231.Parham P, Janeway C. The immune system. 3rd ed. London, NY: Garland Science; 2009. [Google Scholar]
- 232.Taga K, Mostowski H, Tosato G. Human interleukin-10 can directly inhibit T-cell growth. Blood. 1993;81:2964–2971. [PubMed] [Google Scholar]
- 233.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 235.Fujishima S, Hoffman AR, Vu T, Kim KJ, Zheng H, Daniel D, Kim Y, Wallace EF, Larrick JW, Raffin TA. Regulation of neutrophil interleukin 8 gene expression and protein secretion by LPS, TNF-alpha, and IL-1 beta. J Cell Physiol. 1993;154:478–485. doi: 10.1002/jcp.1041540305. [DOI] [PubMed] [Google Scholar]
- 236.Nikolaus S, Bauditz J, Gionchetti P, Witt C, Lochs H, Schreiber S. Increased secretion of pro-inflammatory cytokines by circulating polymorphonuclear neutrophils and regulation by interleukin 10 during intestinal inflammation. Gut. 1998;42:470–476. doi: 10.1136/gut.42.4.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Takemura R, Werb Z. Secretory products of macrophages and their physiological functions. Am J Physiol. 1984;246:C1–C9. doi: 10.1152/ajpcell.1984.246.1.C1. [DOI] [PubMed] [Google Scholar]
- 238.Fink GW, Norman JG. Specific changes in the pancreatic expression of the interleukin 1 family of genes during experimental acute pancreatitis. Cytokine. 1997;9:1023–1027. doi: 10.1006/cyto.1997.0260. [DOI] [PubMed] [Google Scholar]
- 239.Feurino LW, Zhang Y, Bharadwaj U, Zhang R, Li F, Fisher WE, Brunicardi FC, Chen C, Yao Q, Min L. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol Ther. 2007;6:1096–1100. doi: 10.4161/cbt.6.7.4328. [DOI] [PubMed] [Google Scholar]
- 240.Jin C, Ni Q, Zhang Q. [Apoptosis of pancreatic acinar and expression of TNF-alpha mRNA, IL-10 mRNA in rats with acute pancreatitis] Zhonghua Wai Ke Zazhi. 2001;39:626–628. [PubMed] [Google Scholar]
- 241.Altstaedt J, Kirchner H, Rink L. Cytokine production of neutrophils is limited to interleukin-8. Immunology. 1996;89:563–568. doi: 10.1046/j.1365-2567.1996.d01-784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA. 2006;103:12493–12498. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–4760. [PubMed] [Google Scholar]
- 244.Tazawa H, Okada F, Kobayashi T, Tada M, Mori Y, Une Y, Sendo F, Kobayashi M, Hosokawa M. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol. 2003;163:2221–2232. doi: 10.1016/S0002-9440(10)63580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wasiuk A, Dalton DK, Schpero WL, Stan RV, Conejo-Garcia JR, Noelle RJ. Mast cells impair the development of protective anti-tumor immunity. Cancer Immunol Immunother. 2012;61:2273–2282. doi: 10.1007/s00262-012-1276-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Roche WR. Mast cells and tumors. The specific enhancement of tumor proliferation in vitro. Am J Pathol. 1985;119:57–64. [PMC free article] [PubMed] [Google Scholar]
- 247.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 249.Rajarathnam K, Sykes BD, Kay CM, Dewald B, Geiser T, Baggiolini M, Clark-Lewis I. Neutrophil activation by monomeric interleukin-8. Science. 1994;264:90–92. doi: 10.1126/science.8140420. [DOI] [PubMed] [Google Scholar]
- 250.Becker S, Quay J, Koren HS, Haskill JS. Constitutive and stimulated MCP-1, GRO alpha, beta, and gamma expression in human airway epithelium and bronchoalveolar macrophages. Am J Physiol. 1994;266:L278–L286. doi: 10.1152/ajplung.1994.266.3.L278. [DOI] [PubMed] [Google Scholar]