

Summary
Mutations in SURF1 cytochrome c oxidase (COX) assembly protein are associated with Leigh’s syndrome, a human mitochondrial disorder that manifests as severe mitochondrial phenotypes and early lethality. In contrast, mice lacking the Surf1 protein (Surf1−/−) are viable and were previously shown to have enhanced longevity and a greater than 50% reduction in COX activity. We measured mitochondrial function in heart and skeletal muscle, and despite the significant reduction in COX activity, we found little or no difference in reactive oxygen species (ROS) generation, membrane potential, ATP production or respiration in isolated mitochondria from Surf1−/− mice compared to wild-type. However, blood lactate levels are elevated and Surf1−/− mice have reduced running endurance, suggesting compromised mitochondrial energy metabolism in vivo. Decreased COX activity in Surf1−/− mice is associated with increased markers of mitochondrial biogenesis (PGC-1α and VDAC) in both heart and skeletal muscle. While mitochondrial biogenesis is a common response in the two tissues, skeletal muscle have an up-regulation of the mitochondrial unfolded protein response (UPRMT) and heart exhibits induction of the Nrf2 antioxidant response pathway. These data are the first to report induction of the UPRMT in a mammalian model of diminished COX activity. In addition our results suggest that impaired mitochondrial function can lead to induction of mitochondrial stress pathways to confer protective effects on cellular homeostasis.
Loss of complex IV assembly factor Surf1 in mice results in compensatory responses including mitochondrial biogenesis, the nrf2 pathway and the mitochondrial unfolded protein response. This compensatory response may contribute to the lack of deleterious phenotypes under basal conditions.
Keywords: Mitochondrial Unfolded Protein Response, Nrf2, Mitochondrial Biogenesis, Mitochondrial Function, Heme Oxygenase-1, Mitohormesis
Introduction
Surfeit locus protein 1 (Surf1) is a nuclear encoded small hydrophobic protein localized to the mitochondrial inner membrane that aides in the initial assembly of 13 subunits of the cytochrome c oxidase (COX, Complex IV) holoenzyme. Complex IV facilitates the final transfer of electrons in the electron transport chain (ETC) from cytochrome c to molecular oxygen forming water and thus plays a key role in mitochondrial oxidative phosphorylation. In humans, loss of function mutations in Surf1 lead to a devastating disease phenotype characterized by severe neurologic deficits and early lethality [1]. However, Surf1−/− mice engineered to express a truncated and unstable Surf1 protein do not display a deleterious phenotype, despite a significant reduction in complex IV activity. In fact, the complex IV deficient Surf1−/− mice have been reported to exhibit enhanced longevity compared to their wild-type littermates [2]. This phenotype linking mitochondrial dysfunction and increased longevity is consistent with recent studies in yeast, worms [3], and flies [4], in which certain mutations of the ETC can lead to extended lifespan.
We postulated that the loss of Surf1 and the reduction in Complex IV activity might initiate compensatory responses to mitochondrial stress that would be beneficial to cellular homeostasis. Previous studies in the long-lived clk-1, isp-1 and cco-1 C. elegans mitochondrial mutants point to a potential role of the mitochondrial unfolded protein response (UPRMT) [5], mitochondrial biogenesis [6–8], and NF-E2-related factor (Nrf2) activation in mediating longevity in these mutants (see review by Pulliam, et al., 2012)[9]. The UPRMT is an evolutionarily conserved signaling mechanism initiated by mitochondrial stress. In C. elegans, the UPRMT is mediated through the translocation of transcription factors UBL-5/DEV-1 and ATFS-1 (ZC376.7) from the cytosol to the nucleus [10–12]. This results in an increase in the expression of mitochondrial-specific chaperones such as heat shock protein 60 (hsp60) and proteases such as caseinolytic peptidase (ClpP) [13]. These proteins play an important role in mitochondrial protein quality control [14]. A role for this pathway in the maintenance of mitochondrial homeostasis has also been reported in mammalian cell culture and is positively regulated by Chop (C/EBP homology protein). Within twenty base pairs of the Chop DNA binding site, upstream and downstream, are two conserved sequences, MURE1 and MURE2 (Mitochondrial Unfolded Response Element1/2) that positively regulate the expression of UPRMT proteins [15–17]. However, the identity of what binds MURE1 and MURE2 remains unknown.
Mitochondrial biogenesis is another mitochondrial homeostasis mechanism [18]. Mitochondrial biogenesis involves the production of new mitochondria, replication of mitochondrial DNA (mtDNA) and production of nuclear encoded mitochondrial proteins. This process is largely under the control of the transcription co-activator, peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC-1α) [18]. Interestingly, in C. elegans, the lifespan extension mediated by ETC inhibition occurs only when the ETC is inhibited during L3/L4 development [7], and thus coincides with mitochondrial biogenesis [19]. The levels of mtDNA are also increased in the isp-1 and clk-1 mitochondrial mutants, consistent with an increase in mitochondrial biogenesis [6,8]. Thus, it has been postulated that mitochondrial biogenesis might be a significant factor underlying lifespan extension in response to ETC deficits in C. elegans [5].
Nrf2 transcription factor is an integral antioxidant-signaling mechanism. Under basal conditions Nrf2 is rapidly degraded by the proteasome. However, following oxidative stress, Nrf2 localizes to the nucleus where it binds the conserved antioxidant response element (ARE) DNA sequence. Binding of Nrf2 to the ARE results in the upregulation of many phase I and phase II detoxifying enzymes as well as antioxidants such as glutathione S-transferases, peroxiredoxins (prdx), thioredoxins (Trx), and heme-oxygenase 1 (HO-1) [20].
To test whether mitochondrial compensatory responses are up-regulated in the Surf1−/− mice, we measured mitochondrial function and changes in mitochondrial stress responses (UPRMT, mitochondrial proliferation, and Nrf2) in two highly energetic tissues, heart and skeletal muscle, in wild-type and long-lived Surf1−/− mice. We hypothesized that mechanisms associated with longevity in invertebrates might also be induced in a mammalian model of reduced ETC activity. Furthermore, we investigated in vivo physiological changes that result from loss of functional Surf1.
Materials and Methods
Animals
All experiments were performed with the approval by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Health Science Center at San Antonio. Surf1−/− mice, generated as previously described [2] were bred from Surf1+/− heterozygous crosses in a B6D2F1/J (C57/Bl6JxDBA2) background. All wild-type animals were littermate controls of the Surf1−/− animals. Male mice aged 5–7 months were used for all experiments and sacrificed using CO2 asphyxiation.
Mitochondrial Isolation
Heart and hind-limb skeletal muscle mitochondria were isolated using differential centrifugation as we have previously described [21]. Heart and hind-limb skeletal muscles were removed, rinsed and minced in Chappell-Perry Buffer I (100mM KCl, 50mM Tris-HCl, 5mM MgCl2 and 1mM EDTA, pH 7.2) with 1mM ATP (Grade II, Sigma) and 1.5mg protease (Type I: crude, from bovine pancreas, Sigma) per 0.5g tissue. The minced tissue was placed on a shaker for 10 minutes and then homogenized. The homogenate was spun at 600xg for 10 minutes. The supernatant was filtered through a cheesecloth followed by centrifugation at 14,000xg for 10 minutes. The supernatant was discarded and the pellet was resuspended in Chappell-Perry Buffer II (100mM KCl, 50mM Tris-HCl, 1mM MgCl2 and 0.2mM EDTA, pH 7.2) with 0.2mM ATP and bovine serum albumin (100mg/100ml, Sigma) and spun at 7,000xg for 10 minutes. The pellet was resuspended in Chappell-Perry Buffer II with ATP and spun two times at 3,500xg. The final pellet was used for all mitochondrial assays.
Complex Activity Assays
The final mitochondrial pellet was resuspended in ACA/BT buffer (750mM 6-Aminocaprioic acid, 50mM Bistris, pH 7.0) plus 1% n-dodecylmaltoside and 1× protease inhibitor (Cocktail set III, Calbiochem) for 45 minutes with constant agitation at 4°C. The suspension was then spun at 100,000xg for 15 minutes at 4°C. The protein concentration in the supernatant was measured using the Bradford method and then used for the complex activity assays as we have previously described [22].
Complex I Activity Assay
Complex I activity was measured by monitoring the oxidation of nicotinomide adenine dinucleotide (NADH) with a spectrophotometer at 340nm with ubiquinone-2 as the electron acceptor in the presence of diclorophenolindophenol (DCIP) as described previously [22]. To perform the assay, 1ml of respiration buffer (250mM sucrose, 10mM KH2PO4, 1mM EGTA, 10mM Tris, pH 7.4) was placed into a polystyrene cuvette with 100µM NADH, 50µM DCIP, 2mM KCN and 2µM antimycin A. After setting up the blank, 50µM ubiquinone-2 was added followed by 20µg mitochondrial protein and the rate of change in absorbance was measured for one minute (dA/min). As a negative control, complex I activity was inhibited with the addition of 10µM rotenone and the rate was subtracted from the rate of NADH oxidation. The final rate (dA/min) was converted to mM/min/mg by dividing dA/min by 6.22−1mM cm (extinction coefficient for NADH) and then divided by the amount of mitochondrial protein. The final rate was normalized to wild-type control values.
Complex II Activity Assay
Complex II activity was measured by succinate-dependent reduction of DCIP at 600nm using ubiquione-2 as an electron acceptor as previously described [22]. To perform the assay, 1ml of respiration buffer (250mM sucrose, 10mM KH2PO4, 1mM EGTA, 10mM Tris, pH 7.4) was placed into a polystyrene cuvette with 20mM succinate, 50µM DCIP, 2mM KCN, 2µM antimycin A. After setting up the blank, 50µM ubiquinone-2 (Sigma) was added followed by 40µg of mitochondrial protein and the rate of change in absorbance was measured for one minute. As a negative control, activity of complex II was inhibited by the addition of 2mM malonate and the rate was subtracted from the rate of DCIP reduction from complex II activity. The final rate (dA/min) was converted to mM/min/mg by dividing dA/min by 21 mM−1 (extinction coefficient for succinate) and then divided by the amount of mitochondrial protein. The final rate was normalized to wild-type control values.
Complex III Activity Assay
Complex III activity was measured by the reduction of cytochrome c3+ at 550 nm using D-ubiquinol-2 as an electron acceptor as previously described [22]. Preparation of reduced ubiquinol: 100µl of ubiquinone (20mM in ethanol) was reduced with pinch of sodium borohydrate (Sigma). After mixing, 100µl of ethylene glycol was added to stop the reduction of ubiquinone. 2µl of 12M HCl was added and spun to separate the sodium borohydrate from the reduced ubiquinol-2. To perform the assay, 1ml of respiration buffer (250mM sucrose, 10mM KH2PO4, 1mM EGTA, 10mM Tris, pH 7.4) was placed into a polystyrene cuvette with 10µM rotenone, and 2mM KCN. After setting up the blank, 100µM D ubiquinol-2 was added followed by 10µg of mitochondrial protein and the rate of change in absorbance was measured for one minute. As a negative control 2µM antimycin A was added to inhibit the activity of complex III and the rate was subtracted from the rate of cytochrome c oxidation. The final rate (dA/min) was converted to mM/min/mg by dividing dA/min by 21 mM−1 cm (extinction coefficient for cytochrome c) and then divided by the amount of mitochondrial protein. The final rate was normalized to wild-type control values.
Complex IV Activity Assay
Complex IV activity was measured by monitoring the oxidation of cytochrome c2+ at 550nm using a spectrophotometer as previously described [22]. Preparation of reduced cytochrome c2+: 500µl of 8mM cytochrome c3+ (Sigma) was reduced with sodium borohydrate (Sigma). To perform the assay, 1ml of the respiration buffer (250mM sucrose, 10mM KH2PO4, 1mM EGTA, 10mM Tris, pH 7.4) was placed into a polystyrene cuvette with 40µM cytochrome c2+. After setting up the blank, 5µg of mitochondrial protein was added, and rate of cytochrome c2+oxidation was measured at 550nm. As a negative control, complex IV activity was inhibited by the addition of 2mM KCN and the rate of change was then subtracted from the rate of cytochrome c2+ oxidation (dA/min). The rate of was converted to mM/min/mg by dividing dA/min by 21mM−1 cm (extinction coefficient for cytochrome c) and then divided by the amount of mitochondrial protein. The final rate was normalized to wild-type control values.
Mitochondrial Respiration
Mitochondrial oxygen consumption was measured using a Clark electrode (Oxytherm Oxygen Electrode Control Unit, Hansatech Instruments Ltd.) as previously described [23,24]. Briefly, mitochondria were resuspended in respiration buffer (250mM sucrose, 10mM KH2PO4, 1mM EGTA, 10mM Tris, pH 7.4) with 0.3% bovine serum albumin (BSA); glutamate/malate (5mM) was used as the respiratory substrate. State 3 respiration was initiated with the addition of 0.3mM ADP. State 4 respiration was measured as oxygen consumption following the consumption of ADP. Respiratory control ratio (RCR) was measured as state 3/state 4 respiration rates.
ATP Production
ATP production was measured in freshly isolated heart or skeletal muscle mitochondria using the ATP Bioluminescence Assay Kit CLSII (Roche, Cat#1 699 695) and a fluorescent microplate reader at 562nm as previously described [22]. Freshly isolated mitochondria were resuspended in ROS buffer (125mM KCl, 10mM HEPES, 5mM MgCl2, 2mM K2HPO4, pH 7.44) to a final concentration of 0.04µg/µl. In a solid white 96 well plate, 100µl of ROS buffer with mitochondria per well were aliquoted. The complex II-linked substrates used were succinate (5mM) and rotenone (5nM). Complex I-linked substrates glutamate (2.5mM) and malate (2.5mM) for skeletal muscle mitochondria and pyruvate (2.5mM) and malate (2.5mM) for heart mitochondria were used. To each well, 100µl of luciferase reagent containing 0.3mM ADP was added and the kinetic luminescence was determined. The results were then calculated from a standard curve generated with ATP standard provided by the manufacturer.
Membrane Potential
Membrane potential was determined as previously described [25]. Mitochondria were resuspended in ROS buffer (125mM KCl, 10mM HEPES, 5mM MgCl2, 2mM K2HPO4, pH 7.44) at a concentration of 0.05µg/µl with 5µM Safranin O (ICN Biomedical Inc.) and 100µl were aliquoted into a black, flat bottom 96-well microplate. Plates with complete buffer and mitochondria were incubated at 37°C for five minutes before the addition of substrates. For complex I-linked substrates in skeletal muscle, glutamate (5mM) and malate (5mM) were used. For complex I-linked substrates in heart, pyruvate (5mM) and malate (5mM) were used. For complex II-linked substrates succinate (10mM) and rotenone (1µM) were used. Fluorescence of the quench-dye Safranin O was measured using excitation at 485nm and emission at 590nm using a microplate reader and wells with only ROS buffer and Safranin O were used as a negative control.
Hydrogen peroxide (H2O2) production assay
Mitochondrial release of H2O2 was measured as we have previously described [26]. In the presence of horseradish peroxidase, the Amplex Red reagent reacts with hydrogen peroxide to produce the red fluorescent oxidation product, resorufin. The fluorescence of resorufin was measured with fluorescence microplate reader using excitation at 544nm and emission at 590nm. Isolated mitochondria were diluted in Amplex Red reagent buffer (Amplex Red 77.8µM (Invitrogen, cat#a22188), superoxide dismutase (SOD, 37.5units/ml), horse radish peroxidase 1unit/ml in ROS buffer) to a final concentration of 0.4µg/µl. 100ul of diluted mitochondria were used per reaction. Resorufin fluorescence was measured following the addition of succinate (5mM) and rotenone (5nM) or glutamate (2.5mM) and malate (2.5mM). Hydrogen peroxide production was determined by comparing the value to an Amplex Red-H2O2 standard curve.
Superoxide Production
Extramitochondrial superoxide release was measured by EPR using the spin trap, 5-(diisopropoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DIPPMPO, Alexis Biochemicals) as we have previously described [27]. DIPPMPO forms an adduct with superoxide, resulting in the generation of DIPPMPO-OOH, which decays to the DIPPMPO-OH adduct by the action of glutathione peroxidases. EPR measurements were performed using an X-band MS200 spectrometer (Magnetech, Berlin). Mitochondria (1µg/µl) were incubated at 37°C with DIPPMPO (50mM) for 30 minutes in 125mM KCl, 10mM MOPS, 2mM diethylene triamine pentaacetic acid, 5mM MgCl2, 2mM K2HPO4, pH 7.44 in the presence of succinate (24mM), rotenone (2.4µM) and antimycin A. These substrates were used to drive respiration starting at complex II. Although it did not affect the DIPPMPO-OH signal, low levels of catalase (10units/ml) were also added, to prevent the appearance of small additional and unidentified spectrum peaks after extended incubation. For measurements, 40µl of sample was transferred to 50µl capillary tubes and measured at room temperature with the following settings: receiver gain, 5 × 105; microwave power, 20 milliwatt; microwave frequency, 9.55GHz; modulation amplitude, 2G; scan time, 40 s; and scan width, 100 G, with an accumulation of 10 scans. EPR data are expressed as RIU (Relative Intensity Units)/20µg mitochondrial protein. As a negative control, measurements were also in the presence of SOD.
Electron Microscopy
Heart and hind-limb skeletal muscle were excised and fixed in phosphate buffered 4% formaldehyde with 1% glutaraldehyde. The samples were rinsed in 0.1M phosphate buffer and then post-fixed with 1% Zetterqvist’s buffered Osmium Tetroxide for 30 minutes. The samples were dehydrated in 70% alcohol for 10 minutes and then in 95% alcohol for 10 minutes. The samples were then twice dehydrated in 100% alcohol for 10 minutes and then twice dehydrated in propylene oxide for 10 minutes, for each dehydration. The samples were resin infiltrated first in a 1:1 propylene oxide/resin mix for 30 minutes and then in 100% resin for 30 minutes under 25psi vacuum. The samples were then flat-embedded in a BEEM capsule and filled to the top and polymerized at 85°C for 90 minutes. The embedded samples were then sectioned and stained with a drop of uranyl acetate and microwaved for 30 seconds. The section was then gently dipped into distilled water and placed on filter paper. A drop of Reynold’s lead citrate stain was then placed on the section and microwaved for 20 seconds and then rinsed in distilled water. The section was then photographed at 20,000× using a JEOL 1230 transmission electron microscope with AMT imaging software. Five randomly chosen images per animal (n=3) were quantified for mitochondrial number and area using NIS-Elements BR imaging software.
Western Blotting
Heart and gastrocnemius skeletal muscle were homogenized in buffer containing 50mM HEPES, 150mM NaCl, 2mM EDTA, 10% glycerol, 1%Igepal, 1mM MgCl2, 1mM CaCl2 and 1× protease inhibitor. Homogenized samples were spun at 15,000xg for 10 minutes at 4°C and supernatant was collected. Protein concentrations were measured using the Bradford method and diluted to equal concentration. Samples were then diluted in a 1:1 ratio in Laemmli Sample Buffer (Bio-Rad) with 50µl 2-mercaptoethanol per 950µl Laemmli Sample Buffer and boiled for 10 minutes. The final samples were then run on a 4–20% gradient Criterion™ XT precast gel (Bio-Rad) with 1× Tris/Glycine/SDS buffer. Gels were transferred to PVDF membrane using trans-blot SD semi-dry transfer cell (Bio-Rad) in Tris/Glycine/SDS buffer containing 20% methanol. Blots were blocked for one hour in 1% bovine serum albumin (BSA) in TBS buffer containing 0.05% Tween-20 (TBS-T). Primary antibodies used: Hsp60 (StressGen), ClpP (Sigma), Lon (Dr. Luke Szweda Laboratory), Trx2 (AbCam), Chop (Cell Signaling), PGC-1α (AbCam), Tfam (Sigma), Complex II 70kDa subunit (Molecular Probes), VDAC (Cell-Signaling), Cytochrome C (Cell Signaling), Nrf2 (Dr. Scott Plafker Laboratory), Heme Oxygenase-1 (StressGen). Secondary antibodies used were from Vector Laboratories. Western blots were imaged using premium x-ray film from Phenix Research Products.
Quantitative Real-Time PCR
Total RNA was extracted from 50mg of flash-frozen hind-limb skeletal muscle and cardiac tissue using RNeasy Plus kit (Qiagen, cat# 73442). SuperScript II reverse transcriptase and random hexamer primers were used for first strand cDNA synthesis as per manufacturer’s instructions (Life Technologies, cat# 18064071). Quantitative real-time PCR was performed with an ABI Prism using Power SYBR Green PCR Master Mix (Applied Biosystems, cat# 4367659). The calculations were performed by comparative method (2−ΔΔCT) and the following primers were used: Pgc1α Forward 5′-CGGAAATCATATCCAACCAG-3′ Reverse 5′-TGAGGACCGCTAGCAAGTTTG-3′; Gamma-Actin Forward 5’- GCCGCCGCGTCCTT-3’ Reverse 5’- ATGACGAGTGCGGCGATT-3’.
Open-Field Activity Monitoring
Open-field activity monitoring is used to determine total activity as previously described [28]. Mice are placed in a large, clear cage (16”×9” ×5.5”) with a grid size (7×15) for 40 hours with corncob bedding and free access to food and water. The mice are habituated to the apparatus for sixteen hours and data is recorded during the subsequent twenty-four hours to assess activity. The number of beam breaks across the X–Y axis is measured to determine total activity (Kinder Scientific, Poway, CA and Software from MotoMonitor). These values were summed during the 12-hour light and 12-hour dark cycle for each mouse.
Endurance Capacity
Treadmill Running was used to measure endurance capacity as previously described [26] using a mouse treadmill (Columbus Instruments, Columbus, Ohio). Following an acclimation period of 10 minutes without treadmill movement, the treadmill was started at 7m/min for 5 minutes. The speed was increased to 12m/min for no longer than 2.5 hours. Animals were removed from the treadmill when they fell onto the encouragement platform and would not engage the treadmill with physical prodding. The experiment was performed by an individual blinded to the genotype of the animals.
Lactate measurements
Blood lactate was measured using the Lactate Plus lactate meter (NOVA Biomedical) with blood collected from the tail as we have previously described [26].
Grip Strength
A chatillion force gauge (Ametek) connected to a wire mesh was used to measure the grip strength of the animals. Each animal was grabbed by the tail and allowed to grip the wire mesh, once grip was established the animal was pulled back by its tail until the grip was lost. This was repeated 5 times per animal and the maximum grip strength was recorded. The experimenter was blinded to the genotypes of the animal.
2-D Echocardiography
Cardiac function was performed as previously described [29]. Mice were anaesthetized with isofluorane (0.5–2% in a 100% oxygen mix) and placed on an isothermal heat pad. Echocardiograms were performed using a Vevo 770™ High-Resolution In Vivo Imaging System (Visual Sonics), with heart rate continuously monitored throughout the experiment. Mice were given intraperitoneal injections of dobutamine (3µg/kg body weight) and 2-D echocardiography was performed prior to injection of dobutamine, and at 10 and 30 minutes following injection.
Cardiac Glucose Uptake
This experiment was performed as described by [30]. Mice were anaesthetized under 1.2% isofluorane throughout the measurements 0.5mCi of18 FDG dissolved in 1ml of physiologic saline solution was injected through the tail vein. 40 minutes were allowed for18 FDG uptake before scanning. The animal was then moved to the scanner bed (Focus 220 MicroPET, Siemens, Nashville, USA) and placed in the prone position. Emission data was acquired for 20 minutes in a three-dimensional (3D) list mode with intrinsic resolution of 1.5mm. For image reconstruction, 3D PET data was rebinned into multiple frames of 1s duration using a Fourier algorithm. After rebinning the data, a 3D image was reconstructed for each frame using a 2D filtered back projection algorithm. Decay and dead time corrections were applied to the reconstruction process. Because18 FDG is taken up by the whole body of the animal, we chose a “region of interest” (ROI) for our measurements that encompassed the whole heart, as outlined with a white line in Figure7, and determined CMRGlc for whole heart using the mean standardized uptake value (SUV) equation: SUV = (A×W)/Ainj, where A is the activity of the brain, W is the body weight of the mouse, and Ainj is the injection dose of18 FDG.
Figure 7. No difference in cardiac function between wild-type and Surf1−/− mice but enhanced glucose uptake in heart of Surf1−/− mice, in vivo.

For all graphs, white bars indicate wild-type and gray bars indicate Surf1−/− mice. A. Heart rate in beats per minute prior to and 10 and 30 minutes following treatment with dobutamine, n=4–5. B. End diastolic dimension as measured by 2-D echocardiography prior to and 10 and 30 minutes following dobutamine stress treatment. C. Fractional shortening as measured by 2-D echocardiography prior to and 10 and 30 minutes following treatment with dobutamine. D. In vivo cardiac metabolic rate of glucose (CMRGlc) maps of representative wild type and Surf1−/− mice obtained by PET. Circle indicates the location of the heart. Color indicates rate of18 FDG uptake, n=6. E. Quantification of in vivo metabolic glucose uptake. Data are means ± SEM. Significance was determined by 2-tailed t-test, *p<0.05.
Results
Cytochrome c oxidase activity is decreased in skeletal muscle and heart of Surf1−/−mice
To confirm the effect of loss of the Surf1 protein on ETC complex activities, mitochondria were isolated from heart and hind-limb skeletal muscle (gastrocnemius) and the enzymatic activities of complexes I, II, III and IV were measured. Compared to wild-type control littermates, loss of Surf1 resulted in a 71% and 53% decrease in COX activity in heart (Figure 1A) and skeletal muscle (Figure 1B), respectively. The activities of the other ETC complexes were not affected (Figures 1A, 1B).
Figure 1. Complex IV activity is specifically affected in Surf1−/− heart and skeletal muscle.

A. Enzymatic activity of complexes I, II, III, and IV in isolated heart mitochondria from wild-type (white bars) and Surf1−/− (gray bars) mice. Values are normalized to wild-type control for each assay. B. Enzymatic activity of complexes I, II, III, and IV in isolated skeletal muscle (SkM) mitochondria from wild-type (white bars) and Surf1−/− (gray bars) mice. N=5 per experimental group, data are means ± SEM and significance determined by 2-tailed t-test, *p<0.05.
State 3 respiration is decreased in heart, but not skeletal muscle, in mitochondria isolated from Surf1−/− mice
To determine the effect of diminished complex IV activity on mitochondrial respiration, we isolated mitochondria from heart and hind-limb skeletal muscle (gastrocnemius) of Surf1−/− mice and measured mitochondrial state 3 and state 4 respiration using a Clark type electrode. While state 3 respiration was modestly decreased in heart mitochondria (−16%) from Surf1−/− mice, it remained unchanged in skeletal muscle mitochondria compared to wild-type controls (Table 1). There was no significant difference in state 4 respiration between Surf1−/− and wild-type mice in either tissue. Respiratory control ratio (RCR), the ratio of state 3 to state 4 respiration and a measure of coupling of oxygen consumption to phosphorylation, was not affected between Surf1−/− and wild-type mice in heart or skeletal muscle mitochondria (Table 1). Additionally, the ADP to oxygen ratio (P:O ratio) was not significantly different between Surf1−/− and wild-type (Table 1). Together these data suggest there is no difference in the coupling efficiency of the mitochondria, despite a decrease in state 3 respiration in heart mitochondria.
Table 1. Isolated mitochondrial respiration in heart and skeletal muscle from Surf1−/− and wild-type mice.
State 3, state 4, respiratory control ratio (RCR) and ADP to oxygen ratio (P:O) were measured in isolated heart or hind-limb skeletal muscle mitochondria. N=6–10 per experimental group, data are means ± SEM and significance determined by 2-tailed t-test.
| Tissue | Genotype | State 3 | State 4 | RCR (State3/State4) |
ADP:O |
|---|---|---|---|---|---|
| Heart | Wild-type | 325±27 | 58±5 | 5.3±0.5 | 1.98±0.2 |
| Surf1−/− | 264±17 * | 59±6 | 4.8±0.6 | 2.10±0.2 | |
| Skeletal | Wild-type | 548±58 | 53±3 | 10.5±1.3 | 3.38±0.4 |
| Muscle | Surf1−/− | 527±44 | 52±3 | 10.7±1.6 | 2.93±0.4 |
ATP production is unaffected by COX deficiency in isolated mitochondria from heart and skeletal muscle in Surf1−/− mice
A major function of the ETC is production of ATP. Because Surf1−/− mice have a deficit in COX activity, we hypothesized this might result in a decrease in ATP production. To measure ATP production we used a luciferase-based assay with complex I- or complex II- linked substrates. Surprisingly, despite the reduction of COX activity, no significant difference in ATP production was found in isolated heart or skeletal muscle mitochondrial preparations from Surf1−/− mice compared to wild type mice (Figures 2A, 2B). These data are in agreement with our recently published work showing no significant difference in ATP production in isolated brain mitochondria in vitro and no difference in ATP levels in vivo of Surf1−/− brain [30].
Figure 2. Mild reduction of membrane potential in Surf1−/− heart but not skeletal muscle mitochondria and there is no effect on ATP production in either tissue.

A. ATP production in isolated heart mitochondria using either complex I-linked (pyruvate + malate) or complex II-linked (succinate +rotenone) substrates. B. ATP production in isolated skeletal muscle mitochondria using either complex I-linked (glutamate + malate) or complex II-linked (succinate +rotenone) substrates. C. Membrane potential was measured by the change in Safranin O fluorescence using either complex I-linked substrates (pyruvate +malate) or complex II-linked substrates (succinate + rotenone) in isolated heart mitochondria. Data are shown as fluorescent units ×1000. D. Membrane potential was measured by the change in Safranin O fluorescence using either complex I-linked substrates (glutamate +malate) or complex II-linked substrates (succinate + rotenone) in isolated skeletal muscle mitochondria. N=5–10 per experimental group, data are means ± SEM and significance determined by 2-tailed t-test, *p<0.05. pyr=pyruvate, mal=malate, suc=succinate, rot=rotenone
Membrane potential is decreased in heart, but not skeletal muscle mitochondria isolated from Surf1−/− mice
The ETC complexes I, III and IV pump protons from the mitochondrial matrix into the intermembrane space, creating an electrochemical gradient termed the membrane potential. This gradient is important for ATP production, mitochondrial calcium buffering, and protein import into the mitochondria. A previous study using primary neuron cultures from Surf1−/− and control mice did not find any difference in the membrane potential [2]. However, the membrane potential was not measured in other tissues or in isolated mitochondria. We measured membrane potential in heart and skeletal muscle mitochondria using the cationic fluorescent probe Safranin O that is accumulated and quenched inside energized mitochondria [31]. Membrane potential was significantly lower (−18%) in isolated heart mitochondria from Surf1−/− mice compared to wild-type mice (Figures 2C, 2D). This difference was observed using both complex I-linked substrates (pyruvate + malate) and complex II-linked substrates (succinate + rotenone) in heart mitochondria. However, no difference in membrane potential was found in skeletal muscle mitochondria isolated from Surf1−/− and wild-type mice in the presence of substrates for complex I-linked (glutamate + malate) or complex II-linked substrates (succinate + rotenone).
Mitochondrial ROS generation is not increased in heart or skeletal muscle mitochondria from Surf1−/− mice
Generation of reactive oxygen species (ROS) from the mitochondrial ETC complexes I and III can damage proteins, lipids and DNA and is thought to contribute to a number of deleterious phenotypes including aging [32]. We measured H2O2 and superoxide release from isolated mitochondria using the Amplex Red assay and electron paramagnetic resonance (EPR) with the spin trap DIPPMPO, respectively. In heart and skeletal muscle mitochondria, H2O2 production in mitochondria respiring on complex I-linked substrates (pyruvate + malate or glutamate + malate) was not different between Surf1−/− and wild-type mice. However, with complex II-linked substrate and inhibition of reverse electron transfer through complex I using rotenone (succinate + rotenone), both heart and skeletal muscle mitochondria from Surf1−/− mice showed a significant decrease (−23%, and− 7%, respectively) in H2O2 production compared to mitochondria from wild-type mice (Figures 3A, 3B).
Figure 3. ROS production is not increased in mitochondria isolated from Surf1−/− mice.
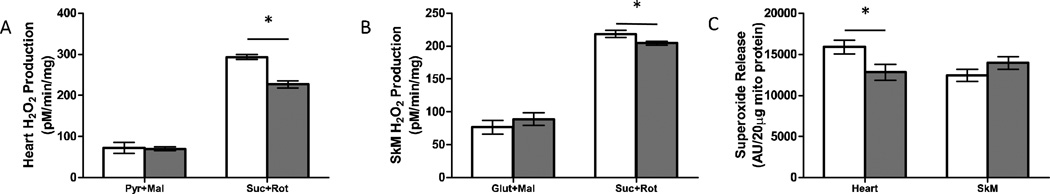
For all graphs pyr=pyruvate, mal= malate, suc=succinate, rot=rotenone, glut=glutamate, AntiA= antimycin A, SkM= skeletal muscle. White bars indicate wild-type and gray bars indicate Surf1−/− mice, n=6–10 for all assays. A. H2O2 production in isolated heart mitochondria assayed with complex I-linked substrates (pyr+mal) or complex II-linked substrates (suc+rot). B. H2O2 production in isolated hind-limb skeletal muscle mitochondria with complex I-linked (glut+mal) or complex II-linked substrates. C. Maximal superoxide production in isolated heart and skeletal muscle mitochondria. Data are shown as mean ± SEM and significance determined by 2-tailed t-test, *p<0.05.
We also measured maximal superoxide production using complex II-linked substrate and inhibitors (succinate + rotenone + antimycin A) using EPR (Figure 3C). This assay allows specific measurement of superoxide anion release from isolated mitochondria. Consistent with the observations from the H2O2 production assays, Surf1−/− heart mitochondria showed a significant decrease (−20%) in extra-mitochondrial superoxide release compared to wild-type mitochondria. However, superoxide release did not differ in skeletal muscle mitochondria from Surf1−/− and wild-type mice.
Surf1−/− mice have an increase in mitochondrial number in heart and skeletal muscle
PGC-1α is a transcriptional co-activator that is an important regulator of mitochondrial biogenesis that responds to a variety of stimuli including calorie restriction or exercise [18]. We hypothesized that diminished complex IV activity might induce mitochondrial biogenesis to compensate for deficits in mitochondrial function. To test this hypothesis, we determined whether PGC-1α levels were changed in Surf1−/− mice. We have previously shown that adipose tissue from Surf1−/− mice displays significantly increased levels of PGC-1α and increased mitochondrial biogenesis [33]. Here we found that PGC-1α protein is significantly upregulated in heart (66%) and skeletal muscle (2.3 fold) of Surf1−/− mice compared to littermate controls (Figure 4C,4D). The mRNA level of PGC-1α was significantly elevated in skeletal muscle (1.1 fold) but unchanged in the heart (Figure 4E). The mitochondrial biogenesis marker mitochondrial transcription factor A (Tfam) was increased in skeletal muscle (1.1 fold) but unchanged in heart. In addition, succinate dehydrogenase (complex II, 70 kDa subunit) in the inner mitochondrial membrane are significantly elevated in heart (43%) and skeletal muscle (70%). Voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane are increased in heart (2.6 fold) and in skeletal muscle (1.7 fold), further supporting an increase in mitochondrial number. In heart, we used electron microscopy (EM) to confirm the changes in mitochondrial biogenesis (Figure 4A). We found a 10% elevation in number and a 53% increase in total area of mitochondria from Surf1−/− heart EM sections (Figure 4B).
Figure 4. Mitochondrial number is increased in Surf1−/− mice heart and skeletal muscle.

A. Representative images of electron micrograph from heart and skeletal muscle from wild-type and Surf1−/− mice. N=3, images shown are 20,000× magnification. B. Quantification of mitochondrial size and number of 5 randomly chosen images per animal (n=3). C,D. PGC-1α, TFAM, porin (VDAC), Complex II (70kDa subunit) and Cytochrome C protein levels from wild-type and Surf1−/− heart (C.) and skeletal muscle (D.) tissue homogenates. Data are means ± SEM of relative density to β-tubulin loading control. Quantifications were done using NIH ImageJ software, n=6. E. Quantitative Real-time PCR of PGC-1α expressed relative to gamma-actin, n=7. Significance was determined by 2-tailed t-test, *p<0.05.
Skeletal muscle from Surf1−/− mice exhibits elevated UPRMT associated proteins
The mitochondrial unfolded protein response (UPRMT) was shown to be required for mediating the longevity phenotype of the C. elegans mitochondrial mutants [5,34]. We hypothesized that the UPRMT may also be upregulated in the Surf1−/− mice. To test our hypothesis we measured protein levels of UPRMT markers in heart and hind-limb skeletal muscle (Figures 5A, 5C). The markers used to measure UPRMT induction include hsp60, ClpP, Lon protease, thioredoxin 2 (Trx2), and transcription factor C/EBP Homologous Protein 10 (Chop). In heart tissue homogenates Lon (77%) and Trx2 (54%) were significantly up-regulated, but hsp60 or ClpP were not. Chop trended toward an increase in Surf1−/− heart but was not statistically significant (54%, p=0.1). However, in skeletal muscle homogenates, hsp60 (51%,), ClpP (57%), Lon (29%), and Chop (22%) were all found to be significantly up-regulated, consistent with the activation of UPRMT.
Figure 5. Induction of proteins involved in the UPRMT and Nrf2 pathway in heart and skeletal muscle from Surf1−/− mice.

Representative western blots and quantification of western blots of UPRMT associated proteins (A,C.) (Hsp60, ClpP, Lon protease, Thioredoxin 2 (Trx2), and Chop) or Nrf2 and heme oxygenase-1 (HO-1) (B,D.). White bars indicate values from wild-type mice and gray bars indicate values from Surf1−/− mice for heart (A,B.) and skeletal muscle (C,D.) tissue homogenates, n=4–6. Data shown are means ± SEM of relative densities of each protein assayed to β-tubulin loading control. Quantifications were done using NIH ImageJ software and significance determined by 2-tailed t-test, *p<0.05.
Surf1−/− heart does not show an induction in the mtUPR, but does show elevated Nrf2 and Heme Oxygenase-1 expression
Nuclear localization, and binding to the antioxidant response element (ARE) of Nrf2 results in the upregulation of genes including antioxidants and heme oxygenase-1 (HO-1). Induction of HO-1 has previously been shown to be cardioprotective and antiapoptotic following heart failure [35] and ischemia/reperfusion injury [36]. In heart tissue homogenates from Surf1−/− mice there was a 68% increase in Nrf2 protein expression compared to wild-type. Consistent with increased Nrf2 expression, HO-1 (82%) was significantly increased in Surf1−/− heart (Figure 5B). However, in skeletal muscle homogenates, the expression of Nrf2 and HO-1 did not differ between WT and Surf1−/− mice (Figure 5D).
In Surf1−/− mice, basal spontaneous activity is not affected; however, endurance capacity is significantly decreased and associated with increased blood lactate in response to moderate exercise
In order to determine whether the decrease in COX activity resulted in physiologic deficits, we measured both basal activity level and endurance capacity in wild-type and Surf1−/−mice. To measure basal activity level, open field activity with laser break counts was used. The number of laser breaks was summed during the 12-hour light and 12-hour dark cycle separately (Figure 6A). No significant differences in cage activity was observed between Surf1−/− and wild-type mice in either the light or the dark cycle suggesting that the Surf1 deletion does not affect basal activity level.
Figure 6. Surf1−/−mice show no difference in basal activity but detriments in grip strength and endurance capacity associated with elevated blood lactate.

A. Activity of wild-type (white bars) and Surf1−/− (gray bars) mice measured during the light and dark cycle over a 24-hour period. Activity was recorded as number of laser breaks and summed for each animal, n=8. Data are means ± SEM. B. Endurance capacity in wild-type (circles) and Surf1−/− (squares) mice. Each point represents a single animal and the time removed due to exhaustion, up to 150 minutes. Data is significant (p=0.0007) as determined by log-rank mantel-cox test; n=15–20. C. Blood lactate levels following moderate exercise. Wild-type and Surf1−/− mice had blood lactate readings measured prior to treadmill running at 12m/min and then measured immediately following 15 and 35 minutes of treadmill running. n=6–9. D. Grip strength for wild-type and Surf1−/− animals in grams force. For each animal, the value for grip strength was the best of five attempts, n=17 animals. Data are means ± SEM and are significant at each time point by 2-tailed t-test,*p<0.05.
To determine if the decrease in COX activity would result in decreased exercise performance, we subjected wild-type and Surf1−/− mice to endurance exercise on a mouse treadmill (Figure 6B). Surf1−/− mice performed significantly worse compared to their wild-type littermates (p<0.001). This suggests that while lower COX activity does not affect basal activity of the animal, endurance capacity is significantly limited.
Surf1−/− mice were previously shown to have a significant increase in blood lactate under basal conditions [2]. Consistent with their findings, we found that Surf1−/− mice had a significant increase in blood lactate under basal conditions (28%). Following treadmill running for 15 and 35 minutes, the Surf1−/− mice have a significantly greater increase in blood lactate (15 min: 55%; 35 min: 72%) compared to wild-type mice (Figure 6C). In addition, Surf1−/− mice also showed a significant decrease in grip strength (−13%) (Figure 6D). Together these data support a limiting effect in vivo of mitochondrial function in the Surf1−/− mice in response to the reduction COX activity.
Elevated glucose uptake and mitochondrial biogenesis may compensate for mitochondrial dysfunction and support normal cardiac function in Surf1−/− mice
Mitochondrial dysfunction in cardiac muscle has previously been shown to adversely affect cardiac function, namely endurance capacity. To determine whether the mild mitochondrial dysfunction in the Surf1−/− mice would adversely affect cardiac function under conditions of stress, we performed 2-D echocardiography before and 10 and 30 minutes after intraperitoneal injection of dobutamine, a β-adrenergic receptor agonist that stresses the heart by elevating heart rate. Heart rate, end diastolic dimension, and fractional shortening, were measured in wild-type and Surf1−/− mice (Figures 7A–7C). We found no significant differences in any of these measurements between wild-type and Surf1−/− mice and all animals displayed a normal response to dobutamine stress. In addition, we found no significant differences in end systolic dimension, septal wall thickness, or posterior wall thickness (data not shown). This suggests that Surf1−/− mice have normal cardiac function despite in vitro decreases in COX activity, mitochondrial respiration and membrane potential in isolated mitochondria. To address this discrepancy, we measured in vivo glucose uptake in the heart of Surf1−/− and wild-type animals. Surf1−/− mice have a significant increase in glucose uptake as determined by PET scan (33%), suggesting a potential mechanism by which Surf1−/− mice hearts compensate for sub-optimal mitochondrial function (Figures 7D and 7E).
Discussion
The loss of the Surf1 complex IV assembly protein in mice was previously shown to lower complex IV assembly, lower complex IV content and cytochrome oxidase activity and was unexpectedly associated with an increased median lifespan (~20% in both males and females) [2]. Because it is well established in C. elegans that ETC inhibition, including inhibition of complex IV, can result in increased lifespan, we reasoned that the Surf1−/− mice would provide an excellent opportunity to investigate potential mechanisms by which ETC inhibition might initiate protective mechanisms in a mammalian model. Here, using mitochondria from two highly energetic tissues, heart and skeletal muscle, we report that loss of Surf1 leads to induction of mitochondrial stress response pathways, including mitochondrial biogenesis, the UPRMT and Nrf2 activation.
Mice with a truncated Surf1 protein were originally generated to study Leigh’s syndrome, a human mitochondrial disorder that stems from loss of function mutations in the Surf1 gene and is characterized by neurologic deficits and early lethality. Surprisingly, mice lacking the Surf1 protein lack any debilitating phenotype and paradoxically have an increase in lifespan. A key difference between human Leigh's syndrome and Surf1−/− mice is the relative decline in COX activity. Leigh's syndrome patients can have a greater than 90% loss of COX activity while Surf1−/− mice have a less severe decline of COX activity ranging from 50–75% of wild-type values across a number of tissues. Although the Surf1−/− mice do show mild changes in mitochondrial function, we propose that a less severe reduction in complex IV activity might actually lead to induction of compensatory pathways that prevent the deleterious phenotypes present in Leigh’s syndrome.
In light of the dramatic deleterious phenotypes of mitochondrial mutations in humans, we were surprised to find that the indices of mitochondrial function we measured in isolated mitochondria from Surf1−/− mice (e.g., generation of ROS, membrane potential, ATP production and respiration) failed to show dramatic changes, despite a substantial decline in complex IV activity. The fact that there was no reduction in ATP production is consistent with previous observations in a complex I mitochondrial mutant mouse model (Ndufs4 knockout mice) that reported no change in ATP levels despite undetectable complex I activity in isolated liver mitochondria measured spectrophotometrically. Unlike Surf1−/− mice, the Ndufs4−/− mice exhibit a severe phenotype resulting in greatly reduced survival [37]. Although we did not find comprehensive mitochondrial dysfunction in the Surf1−/− mice, our results do show some mild alterations in mitochondria isolated from the heart in Surf1−/− mice, including a 16% decrease in state 3 respiration and a 19% decrease in membrane potential. Because of the involvement of complex IV in respiration, we hypothesized that a decrease in COX assembly and Complex IV activity would result in a decline in oxygen consumption. The lower state 3 respiration in heart is consistent with our hypothesis and with a previous study in isolated brain mitochondria from Surf1−/− mice [30]. However, the in vitro decline in state 3 respiration may not be indicative of respiration levels in vivo, highlighted by the fact that there is no difference in whole animal respiration of the Surf1−/− mice compared to wild-type [33]. Surprisingly, in skeletal muscle, indices of mitochondrial function from Surf1−/− mice were virtually indistinguishable from wild-type mice. The mechanism behind the differential responses in heart and skeletal muscle is still unclear, but could be the result of a threshold effect, as complex IV activity was reduced to a greater extent (>70% reduction) in heart than in skeletal muscle (~50%).
In skeletal muscle of Surf1−/− mice, decreased complex IV activity was associated with a significant decrease in grip strength but no change in muscle mass (data not shown), suggesting the muscles may be weaker. This phenotype is consistent with muscle weakness that occurs in humans with complex IV deficiency due to Surf1 mutations [1]. The Surf1−/− mice also show a pronounced reduction in endurance capacity that is associated with elevated levels of blood lactate prior to and after moderate exercise. Blood lactate during exercise is predominately produced from skeletal muscle and indicates a greater reliance on anaerobic glycolysis [38]. Surprisingly, despite a decrease in grip strength and endurance capacity in the Surf1−/− mice, there is no significant change in the overall basal activity level of the Surf1−/− mice, suggesting that the mitochondrial limitations are evident only under conditions of physiological challenge. These results and the data in heart mitochondrial function in vivo and in vitro in the Surf1−/− mice highlight the discrepancy between in vitro measures in isolated mitochondria and physiological parameters measured in vivo. Despite only mild alterations in Surf1−/− skeletal muscle mitochondrial function in vitro, exercise tolerance was clearly limited in vivo.
Our previous data suggest that the reduction in complex IV activity results in up-regulation of compensatory responses that are actually beneficial. For example, in a recent study we reported that in brain loss of Surf1 leads to decreased respiration and increased ROS generation in isolated brain mitochondria. However, these mild deficits were associated with elevated brain glucose metabolism, increased cerebral blood flow and surprisingly enhanced memory [30]. We have also reported that loss of Surf1 lead to key metabolic changes resulting in reduced adiposity, increased insulin sensitivity and induction of mitochondrial biogenesis in adipose tissue [33]. These findings suggest that mild mitochondrial dysfunction from loss of Surf1 may in fact have positive effects overall, and is consistent with the mitohormesis hypothesis that suggests mild mitochondrial dysfunction could be beneficial because it results in compensatory responses that overcome the deleterious effects [39].
Consistent with this idea, our current findings show that a number of stress response pathways including mitochondrial biogenesis, the UPRMT, and the Nrf2 pathway, pathways known to respond to mitochondrial stress, are elevated in Surf1−/− mice. For example, mitochondrial biogenesis has previously been shown to be elevated in response to energy stress conditions such as calorie restriction and endurance exercise in mice [18]. In C. elegans mitochondrial mutants, lifespan extension is limited to inhibition of the ETC during the L3/L4 stage of development [7] during which mitochondrial biogenesis occurs [19], supporting a potential link between mitochondrial biogenesis and lifespan extension. This is consistent with elevated levels of PGC-1α in response to caloric restriction [40] which is linked to increased lifespan in a number of species. Mitochondrial biogenesis in mice overexpressing PGC-1α specifically in muscle is also associated with changes in insulin sensitivity and increased lifespan, further supporting a role for mitochondrial biogenesis in longevity[41,42]. In the Surf1−/− mice, markers of mitochondrial biogenesis are elevated in heart and skeletal muscle compared with wild-type mice. These data suggest that increasing the number of mitochondria is an important compensatory response to energy stress caused by Surf1 deficiency.
Nuclear factor-erythroid 2 p45-related factor 2 (Nrf2) is a transcription factor that coordinates the expression of ~200 genes involved in drug detoxification, endogenous antioxidant production, NADPH regeneration, and metabolism. Under basal conditions, Nrf2 is constitutively targeted for degradation by the proteasome. Following stress, however, Nrf2 binds the antioxidant response element resulting in the upregulation of cytoprotective genes. Among the genes upregulated by Nrf2 is heme oxygenase-1 (HO-1), which catalyzes the degradation of heme to ferrous iron, carbon monoxide, and biliverdin. These products are important in mediating the anti-oxidant, anti-inflammatory and anti-apoptotic effects of HO-1 upregulation. Overexpression of HO-1 specifically in heart has been shown to be cardioprotective in response to ischaemia/reperfusion injury [36].Because of the role of Nrf2 and HO-1 in antioxidant defense and cardioprotection, we asked whether Nrf2 and HO-1 were altered in the Surf1−/− mice. Interestingly, protein levels of Nrf2 and heme-oxygenase 1 (HO-1) are increased in heart. The combined increase in the mitochondrial number, HO-1, and 33% enhanced glucose uptake we found in heart may compensate for the decline in function of the individual mitochondrion and allows for normal heart physiologic function.
Initiation of the UPRMT results in the upregulation of mitochondrial specific chaperones and proteases that aim to maintain mitochondrial proteostasis by refolding or degrading unfolded proteins. The UPRMT was shown to be required for the lifespan extension of the cco-1 complex IV mitochondrial mutant in C. elegans [5]. Furthermore, disruption of mitochondrial ribosomal protein S5 in C. elegans resulted in decreased mitochondrial respiration and induced the UPRMT [34]. This compensatory response was attributed to a mitonuclear protein imbalance and also resulted in enhanced longevity of C. elegans, and showed similar increases in mammalian cell culture. These data, along with the data from Surf1−/− animals, add further evidence to the UPRMT being an evolutionarily conserved longevity response and merits further investigation. We find that some but not all proteins involved in the UPRMT are elevated in the heart. The most commonly used markers for UPRMT induction are Hsp60 and ClpP, however neither were upregulated in the heart of Surf1−/− mice. At this point it is unclear whether the increase in the UPRMT in response to COX deficiency is beneficial to skeletal muscle function. This tissue specificity of the UPRMT induction has important implications and merits further research into this pathway. While it may be important in maintaining the mitochondrial function under conditions of mitochondrial dysfunction, it is unclear what the physiological outcomes of this response are or what affect the UPRMT has on the lifespan of mammalian mitochondrial mutants. These results are significant because they provide evidence for an evolutionarily conserved stress response between invertebrates and mammals (UPRMT). Furthermore, this is the first report of the UPRMT in a mammalian in vivo model of mitochondrial ETC insufficiency. Studies are ongoing to define the potential role of the UPRMT in the longevity of the Surf1−/− mouse.
Acknowledgements
We received assistance from the Cardiac proteomics core (Merry Lindsey Laboratory) and Barbara Hunter from the electron microscopy core at UTHSCSA. We thank Drs. Luke Szweda and Scott Plafker, Oklahoma Medical Research Foundation, for providing the Lon protease and Nrf2 antibodies, respectively.
Funding: This work was supported by an Ellison Medical Foundation Senior Scholar award to Holly Van Remmen and the Biology of Aging Training Grant (T32 AG021890) to Daniel Pulliam. In vivo imaging studies were supported by NIH grant K01AG040164 and American Federation for Aging Research grant #A12474 to Ai-Ling Lin.
References
- 1.Wedatilake Y, Brown R, McFarland R, Yaplito-Lee J, Morris AA, Champion M, Jardine PE, Clarke A, Thorburn DR, Taylor RW, et al. SURF1 deficiency: a multi-centre natural history study. Orphanet. J. Rare Dis. 2013;8:96. doi: 10.1186/1750-1172-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Human Molecular Genetics. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 3.Wong A, Boutis P, Hekimi S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics. 1995;139:1247–1259. doi: 10.1093/genetics/139.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila Life Span by RNAi of the Mitochondrial Respiratory Chain. Curr. Biol., Elsevier Ltd. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cristina D, Cary M, Lunceford A, Clarke C, Kenyon C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000450. doi: 10.1371/journal.pgen.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007;5:e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell. 2010;9:433–447. doi: 10.1111/j.1474-9726.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- 9.Pulliam DA, Bhattacharya A, Van Remmen H. Mitochondrial Dysfunction in Aging and Longevity: A Causal or Protective Role? Antioxid. Redox Signal. 2012;19:1373–1387. doi: 10.1089/ars.2012.4950. [DOI] [PubMed] [Google Scholar]
- 10.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 11.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J. Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 14.Baker MJ, Tatsuta T, Langer T. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of Genes Activated by the Mitochondrial Unfolded Protein Response (mtUPR) and Cognate Promoter Elements. PLoS ONE. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE. 2007;2:e835. doi: 10.1371/journal.pone.0000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- 19.Tsang WY, Lemire BD. Mitochondrial genome content is regulated during nematode development. Biochem. Biophys. Res. Commun. 2002;291:8–16. doi: 10.1006/bbrc.2002.6394. [DOI] [PubMed] [Google Scholar]
- 20.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Muller FL. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. Journal of Biological Chemistry. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 22.Pérez VI, Lew CM, Cortez LA, Webb CR, Rodriguez M, Liu Y, Qi W, Li Y, Chaudhuri A, Van Remmen H, et al. Thioredoxin 2 haploinsufficiency in mice results in impaired mitochondrial function and increased oxidative stress. Free Radic. Biol. Med. 2008;44:882–892. doi: 10.1016/j.freeradbiomed.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 23.Bhattacharya A, Lustgarten MS, Shi Y, Liu Y, Jang YC, Pulliam D, Jernigan AL, Van Remmen H. Increased mitochondrial matrix-directed superoxide production by fatty acid hydroperoxides in skeletal muscle mitochondria. Free Radic. Biol. Med. 2011;50:592–601. doi: 10.1016/j.freeradbiomed.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker D, Walker R. The use of the oxygen electrode and fluorescence probes in simple measurements of photosynthesis. 1987 [Google Scholar]
- 25.Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. Journal of Neurochemistry. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 26.Lustgarten MS, Jang YC, Liu Y, Muller FL, Qi W, Steinhelper M, Brooks SV, Larkin L, Shimizu T, Shirasawa T, et al. Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. A.J.P.: Cell Physiology. 2009;297:C1520–C1532. doi: 10.1152/ajpcell.00372.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lustgarten MS, Bhattacharya A, Muller FL, Jang YC, Shimizu T, Shirasawa T, Richardson A, Van Remmen H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012;422:515–521. doi: 10.1016/j.bbrc.2012.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, Diaz V, Sloane L, Maslin K, Treaster S, et al. Rapamycin extends life and health in C57BL/6 mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:119–130. doi: 10.1093/gerona/glt056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo SM, Dai Q, Zhang J, Jin Y-F, Lindsey ML. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc. Res. 2012;96:444–455. doi: 10.1093/cvr/cvs275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin A-L, Pulliam DA, Deepa SS, Halloran JJ, Hussong SA, Burbank RR, Bresnen A, Liu Y, Podlutskaya N, Soundararajan A, et al. Decreased in vitro mitochondrial function is associated with enhanced brain metabolism, blood flow, and memory in Surf1-deficient mice. J. Cereb. Blood Flow Metab. 2013;33:1605–11. doi: 10.1038/jcbfm.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akerman KE, Wikström MK. Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 1976;68:191–197. doi: 10.1016/0014-5793(76)80434-6. [DOI] [PubMed] [Google Scholar]
- 32.Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech. Ageing Dev. 2004;125:811–826. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 33.Deepa SS, Pulliam D, Hill S, Shi Y, Walsh ME, Salmon AB, Sloane L, Zhang N, Zeviani M, Viscomi C, et al. Improved insulin sensitivity associated with reduced mitochondrial complex IV assembly and activity. The FASEB Journal. 2013;27:1371–1380. doi: 10.1096/fj.12-221879. [DOI] [PubMed] [Google Scholar]
- 34.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature, Nature Publishing Group. 2014;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang G, Hamid T, Keith RJ, Zhou G, Partridge CR, Xiang X, Kingery JR, Lewis RK, Li Q, Rokosh DG, et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation. 2010;121:1912–1925. doi: 10.1161/CIRCULATIONAHA.109.905471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vulapalli SR, Chen Z, Chua BHL, Wang T, Liang C-S. Cardioselective overexpression of HO-1 prevents I/R-induced cardiac dysfunction and apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H688–H694. doi: 10.1152/ajpheart.00133.2002. [DOI] [PubMed] [Google Scholar]
- 37.Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabrera ME, Saidel GM, Kalhan SC. Lactate metabolism during exercise: analysis by an integrative systems model. Am. J. Physiol. 1999;277:R1522–R1536. doi: 10.1152/ajpregu.1999.277.5.R1522. [DOI] [PubMed] [Google Scholar]
- 39.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. Elsevier Inc. 2011;51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Anderson RM, Barger JL, Edwards MG, Braun KH, O'Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA. 2009;106:20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]