

Abstract
Dyskeratosis congenita (DC) is an inherited bone marrow failure (BMF) syndrome characterized by the classic triad of abnormal skin pigmentation, nail dystrophy, and oral leukoplakia. However, patients usually develop BMF and are predisposed to cancer, with increased risk for squamous cell carcinoma and hematolymphoid neoplasms. DC is a disease of defective telomere maintenance and is heterogeneous at the genetic level. It can be inherited in X-linked, autosomal dominant, or autosomal recessive patterns. Mutations in at least ten telomere- and telomerase-associated genes have been described in DC. There are no targeted therapies for DC and patients usually die of BMF due to a deficient renewing capability of hematopoietic stem cells. Allogeneic hematopoietic stem cell transplantation is the only curative treatment for BMF.
Keywords: dyskeratosis congenita, diagnosis, genetics, clinical, treatment
Introduction
Dyskeratosis congenita (DC) is an inherited bone marrow (BM) failure (BMF) syndrome characterized by abnormal skin pigmentation, nail dystrophy, oral premalignant leukoplakia, BMF, and cancer predisposition, with increased risk for squamous cell carcinoma and hematolymphoid neoplasms.1 The disease was first described by Zinsser in 1906 and was recognized as a clinical entity by Engman (1926) and Cole (1930).2 DC patients suffer premature morbidity most commonly from BMF, which affects 80%–90% of cases by age 30 years and is the leading cause of death.3,4 Pulmonary fibrosis and cancer are severe complications during the evolution of the disease.4
DC is heterogeneous at the genetic level, depending on the affected gene. DC can be inherited in X-linked, autosomal dominant (AD), or autosomal recessive (AR) patterns.2 Mutations in at least ten telomere- and telomerase-associated genes have been linked to DC, although the genetic basis of the disease is still undetectable in approximately 30%–40% of DC cases. DC is a disease of defective telomere maintenance, and patients with DC have premature telomere shortening and subsequent replicative senescence, leading to premature stem cell exhaustion and tissue failure.5,6 Telomere maintenance is closely associated with life events, including aging and cancer predisposition, which is the reason why DC has been receiving increased attention. However, mutations in telomerase and telomere components have also been identified in patients with aplastic anemia, pulmonary fibrosis, and liver diseases.5
There are no targeted therapies for DC. DC patients usually die of BMF due to deficient renewing capability of hematopoietic stem cells (HSCs).7 Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the only curative treatment for BMF in patients with DC.8 Historically, HSCT was associated with significant morbidity and mortality for these patients. However, the utilization of new transplant regimens with better tolerance is resulting in improved outcomes.3
Here we present a review of the clinical aspects, role of telomerase, inheritance of DC, genetic anticipation, diagnosis, and treatment of this disorder.
Clinical aspects
Epidemiology and clinical manifestations
DC is a rare disease with an estimated annual incidence of <1 in 1 million, with multiple and variable clinical manifestations.9,10 Genotype–phenotype correlations are complex due to several factors, including a variety of underlying hypomorphic gene mutations, disease anticipation, and genetic and environmental modifier effects.11
The group of diseases referred to by the term “dyskeratosis congenita” has expanded considerably since its initial description. The classic and initial form is usually characterized by the mucocutaneous triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia (Figure 1). Subsequently, new cases were identified and showed that DC could affect multiple systems of the body, particularly and severely the BM.9 DC occurs mostly in males and clinically manifests between 5 years and 12 years.12 Patients with DC have been shown to have disease diversity in terms of age at onset, symptoms, and severity. Even if the patients have the same gene mutation, the manifestations of the disease are variable, which is why sometimes it is difficult to make a correct diagnosis.13 Due to the heredity pattern, females may have less severe clinical features.14
Figure 1.
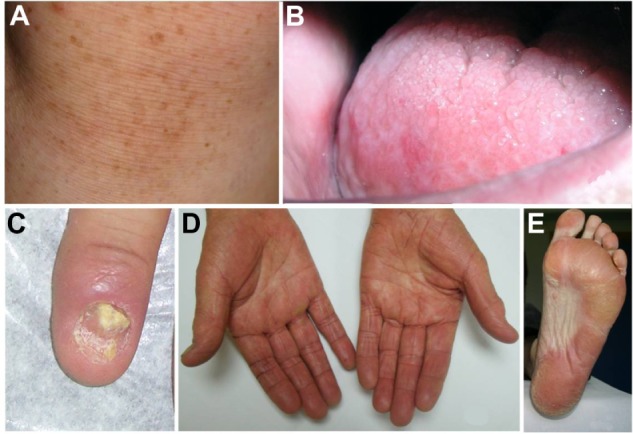
Clinical findings.
Notes: (A) Abnormal skin pigmentation. (B) Leukoplakia. (C) Nail dystrophy. (D and E) Hyperkeratosis and hyperpigmentation of the palms and soles.
Approximately 90% of patients exhibit nail dystrophy, which affects the fingernails first and then the toenails in most cases. Nail dystrophy begins with ridging and longitudinal splitting and progresses, resulting in small, rudimentary, or absent nails. Mucosal leukoplakia is a pathognomonic feature and occurs in approximately 80% of patients. It typically involves the buccal mucosa, tongue, and oropharynx. The treatment of these lesions is symptomatic. However, approximately 30% of the leukoplakic areas progress to malignant transformation with the development of a squamous cell carcinoma in 10–30 years, so they require frequent monitoring14 and biopsy of suspicious areas in order to detect a possible oral cancer.
In DC, severe periodontal destruction occurs due to anomalies in ectodermal derived structures and a poor response in the patient caused by neutropenia. Patients may have gingival inflammation, bleeding, recession, and bone loss that simulate juvenile periodontitis.12 In addition, there may be defects in decreased root/crown ratio and mild taurodontism. The evidence of multiple permanent teeth with decreased root/crown ratios may suggest a diagnosis of DC. These patients also may have more incidence of and more severe periodontal disease.14
The recognition of DC as a multisystem disorder includes additional “minor” features like intrauterine growth retardation; developmental delay; microcephaly; abnormalities involving the eyes and hair, including premature greying; excessive sweating; short stature; hypogonadism; enteropathy; liver disease; esophageal and urethral stenosis; osteoporosis; and avascular necrosis of the hips and shoulders.3 Ophthalmic manifestations of DC include blepharitis, conjunctivitis, nasolacrimal duct obstruction, ectropion, entropion, and trichiasis. Retinal changes are rare and include hemorrhages, nerve fiber layer infarction, arteriosclerosis, macular edema, preretinal fibrosis, and optic atrophy.15
These heterogeneous clinical manifestations affect patients in different ways with a variable percentage. However, the classical features are the most frequent (Table 1) with other somatic features indicated.9,11
Table 1.
Multisystem clinical features of dyskeratosis congenita
| Major clinical features | Approximate % of patients |
|---|---|
| Mucocutaneous triad | |
| Abnormal skin pigmentation | 90 |
| Nail dystrophy | 90 |
| Leukoplakia | 80 |
| Bone marrow failure | 90 |
Clinical variants of DC
Clinical variants of DC have been described: Revesz syndrome, Hoyeraal–Hreidarsson syndrome (HHS), and Coats plus disease or cerebroretinal microangiopathy with calcifications and cysts.16 The defining feature of Revesz syndrome is bilateral exudative retinopathy, occurring in most cases in association with intracranial calcification and more classical features of DC such as BMF, mucocutaneous abnormalities, intrauterine growth retardation, cerebellar hypoplasia, and developmental delay.
HHS is a severe form of DC characterized by intrauterine growth retardation, microcephaly, cerebellar hypoplasia, moderate to severe mental retardation, progressive immunodeficiency, and pancytopenia. A diagnosis of HHS could be made if the patient has four or more of these or has cerebellar hypoplasia and additional features of DC. In these cases, the mucocutaneous features of DC and cerebral calcifications may also be present.3
Summary of the evolution of DC
During the evolution of the disease, the abnormal skin pigmentation and nail changes usually appear first, often below the age of 10 years, and the patients usually develop BMF below the age of 20 years. Up to 80% of patients show signs of BMF by the age of 30 years. However, sometimes the manifestation of the disease is ambiguous, and BMF or another abnormality in another system could appear before the classic mucocutaneous features. This is being recognized in more cases with the advances in genetic diagnosis techniques.9
DC-related BMF
The BM findings in DC are variable and range from normal to different severity of aplasia depending on the stage of the disease. Sometimes it is indistinguishable from aplastic anaemia due to other causes.17 Aplastic anemia occurs in patients with a median age of presentation of 11 years. The anemia is macrocytic. Fetal hemoglobin levels are increased.
Approximately 90% have peripheral cytopenia of one or more lineages.14 Initially, the patient usually presents thrombocytopenia, and during the evolution of the disease this becomes more global and they develop severe BMF. BMF (in up to 80% of patients) is one of the major causes of premature mortality in DC due to opportunistic infections because of the significant reduction in mature blood cells.9,12 The BM abnormalities can progress in different forms with the appearance of myelodysplasia in one or more lineages or leukemia.9 It has been reported that patients with DC have an observed expected ratio of 2,663 to develop myelodysplasia (95% confidence interval 858–6,215), with a mean age of onset of 35 years.18
DC-related cancer and malignant transformation
Due to the etiology of DC, patients show signs of premature aging and they are more susceptible to developing cancers.7 There is the presence of an excessive telomere shortening in this patient population, which in the absence of a deoxyribonucleic acid (DNA) damage response may lead to genomic instability and a predisposition for malignant transformation.19
Electron microscopy studies revealed that cells in DC have an embryonic immature nucleus and have a predisposition to undergo malignant transformation. In addition, the barrier zone of epithelium is less effective in DC than in the normal epithelium, so there is an increased permeability of noxious substances and carcinogens to the germinal layers. Therefore, an increased malignant transformation rate is seen in leukoplakic areas, so they need to be monitored periodically.12
In addition, individuals with DC have a 40%–50% cumulative incidence of risk of malignancy by age 50 years. For example, squamous cell carcinoma of the tongue, Hodgkin’s lymphoma, adenocarcinoma of the gastrointestinal tract, and bronchial and laryngeal carcinoma can develop. The skeletal, gastrointestinal, and genitourinary systems also may be affected.14
Pulmonary fibrosis and hepatic complications
In childhood, BMF is the most frequent complication of DC, whereas pulmonary fibrosis is a frequent cause of mortality in adults.20 Pulmonary fibrosis, characterized by loss of lung epithelium, prominent fibrosis, and impaired gas exchange,21,14 and abnormalities of pulmonary vasculature are seen in about 80% of cases.14
Hepatic complications have been described, too. Some studies reported that loss-of-function telomerase gene variants are risk factors for sporadic cirrhosis. However, there is not a definitive conclusion that can explain the appearance of hepatic complications in DC. The possible hepatic complications can show diverse histological liver features such as necrosis, fibrosis, cirrhosis, inflammation, and hyperplasia, although hepatic lesions are not a frequent complication in DC (approximately 7%).11
Telomerase
Telomere is the end of a chromosome. Both ends of all chromosomes end with six rich repeats in the 5′-3′ strand in every eukaryote. Every vertebrate has (TTAGGG)n repeats. In humans, (TTAGGG)n repeats are about 15–20 kb in length at birth and then about 8–10 kb in adults, but the length varies among chromosomes. Telomeres protect chromosome ends from DNA degradation, DNA repair mechanisms, and fusion.19,22,23
Telomeres in mammals are formed by tandem repeats of the TTAGGG sequence, which is bound by a six-protein shelterin complex that protects chromosomes and regulates telomerase activity at the ends of chromosomes.22 A DNA damage response at chromosome ends is triggered by excessive telomere shortening and severe telomere uncapping, recognized as double-strand breaks. Depending on the integrity of the DNA damage response, dysfunctional telomeres can lead to either cancer or aging pathologies. The shelterin complex has been proposed to modulate telomerase activity at chromosome ends. The shelterin complex is composed of six core proteins, telomeric repeat binding factor 1 (TRF1; also known as TERF1) and TRF2 (also known as TERF2), TRF1-interacting nuclear factor 2 (TIN2), protection of telomeres (POT1), TIN2-interacting protein 1 (TPP1) (also known as TINT1, PToP, and PIP1), and RAP1. A role beyond its role in telomeres has been shown for RAP1, a shelterin component. RAP1 in mammals is involved in subtelomeric gene silencing and transcriptional regulation, and it also acts as an essential modulator of the nuclear factor-κB (NF-κB)-mediated pathway. Cells and mice deficient for telomerase or some of the shelterin proteins support a model in which telomere dysfunction, either owing to the loss of telomeric repeats or owing to the loss of the telomere protective structure, causes genome instability and thereby affects tumorigenesis. It is conceivable that the expression or function of these proteins is differentially regulated during embryogenesis, as well as during nuclear reprogramming, to contribute to telomere rejuvenation. Shelterin components are mediators of telomere length and are required for proper telomere capping. Although the regulation of shelterin components in stem cells and during nuclear reprogramming is still unexplored, some recent evidence suggests shelterin components as key factors in “stemness”. Indeed, some cases of premature aging in human syndromes have been linked to shelterin mutations, such as in TRF1, TRF2, and TIN2 (also known as TINF2). Further investigation is needed to fully establish the molecular roles and regulation of shelterin components during embryonic development, aging, and nuclear reprogramming.22
Telomere length is a marker for cell aging, senescence, replicative capacity and shorten with every cell division. In human cells, defects in telomeres represent a unique form of DNA damage in which repair mechanisms are very limited. In the pathologies with abnormal shortened telomeres, determinate cells become senescent and ultimately die. But, in other situations, cells with critically short telomeres may bypass senescence and continue to divide, despite the presence of genetic instability, which results in malignant transformation of them. This is often associated with alterations in the p53 or Rb pathways.9,24 Telomeres shorten with age and, as a consequence, cell proliferation is limited.22,25
The well-known function of telomerase is elongation of telomeres so that cells can increase their replicative capacity, sometimes indefinitely. Telomerase is a ribonucleic acid (RNA)-dependent DNA polymerase that synthesizes telomeric repeats. This enzyme complex has a catalytic protein with telomere-specific reverse transcriptase (TERT) activity, an internal RNA template strand (TERC), and a group of associated proteins. The lifelong maintenance of multiple cells, including HSCs, depends on the correct function of telomerase.7,26 Telomerase is mainly restricted to cells such as germ cells, stem cells, and their immediate progeny, activated T-cells and monocytes.9,22
Telomerase is expressed in 80%–90% of human tumors,27 where it appears critical for endowing cancers with unlimited growth potential. However, whether telomerase is upregulated during carcinogenesis or is expressed in the cell of origin for cancers remains unresolved. Diverse studies postulate that the mechanisms underlying this strong cancer predisposition may relate to cycles of chromosome fusion–bridge–breakage and chromosomal rearrangements driven by telomere dysfunction and the ability of uncapped telomeres to promote tetraploidization and aneuploidy.21,22 In addition, it has been observed that chronic oxidative stress can damage telomeres. Telomere DNA is susceptible to oxidative stress because it is single stranded, and the sequence also has high content of guanine.28
Inheritance of DC
DC can be inherited in one of three forms, X-linked, AD, or AR. De novo germline mutations are also relatively frequent in DC, and, to date, about 70% of DC patients have an identifiable germline mutation.3,16,29
All mutations identified to date in DC patients affect telomerase components or telomere-stabilizing components with the alteration of the normal function of the enzyme. Therefore, these defects in telomere biology affect the renewing capabilities of HSCs.7 The result of these mutations is the presence of very short telomeres. In addition, some studies have suggested that in the disease pathology it is important that cells with DC-relevant mutations have an increased DNA damage response, which has previously been ignored.30
To date, ten genes (DKC1, TERC, TERT, TINF2, NOP10, NHP2, TCAB1, C16orf57, RTEL1) have been identified; nine of these are important in telomere maintenance (Table 2).6 The first DC-associated gene, X-linked DKC1, was discovered by linkage analysis in 1998.16,18 With the identification of mutations in DKC1, the first diagnostic tests became available. DKC1 (DC gene 1 at Xq28) mutations are the most frequent,9 appearing in 30% of DC patients. The DKC1 gene encodes the nucleolar protein dyskerin, which is ubiquitously expressed, and this protein is involved in telomere maintenance and ribosomal biogenesis. Mutation in DKC1 results in dysfunction of dyskerin.15,26
Table 2.
Genetic abnormalities associated with dyskeratosis congenita (DC)
| Gene | Chromosome | Product | Function | Mutation comments | Inheritance | % of cases |
|---|---|---|---|---|---|---|
| DKC1 | Xq28 | Dyskerin | Telomere maintenance and ribosomal biogenesis | Concentration of mutations in N-terminus. Many mutations result in exon 15 loss | XLR | Approximately 40% |
| TERT | 5p15.33 | TERT | Telomere maintenance | Located throughout gene | AD, AR | <5% |
| TERC | 5q21–3q28 | TERC | Telomere maintenance | Located throughout gene. No DC mutations in N-terminus | AD | Approximately 5% |
| TINF2 | 14q12 | TIN2 | Telomere maintenance | All mutations concentrated in exon 6a | AD, S | <1% |
| NOP10 | 15q14 | NOP10 | Telomere maintenance | Located in exon 2 | AR | <1% |
| NHP2 | 22q13.2 | NHP2 | Ribosomal biogenesis | All mutations concentrated in exon 4 | AR | <1% |
| TCAB1 | 17p13.1 | TCAB1 | Telomere maintenance | Located in exon 2, 7, 8, and 9 | AR | <1% |
| C16orf57 | C16orf57 | Located in exon 7 | AR | 2% | ||
| Uncharacterizeda | 40% |
Note:
These are likely to represent more than one genetic locus and include the genetically heterogeneous AR DC.
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; S, sporadic; XLR, X-linked recessive.
Subsequently, mutations in the TERT gene have been reported in 1%–7% of cases, being heterozygous mutations in the AD variant of DC, or biallelic mutations in the recessive variant of DC.10 TERT encodes the enzymatic component of the telomerase complex. Patients with TERT mutations have a very variable clinical presentation, ranging from a nearly DC phenotype to just aplastic anemia.9 However, approximately 3% of sporadic aplastic anemia and idiopathic pulmonary fibrosis can also carry mutations in TERC and TERT.31 These findings unite this wide spectrum of different diseases under the unique concept of DC-associated telomere biology disorders.16
It has been described how AD mutations in TINF2, which encodes the shelterin protein TIN2, were suggested to cause the disease in <11% of patients,10 with high penetrance and an early age of onset.18 The shelterin complex determines the structure of the telomeric terminus, generates T-loops, and controls the synthesis of telomeric DNA by telomerase. If the shelterin has defects in its function, there is an inadequate damage response, and chromosome ends are incorrectly processed by the DNA repair pathways. The shelterin complex is composed of six proteins: TRF1, TRF2, TIN2, TERF2-interacting protein (RAP1), TPP1, and POT1. TIN2 maintains the composition and protein interaction of the different components of the shelterin complex. Patients with TIN2 mutations develop severe forms of the disease with very short telomere lengths.9
NOP10 is an AR DC gene. Patients with mutations in NOP10 have reduced telomere length and reduced TERC levels. Biallelic mutations in NHP2 have been identified in a group of AR DC patients, too. Both NOP10 and NHP2 are components of the H/ACA ribonucleoprotein complex (H/ACA RNP). This complex is composed of an RNA molecule and four proteins: dyskerin, GAR1, NOP10, and NHP2. These four proteins are highly conserved and have been implicated in ribosome biogenesis, pre-mRNA splicing, and telomere maintenance. In DC, mutations have been discovered in all the components of the H/ACA RNP complex, except for GAR1.9,11
TCAB1 is another AR DC gene. TCAB1 is a telomerase holoenzyme protein that facilitates trafficking of telomerase to Cajal bodies, the nuclear sites of nucleoprotein complex modification and assembly. Compound heterozygous mutations in TCAB1 cause alteration of the nuclear localization of telomerase, so it cannot elongate telomeres, thereby resulting in short telomeres.9,11
Biallelic mutations have been described in the C16orf57 gene. The peculiarity of the patients with these mutations is that they have normal length telomeres, so they represent a biologically different subtype of the disease. The precise function of the C16orf57 gene remains unknown. Clinically, patients with C16orf57 mutations can be identical or similar to those with genetic defects in telomere length maintenance.9,10,16,26 A possible explanation for this is that C16orf57 mutations result in an independent telomere length defect that causes a telomere dysfunction, resulting in a cellular and tissue pathology similar to that in the context of DC with critically short telomeres.3
In addition, there has been the discovery of an intronic splice variant in Apollo (encoded by DCLRE1B) in a patient with HHS and normal telomeres. On account of this finding, the role of Apollo in telomere biology is currently under investigation.16,32
Biallelic mutations of the conserved telomere maintenance component gene have also been described. This gene was originally described as causative gene of the Coats plus syndrome, which is a form of cerebroretinal microangiopathy with calcifications and cysts. However, the frequency of mutations of these new genes has not been established for DC.8,33
Recently, RTEL1 germline mutations have been reported in patients with a severe variant of DC, HHS, performing techniques like whole genome linkage analyses and exome sequencing.34 Most of the RTEL1 mutations appear to be AR, but AD mutations have been reported.16 RTEL1 (regulator of telomere elongation helicase 1) is an essential DNA-helicase, with implication in telomere length regulation and in DNA repair, processes that are critical for the maintenance of genome integrity.34 RTEL1 also functions as an antirecombinase, in that it disrupts D-loop formation during homologous recombination and is essential for the disassembly of T-loops during DNA replication. In mice it has been shown that it participates in genome stability because its knockout causes embryonic death, telomere loss, and chromosome fusions.35 However, its mechanism of action is still unknown and whether it regulates telomere length in human cells.25
Genetic anticipation
In addition to the different modes of DC inheritance, genetic anticipation has been reported in TERC, TERT, and TINF2 pedigrees.36–38 This phenomenon is marked by increasing severity in the clinical phenotype and shorter telomeres with each successive generation. Haploinsufficiency has been described for telomerase in families with mutations in the telomerase RNA component, like a mechanism that produces a progressive telomere shortening with anticipation.36
The phenotype characteristics of DC are variable even between family members with the same mutation, and they can present differences in the penetrance, severity, and development of clinical features. According to the inheritance of the disease, the variability in healthy individuals depends more on telomere length of paternal chromosomes in the zygote than on telomere length of maternal chromosomes.
The importance of disease anticipation is reflected in the possible situation that a mutation-positive person with an unaffected phenotype can have affected sons.38 However, we cannot rule out the presence of other disease-modifying factors. Correlations between clinical manifestations of DC and genetic mutations are complicated by the presence of disease heterogeneity, incomplete penetrance, and genetic anticipation.39
Diagnosis
The clinical diagnosis of DC is based on the presence of the four major features of the disease, which include the mucocutaneous triad and BMF, as well as the presence of multisystem features of the disease: epiphora, developmental delay or mental retardation, pulmonary disease, periodontal disease, esophageal stricture, premature hair greying or loss, hiperhidrosis, or development of malignant lesions. To make a correct clinical diagnosis of DC requires at least two of four major features and at least two multisystem features, and sometimes it is difficult because of the disease heterogeneity.9
The pediatric age group often present DC as a multisystem disorder, whereas adult patients generally present a very variable phenotype, not necessarily with the classical features of DC, and being a carrier of telomerase mutations usually acts as a risk factor. Therefore, DC and related disorders represent a very wide clinical and genetic spectrum.10
It is very important for clinical experts to make a correct diagnosis of the disease because of its heterogeneity. A delay or a mistake in diagnosis can lead to inappropriate and inadequate management of the symptoms and sometimes to increased morbidity and mortality. Some centers test the length of telomere, even if the patient presents just one major feature.
After demonstrating a shortening in the length of telomerase it is suitable to perform a genetic study. DC mutation testing is available clinically. However, only about 50% of these patients will have a positive test for a mutation in one of the known predisposition genes.10
Three methods are commonly used for measuring telomere length, including Southern blot, real-time polymerase chain reaction, and flow cytometry and fluorescence in situ hybridization. Of these, measurement of telomere length peripheral white blood cells by flow cytometry and fluorescence in situ hybridization was found to be both sensitive and specific for identifying patients with DC.39
For diagnosis of DC, patients need to have telomere length below the first percentile of telomere length found in healthy controls.9 To generate percentiles of normal telomere length, 400 healthy controls were used. The diagnosis was based on telomere length in total leukocytes and in leukocyte subsets (granulocytes, total lymphocytes, naïve T-cells, memory T-cells, B-cells, and natural killer cells) on cells from people with DC, their relatives, and people with other inherited BMF syndromes.37
Telomere length less than the first percentile for age in lymphocytes is 97% sensitive and 91% specific for DC in comparison with healthy relatives of people with DC or people with non-DC inherited BMF syndromes. In individuals with complex or atypical DC, it is recommended to analyze the six-cell panel more than the two-panel test of total lymphocytes and granulocytes.37 The technique requires being automated and evaluated by an expert.
Needless to say, abnormally short telomere lengths do not appear to be restricted to the hematopoietic compartment in DC. For example, primary skin fibroblasts in patients with DC have relative short telomere length, although normal distribution curves have not been established for cells other than HSCs.3
It should be noted that short telomeres are not specific for patients with DC. Patients with other BMF syndromes such as Shwachman–Diamond syndrome and Fanconi anemia can show alterations in telomeres length, too, although telomere length in patients with DC is shorter than in patients with other BMF syndromes.8 In Shwachman–Diamond syndrome almost all those affected are children, and they present hematologic abnormalities with cytopenias and a predisposition to develop myelodysplastic syndrome and acute myelogenous leukemia, like in DC. Fanconi anemia has an increased risk of malignancy with physical abnormalities like abnormal skin pigmentation, malformations in different systems, hearing loss, hypogonadism, and developmental delay. Features similar to those in DC are progressive BMF with pancytopenia, like in DC. The incidence of hematologic malignancies is 10%–33% and of solid tumors, particularly of the head and neck, skin, gastrointestinal tract, and genital tract, it is 28%–29%.40
Therefore, genetic analysis for DC genes should be performed in individuals with very short telomeres.10 Even so, telomere length cannot predict the presence or absence of DC. At the opposite end there have been rare cases of patients positive for a determinate mutation who show normal telomere length. This suggests that mutation alone does not sufficiently explain the reduction of telomere length.8
In cases with a genetic test positive for DKC1 and TINF2 mutations there is a high risk (>90%) of developing the disease. However, it is different in people with TERC and TERT mutations. Not all individuals carrying mutations in the TERT gene will develop the disease, suggesting that the mutation is more a risk factor than an etiologic factor for the disease. In female carriers of X-linked DC (DKC1), there is usually no medical problem, but they can have affected boys.10
Genetic counseling is the main preventive measure. Prenatal testing for pregnancies at increased risk is possible for DC if a specific mutation in the family under study is known.41
Treatment
DC is a multisystem disorder, so it is important to monitor many systems of the body. BMF is one of the most common and severe complications and is the principal cause of death, followed by predisposition to malignancy and fatal pulmonary failure. BMF in patients with DC does not respond to immunosuppressive therapy (antithymocyte globulin and cyclosporine).42
Telomerase-based therapeutics used in combination with ongoing cancer therapeutics have advantages, disadvantages, promises, and challenges, with ongoing clinical trials.43–45
It has been demonstrated that about 50%–70% of patients will respond to androgens, but patients have to be monitored carefully for abnormalities in cholesterol, triglycerides, and liver function and for the development of liver lesions like adenomas.10,38,41 In a recent study,46 70% of DC patients treated with androgens had a hematological response with red blood cell and/or platelet transfusion independence. However, all of them had at least one significant lipid abnormality. In addition, other treatment-related findings detected were a significant decrease in thyroid binding globulin, accelerated growth in prepubertal children, and splenic peliosis. In this study, liver enzymes were elevated in both androgen-treated and untreated patients, suggesting underlying liver involvement in DC.46
Patients taking androgens should not use growth factors such as granulocyte colony-stimulating factor or erythropoietin, because splenic peliosis and rupture have been described in some cases.10,38,41 Telomerase can be modulated by sex hormones. That is a possible reason why androgens could have actions in BMF.6 Approximately 70% of patients can respond to oxymetholone. Sometimes this response can be maintained for years and involve all lineages,9 but it has the adverse effect of being potently masculinizing, so in female patients this could be a problem. Beneficial activities were reported in DC patients treated with danazol, a synthetic androgen derivative also known as 17α-ethinyl testosterone. It has predominantly antigonadotropic and antiestrogenic activities with less masculinizing side effects.6
Allo-HSCT is the only curative treatment for BMF in patients with DC. However, severe transplant-related toxicities have been described due to underlying organ impairment, particularly when using myeloablative conditioning regimen doses of chemotherapy or chemoradiotherapy.9
Commonly observed transplant-related problems include graft failure, graft versus host disease, sepsis, and, more importantly, the increased propensity to develop organ toxicity like pulmonary fibrosis, hepatic cirrhosis, and veno-occlusive disease, among others. These complications induce shorter survival of people with DC with HSCT.41 These complications are probably related to the presence of total body irradiation, busulfan, or high cyclophosphamide doses in the conditioning regimens in a patient population sensitive to DNA damage. The introduction of nonmyeloablative protocols for related and unrelated donor procedures could achieve engraftment with less toxicity. Reduced-intensity conditioning (RIC) procedures in the context of matched related donors are associated with favorable outcomes without pulmonary complications during short-term follow-up.47
Introduction of fludarabine as an integral component of RIC regimens for allo-HSCT, due to its strong T-cell immunosuppressive effect, represented an ideal option for RIC allo-HSCT in patients with BMF syndromes such as DC.48 Although the use of reduced-intensity regimens has resulted in successful engraftment and lower toxicity, pulmonary fibrosis may develop in these patients at any time in the course of the disease as part of disease pathogenesis. Germline mutations in TERT or TERC can cause both apparently isolated aplastic anemia and pulmonary fibrosis. HSCT does not correct abnormalities related to the underlying genetic defects in DC, and the pulmonary complications may indeed be accelerated after HSCT. Pulmonary fibrosis in patients with DC may reflect cell apoptosis resulting from critically short telomeres in rapidly dividing lung cells.42 Moreover, neurological complications have been observed after reduced-intensity HSCT treatment.49
Actually, there are no standard protocols available for HSCT in DC patients. The development of the disease after HSCT has been reported in different cohorts of patients (Table 3).42,50–54
Table 3.
Reported cases and case series for hematopoietic stem cell transplantation in dyskeratosis congenita
| Number of patients | Type of donor | Conditioning regimens | GVHD | Follow-up, surviving patients | Cause of death | Reference |
|---|---|---|---|---|---|---|
| 1 | MUD (10/10 match) | RIC Flu/CY/ATG | Acute | Alive at 24 months | O’Connell et al50 | |
| 34 | HLA-identical sibling (N=16) Other relative (N=2) MUD (N=9) Mismatched unrelated donor (N=7) |
Cyclophosphamide + TBI <500 cGy (N=6) TBI 200 cGy + Flu (N=3) Melphalan + Flu (N=3) Busulfan + Flu (N=1) Cyclophosphamide + Flu (N=4) Cyclophosphamide + busulfan (N=6) Cyclophosphamide only (N=10) Not reported (N=1) |
Acute (N=8) Chronic (N=11) |
Alive (N=14) at 10 years | GVHD (N=1) Graft failure (N=8) Pulmonary failure (N=5) Infection (N=2) ARDS (N=1) Encephalopathy (N=1) Hemorrhage (N=1) Unknown (N=1) |
Gadalla et al42 |
| 1 | MRD | NMA Flu/CY/ATG (rabbit) | Acute | Alive at 15 months | Coliţă et al47 | |
| 8 | UCB (N=2) MUD (N=6) |
Myeloablative (N=5) RIC (N=3) |
Acute (N=2) | Alive (N=1) at 1 year | Sepsis (N=1) GVHD (N=1) Hemorrhage (N=2) Graft failure (N=2) Unknown (N=1) |
Bizzetto et al51 |
| 6 | UCB (N=1) MUD (N=5) |
NMA Flu/CY Alemtuzumab/TBI 200 cGy |
Acute (N=2) | Alive (N=4) at 26.5 months | Sepsis (N=2) | Dietz et al52 |
| 1 | MUD (10/10 match) | RIC Flu/ATG (rabbit) | Chronic | Alive at 38 months | Vuong et al53 | |
| 2 | MUD (6/6 match) T-cell depleted |
RIC Flu/CY/ATG (equine) | Alive (N=2) at 15 months | Dror et al54 |
Abbreviations: ARDS, acute respiratory distress syndrome; ATG, antithymocyte globulin; Flu, fludarabine; GVHD, graft versus host disease; HLA, human leukocyte antigen; MRD, matched related donor; MUD, matched unrelated donor; NMA, nonmyeloablative; RIC, reduced-intensity conditioning; TAI, thoracoabdominal irradiation; UCB, umbilical cord blood; CY, cyclophosphamide; TBI, total body irradiation.
There is an increased predisposition of DC patients to malignancy, especially after HSCT, and to developing post-transplant complications. Thus, it is preferable to avoid agents that are known to cause DNA damage, such as high-dose irradiation, during conditioning.41
To select human leukocyte antigen-matched sibling donors, it is important to note that affected heterozygotes might not have obvious manifestations of the disease. That is the reason why potential related HSCT donors should be tested either for the mutation if it is known or for telomere length if the mutation is not known. In the absence of a suitable matched sibling donor, HSCT from an unrelated donor could be considered.41
In addition, the fact that DC patients have an increase in transplant-related morbidity is consistent with the result of a recent study that reported that DC lymphocytes expressed a “stress phenotype” with an increase in apoptosis, DNA damage response protein expression, and reactive oxygen species under in vitro stimulated culture conditions. This consequence was partially corrected by the antioxidant N-acetyl cysteine. This study mentions cutaneous improvements in several DC patients treated with oral and topical vitamin E. These findings suggest that oxidative stress may play a role in the pathogenesis of DC and that pharmacologic intervention to correct this pro-oxidant imbalance may prove useful in the clinical setting.50
Sirtuins are a family of seven proteins in humans (SIRT1–SIRT7) that are involved in cellular pathways related to skin structure and function, including aging, ultraviolet-induced photoaging, inflammation, epigenetics, cancer, and a variety of cellular functions, including cell cycle, DNA repair, and proliferation. SIRT6 is a possible target in DC. SIRT6 protects telomeric chromatin from senescence and age-induced chromosomal abnormalities such as end-to-end chromosomal fusions via deacetylation of histones at replicating telomeres. The research on the role of sirtuins is in its early stages. Sirtuin modulators are already being evaluated in clinical trials for a wide variety of human diseases like DC.51
Recently, Agarwal et al52 established that using induced pluripotent stem cells derived from a patient with DC contributed to restoring telomere maintenance. They saw that the RNA component of telomerase is upregulated in the pluripotent state to a degree sufficient to overcome limitations to telomere maintenance in X-linked and AD DC. So drugs that activate the TERC locus may favor telomere maintenance over premature attrition in stem cell compartments where TERT is present but telomerase activity is limiting, such as HSCs, and thus might serve as therapeutic agents for BMF. In addition, in DC patients, certain cell types whose transcriptional programs share similarities with pluripotent cells, such as germ cells and cancer cells, may also upregulate TERC to permit germline propagation of mutations and malignant proliferation.52
DC prognosis can vary considerably from death in infancy (usually due to BMF) to that in the seventh decade. The major causes of death relate to BMF, cancer, and lung disease, particularly pulmonary fibrosis. BMF develops progressively with time. Up to 80% of patients will have BMF by the age of 30 years. Cancer (hematological and nonhematological) usually develops after the third decade. The most frequent solid malignancies are head and neck squamous cell carcinomas.10
Overall summary, future directions, and perspective commentary
We review the historical and current understanding of DC, including clinical aspects, role of telomerase, inheritance of DC, genetic anticipation, diagnosis, and treatment. Much has been discovered in recent years, yet continued additional deeper investigation is warranted in the area and on the role of telomeres, genetic instability, shelterin components, the predisposition of cancer development, the role of transplantation, and clinical trials with telomerase therapeutics and vaccines in genetic variant syndromes.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bohn OL, Whitten J, Spitzer B, et al. Posttransplant lymphoproliferative disorder complicating hematopoietic stem cell transplantation in a patient with dyskeratosis congenita. Int J Surg Pathol. 2013;21(5):520–525. doi: 10.1177/1066896912468214. [DOI] [PubMed] [Google Scholar]
- 2.Mason PJ, Bessler M. The genetics of dyskeratosis congenita. Cancer Genet. 2011;204(12):635–645. doi: 10.1016/j.cancergen.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson ND, Bertuch AA. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat Res. 2012;730(1–2):43–51. doi: 10.1016/j.mrfmmm.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alder JK, Parry EM, Yegnasubramanian S, et al. Telomere phenotypes in females with heterozygous mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum Mutat. 2013;34(11):1481–1485. doi: 10.1002/humu.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishio N, Kojima S. Recent progress in dyskeratosis congenita. Int J Hematol. 2010;92(3):419–424. doi: 10.1007/s12185-010-0695-5. [DOI] [PubMed] [Google Scholar]
- 6.Islam A, Rafiq S, Kirwan M, et al. Haematological recovery in dyskeratosis congenita patients treated with danazol. Br J Haematol. 2013;162(6):854–856. doi: 10.1111/bjh.12432. [DOI] [PubMed] [Google Scholar]
- 7.Alcaraz-Perez F, Garcia-Castillo J, Garcia-Moreno D, et al. A non-canonical function of telomerase RNA in the regulation of developmental myelopoiesis in zebrafish. Nat Commun. 2014;5:3228. doi: 10.1038/ncomms4228. [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi H, Nakanishi K, Kojima S. Inherited bone marrow failure syndromes in 2012. Int J Hematol. 2013;97(1):20–29. doi: 10.1007/s12185-012-1249-9. [DOI] [PubMed] [Google Scholar]
- 9.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–486. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 10.Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: dyskeratosis congenita. EJHG. 2011;19(11):1–11. doi: 10.1038/ejhg.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361(24):2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinha S, Trivedi V, Krishna A, Rao N. Dyskeratosis congenita-management and review of complications: a case report. Oman Med J. 2013;28(4):281–284. doi: 10.5001/omj.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beier F, Foronda M, Martinez P, Blasco MA. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood. 2012;120(15):2990–3000. doi: 10.1182/blood-2012-03-418038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karunakaran A, Ravindran R, Arshad M, Ram MK, Laxmi MK. Dyskeratosis congenita: a report of two cases. Case Rep Dent. 2013;2013:845125. doi: 10.1155/2013/845125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finzi A, Morara M, Pichi F, Veronese C, Ciardella AP. Vitreous hemorrhage secondary to retinal vasculopathy in a patient with dyskeratosis congenita. Int Ophthalmol. 2013 Oct 10; doi: 10.1007/s10792-013-9867-7. Epub. [DOI] [PubMed] [Google Scholar]
- 16.Khincha PP, Savage SA. Genomic characterization of the inherited bone marrow failure syndromes. Semin Hematol. 2013;50(4):333–347. doi: 10.1053/j.seminhematol.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leguit RJ, van den Tweel JG. The pathology of bone marrow failure. Histopathology. 2010;57(5):655–670. doi: 10.1111/j.1365-2559.2010.03612.x. [DOI] [PubMed] [Google Scholar]
- 18.Nickels EM, Soodalter J, Churpek JE, Godley LA. Recognizing familial myeloid leukemia in adults. Ther Adv Hematol. 2013;4(4):254–269. doi: 10.1177/2040620713487399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gramatges MM, Bertuch AA. Short telomeres: from dyskeratosis congenita to sporadic aplastic anemia and malignancy. Transl Res. 2013;162(6):353–363. doi: 10.1016/j.trsl.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parry EM, Alder JK, Lee SS, et al. Decreased dyskerin levels as a mechanism of telomere shortening in X-linked dyskeratosis congenita. J Med Genet. 2011;48(5):327–333. doi: 10.1136/jmg.2010.085100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batista LF, Artandi SE. Understanding telomere diseases through analysis of patient-derived iPS cells. Curr Opin Genet Dev. 2013;23(5):526–533. doi: 10.1016/j.gde.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011;11(3):161–176. doi: 10.1038/nrc3025. [DOI] [PubMed] [Google Scholar]
- 23.Blasco MA. Telomerase beyond telomeres. Nat Rev Cancer. 2002;2(8):627–633. doi: 10.1038/nrc862. [DOI] [PubMed] [Google Scholar]
- 24.Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer. 2008;8(6):450–458. doi: 10.1038/nrc2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng Z, Glousker G, Molczan A, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci U S A. 2013;110(36):E3408–E3416. doi: 10.1073/pnas.1300600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carrillo J, Gonzalez A, Manguan-Garcia C, Pintado-Berninches L, Perona R. p53 pathway activation by telomere attrition in X-DC primary fibroblasts occurs in the absence of ribosome biogenesis failure and as a consequence of DNA damage. Clin Transl Oncol. 2014;16(6):529–538. doi: 10.1007/s12094-013-1112-3. [DOI] [PubMed] [Google Scholar]
- 27.Gadalla SM, Savage SA. Telomere biology in hematopoiesis and stem cell transplantation. Blood Rev. 2011;25(6):261–269. doi: 10.1016/j.blre.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Rivero G, Ruiz-Torres MP, Diez-Marques ML, et al. Telomerase deficiency promotes oxidative stress by reducing catalase activity. Free Radic Biol Med. 2008;45(9):1243–1251. doi: 10.1016/j.freeradbiomed.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 29.Walne AJ, Bhagat T, Kirwan M, et al. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica. 2013;98(3):334–338. doi: 10.3324/haematol.2012.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirwan M, Beswick R, Walne AJ, et al. Dyskeratosis congenita and the DNA damage response. Br J Haematol. 2011;153(5):634–643. doi: 10.1111/j.1365-2141.2011.08679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parry EM, Alder JK, Qi X, Chen JJ, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117(21):5607–5611. doi: 10.1182/blood-2010-11-322149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shtessel L, Ahmed S. Telomere dysfunction in human bone marrow failure syndromes. Nucleus. 2011;2(1):24–29. doi: 10.4161/nucl.2.1.13993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keller RB, Gagne KE, Usmani GN, et al. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr Blood Cancer. 2012;59(2):311–314. doi: 10.1002/pbc.24193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faure G, Revy P, Schertzer M, Londono-Vallejo A, Callebaut I. The C-terminal extension of human RTEL1, mutated in Hoyeraal-Hreidarsson syndrome, contains Harmonin-N-like domains. Proteins. 2014;82(6):897–903. doi: 10.1002/prot.24438. [DOI] [PubMed] [Google Scholar]
- 35.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92(3):448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 2005;102(44):15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vulliamy TJ, Dokal I. Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie. 2008;90(1):122–130. doi: 10.1016/j.biochi.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 38.Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753–764. doi: 10.1097/GIM.0b013e3181f415b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballew BJ, Yeager M, Jacobs K, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet. 2013;132(4):473–480. doi: 10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savage S. Dyskeratosis congenita. In: Pagon R, Adam M, Ardinger H, et al., editors. Dyskeratosis Congenita. Seattle, WA: University of Washington, Seattle; 2013. [Google Scholar]
- 41.Ayas M, Nassar A, Hamidieh AA, et al. Reduced intensity conditioning is effective for hematopoietic SCT in dyskeratosis congenita-related BM failure. Bone Marrow Transplant. 2013;48(9):1168–1172. doi: 10.1038/bmt.2013.35. [DOI] [PubMed] [Google Scholar]
- 42.Gadalla SM, Sales-Bonfim C, Carreras J, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013;19(8):1238–1243. doi: 10.1016/j.bbmt.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8(3):167–179. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 44.Neumann AA, Reddel RR. Telomere maintenance and cancer – look, no telomerase. Nat Rev Cancer. 2002;2(11):879–884. doi: 10.1038/nrc929. [DOI] [PubMed] [Google Scholar]
- 45.Lieberman L, Dror Y. Advances in understanding the genetic basis for bone-marrow failure. Curr Opin Pediatr. 2006;18(1):15–21. doi: 10.1097/01.mop.0000192520.48411.fa. [DOI] [PubMed] [Google Scholar]
- 46.Khincha PP, Wentzensen IM, Giri N, Alter BP, Savage SA. Response to androgen therapy in patients with dyskeratosis congenita. Br J Haematol. 2014;165(3):349–357. doi: 10.1111/bjh.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colita A, Tanase A, Varady Z, Colita AD, Arion C. Fludarabine, low-dose cyclophosphamide and rabbit antithymocyte globulin allowed stable engraftment after allogeneic peripheral blood stem cell transplantation for poly-transfused dyskeratosis congenita patient: case report. Transplant Proc. 2013;45(7):2849–2853. doi: 10.1016/j.transproceed.2013.02.137. [DOI] [PubMed] [Google Scholar]
- 48.Kharfan-Dabaja MA, Otrock ZK, Bacigalupo A, Mahfouz RA, Geara F, Bazarbachi A. A reduced intensity conditioning regimen of fludarabine, cyclophosphamide, antithymocyte globulin, plus 2 Gy TBI facilitates successful hematopoietic cell engraftment in an adult with dyskeratosis congenita. Bone Marrow Transplant. 2012;47(9):1254–1255. doi: 10.1038/bmt.2011.257. [DOI] [PubMed] [Google Scholar]
- 49.Isoda T, Mitsuiki N, Ohkawa T, et al. Irreversible leukoencephalopathy after reduced-intensity stem cell transplantation in a dyskeratosis congenita patient with TINF2 mutation. J Pediatr Hematol Oncol. 2013;35(4):e178–e182. doi: 10.1097/MPH.0b013e318279e5ca. [DOI] [PubMed] [Google Scholar]
- 50.Pereboeva L, Westin E, Patel T, et al. DNA damage responses and oxidative stress in dyskeratosis congenita. PloS one. 2013;8(10):e76473. doi: 10.1371/journal.pone.0076473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serravallo M, Jagdeo J, Glick SA, Siegel DM, Brody NI. Sirtuins in dermatology: applications for future research and therapeutics. Arch Dermatol Res. 2013;305(4):269–282. doi: 10.1007/s00403-013-1320-2. [DOI] [PubMed] [Google Scholar]
- 52.Agarwal S, Loh YH, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464(7286):292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]