

Abstract
Tetrahydroisoquinoline (THIQ)-based “chimeric” microtubule disruptors were optimised through modification of the N-benzyl motif, in concert with changes at C3 and C7, resulting in the identification of compounds with improved in vitro antiproliferative activities (e.g. 15: GI50 20 nM in DU-145). The broad anticancer activity of these novel structures was confirmed in the NCI 60-cell line assay, with 12e,f displaying MGM values in the 40 nM region. In addition, their profiles as inhibitors of tubulin polymerisation and colchicine binding to tubulin were confirmed. Compound 15, for example, inhibited tubulin polymerisation with an IC50 of 1.8 μM, close to that of the clinical drug combretastatin A-4, and also proved effective at blocking colchicine binding. Additionally, compound 20b was identified as the only phenol in the series to date showing both better in vitro antiproliferative properties than its corresponding sulfamate and excellent antitubulin data (IC50 = 1.6 μM). Compound 12f was selected for in vivo evaluation at the NCI in the hollow fibre assay and showed very good activity and wide tissue distribution, illustrating the value of this template for further development.
Keywords: chimeras, colchicine binding, microtubule disruptors, tetrahydroisoquinolines, tubulin assembly
Introduction
Chimeras in mythology are creatures composed from the elements of multiple animals or animals and man. Small or large molecules that combine the essential features of two or more separate entities are also termed chimeras. The chimeric fusion protein bcr-abl,[1] derived from the naturally occurring oncogenic gene fusion that drives the growth of chronic myelogenous leukemia,[2] serves as an example of the latter. A long-standing interest in the development of microtubule disruptors as anticancer agents led us to explore whether, through combining the key pharmacophore elements of two series of colchicine site binders, we might generate “chimeric” small molecules with similar activity. In a preliminary report[3] we outlined how, by introducing the A,B-ring elements of the pharmacophore from a series of steroidal microtubule disruptors 1[4–9] (e.g., 2-methoxyestradiol-3,17-O,O-bis-sulfamate, 2-MeOE2bis-MATE, STX140, 1a) into a tetrahydroisoquinoline (THIQ) motif and connecting this to a trimethoxyaryl motif commonly found in a range of colchicine site binding natural products (e.g., colchicine 2), we could generate novel “chimeric” microtubule disruptors 3,4 (Figure 1).
Figure 1.
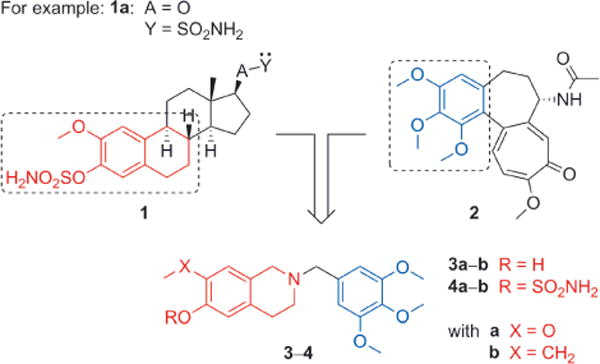
Generation of small-molecule chimeric microtubule disruptors (Y=H-bond acceptor).
These chimeras, and their sulfamates 4a,b in particular, are notable for their excellent physicochemical properties, their in vitro and in vivo activity against various cancer cell lines, including drug-resistant types, and also their structural simplicity when compared to existing clinical agents. One notable advantage is the synthetic ease with which these compounds can be constructed. In parallel work, we also applied the same THIQ core to generate steroidomimetic microtubule disruptors that exhibit a distinct structure–activity relationship (SAR) to the chimeras, yet share their favourable physicochemical properties.[10] In the present study, we explore optimisation of our chimeric system through modification of the trimethoxyaryl component, knowing that in some colchicine site binding microtubule disruptor series replacement of this motif with alternate trisubstituted systems can have a dramatic effect on potency.[11] We therefore synthesised candidate chimeric microtubule disruptors in which one or more of the methoxy groups are exchanged for an alternate functionality.
Results and Discussion
Chemistry
Having previously established synthetic approaches to protected 6-hydroxy-7-methoxy and 6-hydroxy-7-ethyl THIQs in preliminary studies, the logic was already in hand.[3] The major modification over foregoing work was introduction of an appropriately substituted benzyl motif at N2, followed by sequential deprotection and sulfamoylation of the 6-hydroxy group. This was achieved by transforming THIQs 5[3] and 6a,b[12, 13] into the corresponding functionalised N-benzylated compounds 8a–d[12] and 10a–f using various direct N-benzylation methods or N′-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDCI) coupling with the corresponding benzoic acids and subsequent reduction of product with lithium aluminium hydride (LiAlH4). The protected phenols were either treated with hydrogen and palladium on carbon (Pd/C; for 8a–d) or tetra-n-butylammonium fluoride (TBAF; for 10a–f), furnishing phenols 11a–j in good yields. Subsequent treatment of 11a–j with sulfamoyl chloride in N,N-dimethylacetamide (DMA)[14] gave the corresponding sulfamates 12a–j (Scheme 1).
Scheme 1.

Synthesis of 7-methoxy-THIQs. Reagents and conditions: a) ArCH2Cl, Et3N, EtOH, 130 °C, MW; b) ArCH2Br, DIPEA, DMF, 80 °C; c) ArCO2H, EDCI, CH2Cl2/THF, 25 °C; d) LiAlH4, THF, reflux; e) H2, Pd/C, THF/MeOH, 25 °C; f) TBAF, THF, 25 °C; g) H2NSO2Cl, DMA, 25 °C.
Compound 15 was elaborated in two synthetic steps by direct N-benzylation of the unprotected phenol 13[3] with 3-bromo-4,5-dimethoxybenzyl bromide[15] and diisopropylethylamine (DIPEA) in N,N-dimethylformamide (DMF) and subsequent treatment of 14 with sulfamoyl chloride in DMA to give the corresponding sulfamate 15 in moderate overall yield (Scheme 2).
Scheme 2.

Synthesis of 7-ethyl-THIQs. Reagents and conditions: a) 3-Bromo-4,5-dimethoxybenzyl bromide, DIPEA, DMF, 80 °C, 20 h, 40%; b) H2NSO2Cl, DMA, 25 °C, 20 h, 47%.
We also modified the potential hydrogen bonding effects around C6, while retaining the C7-methoxy and the N-2-(3′,4′,5′-trimethoxybenzyl) groups unchanged, to establish if in vitro activity could be further improved. Compound 17a was synthesised from the commercially available 6,7-dimethoxy-THIQ salt 16, 3,4,5-trimethoxybenzyl chloride and DIPEA in DMF. Compounds 17b,c were accessed from phenol 3a by treatment with acetic anhydride or methanesulfonyl chloride, respectively (Scheme 3).
Scheme 3.

Synthesis of C6-modified THIQs. Reagents and conditions: a) 3,4,5-Trimethoxybenzyl chloride, Et3N, EtOH, 130 °C, MW, 1 h, 62%; b) Ac2O, Et3N, CHCl3, 25 °C, 24 h, 79%; c) CH3SO2Cl, pyridine, 25 °C, 73%.
Another objective was to study the effect of deletion of the group at C7 since this had not previously been explored. A range of functionalised benzyl groups was then introduced at N2, as described above, starting from compounds 18a–c.[16, 17] Direct benzylation methods, or by coupling with the corresponding benzoic acids or benzoyl chlorides and successive reduction of product with LiAlH4, afforded compounds 20a–i, usually in moderate yield. An extension of the linker between the THIQ core and the aryl motif connected to it was also achieved by the same strategy and gave 20j in good overall yield (Scheme 4).
Scheme 4.

Synthesis of C7-hydrogen-substituted THIQs. Reagents and conditions: a) ArCH2Cl, Et3N, EtOH, 130 °C, MW; b) ArCH2Br, DIPEA, DMF, 80 °C; c) ArCO2H, EDCI, Et3N, CH2Cl2/THF, 25 °C; d) LiAlH4, THF, 25 °C; e) ArCOCl, Et3N, CH2Cl2, 25 °C; f) H2, Pd/C, THF/MeOH, 25 °C, 1.5 h, 74%; g) H2NSO2Cl, DMA, 25 °C, 24 h, 88%.
Biology
Compounds were evaluated for their ability to inhibit DU-145 (prostate cancer) and MDA MB-231 (breast cancer) cell proliferation and compared to the first generation chimeras 3a and 4a and to 2-methoxyestradiol-3,17-O,O-bis-sulfamate (2-MeOE2bisMATE; 1a) and paclitaxel (Taxol) as benchmark drugs (Table 1). The results obtained across the two cell lines are in close agreement and therefore only the DU-145 data are used for SAR discussion herein. As reported previously, in both the trimethoxybenzyl phenol 3a and 11a–c and sulfamate series 4a and 12a–c, the 3′,4′,5′-trimethoxy system proves optimal (4a GI50=297 nm), although the 2′,4′,5′-trimethoxy sulfamate 12b also displays good activity (GI50 =660 nm).[12] Deletion of the 5′-methoxy group of 4a to give the 3′,4′-dimethoxy compound 12d results in a near 30-fold reduction in activity, thus illustrating the highly preferred status of trisubstitution in this series of chimeras.[12] Replacement of the 5′-methoxy with either chlorine in 12e, or bromine in 12f, however, delivers a dramatic 8- to 10-fold increase in antiproliferative activity, with the corresponding phenols 11e,f also exhibiting significant activity. The 3′,4′-dimethoxy-5′-bromo derivatives 11f and 12f are exceptionally active. Substitution of the 4′-methoxy of 4a with an ethoxy group also delivers improved activity for phenol 11g and sulfamate 12g, revealing a degree of steric flexibility at this position in the chimeric series in strong contrast to the steroidomimetic series.[12, 13] Replacement of the 3′,4′,5′-trimethoxybenzyl group with a 3′,4′,5′-triethoxybenzyl group results in a >20-fold reduction in activity. Similarly, the 3′,4′,5′-triethyl compounds 11i and 12i are significantly less active than 3a and 4a, respectively.
Table 1.
Antiproliferative [μM] activity of THIQs against DU-145 human prostate and MDA MB-231 human breast cancer cells in vitro.[a]
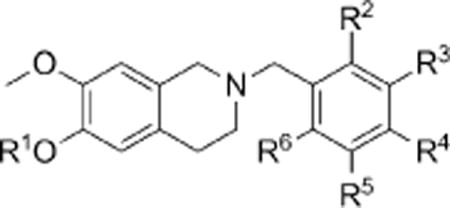
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | R4 | R5 | R6 | DU-145 | MDA MB-231 |
| Taxol | N/A | N/A | N/A | N/A | N/A | N/A | 0.004 | 0.002 |
| 1a | N/A | N/A | N/A | N/A | N/A | N/A | 0.34 | 0.28 |
| 3a | H | H | OMe | OMe | OMe | H | 0.65 | 0.62 |
| 4a | SO2NH2 | H | OMe | OMe | OMe | H | 0.297 | 0.329 |
| 11a | H | OMe | OMe | OMe | H | H | > 100 | > 100 |
| 12a | SO2NH2 | OMe | OMe | OMe | H | H | 17 | 11.9 |
| 11b | H | OMe | H | OMe | OMe | H | 7.8 | 4.17 |
| 12b | SO2NH2 | OMe | H | OMe | OMe | H | 0.66 | 0.491 |
| 11c | H | OMe | H | OMe | H | OMe | > 100 | > 100 |
| 12c | SO2NH2 | OMe | H | OMe | H | OMe | > 100 | > 100 |
| 11d | H | H | OMe | OMe | H | H | > 100 | > 100 |
| 12d | SO2NH2 | H | OMe | OMe | H | H | 8.54 | 3.15 |
| 11e | H | H | OMe | OMe | Cl | H | 0.9 | 0.4 |
| 12e | SO2NH2 | H | OMe | OMe | Cl | H | 0.04 | 0.04 |
| 11f | H | H | OMe | OMe | Br | H | 0.3 | 0.2 |
| 12f | SO2NH2 | H | OMe | OMe | Br | H | 0.03 | 0.03 |
| 11g | H | H | OMe | OEt | OMe | H | 0.3 | 0.3 |
| 12g | SO2NH2 | H | OMe | OEt | OMe | H | 0.1 | 0.07 |
| 11h | H | H | OEt | OEt | OEt | H | > 100 | > 100 |
| 12h | SO2NH2 | H | OEt | OEt | OEt | H | 8.5 | 4.4 |
| 11i | H | H | Et | Et | Et | H | 63 | 26 |
| 12i | SO2NH2 | H | Et | Et | Et | H | 1.8 | 5.7 |
Having established that the 3′,4′-dimethoxy-5′-bromobenzyl derivative is the most active of the various trisubstituted benzyl derivatives, we combined this motif with THIQ core modifications at C3 and C7 that had delivered enhanced activity in preceding studies. Introduction of a methyl group at C3 had delivered a modest enhancement in antiproliferative activity for the 3′,4′,5′-trimethoxybenzyl THIQs,[13] while replacement of the C7-methoxy group with an ethyl group had afforded a near 10-fold improvement in activity.[3] As can be seen in Table 2, when such modifications were made to the THIQ core bearing the 3′,4′-dimethoxy-5′-bromobenzyl group at N2, only relatively modest activity changes resulted with a slight improvement and a slight reduction in activity resulting from C7 and C3 modification, respectively. Note that the potential effect of individual enantiomers on the biological activity of 11j and 12j has not been pursued. Nonetheless, the C7 ethyl derivative 15 (GI50=20 nM) is in vitro the most active compound discovered in this series of chimeras to date. We then focused our attention on the C6 position. Changing the nature of the motif at C6 has a clear effect on in vitro antiproliferative activity (Table 2). Compounds bearing unhindered hydrogen-bond acceptors, such as in 3a (see Table 1), prove much more active than hindered ones; for example, 17a is 120-fold less active (GI50=77.6 μM in MDA MB-231) than 3a. However, projection of a group bearing an unhindered hydrogen-bond acceptor at C6 proves positive with, for example, the sulfamoyl group of 4a and the carbonyl and sulfonyl groups of 17b,c delivering a ca. two- to three-fold better antiproliferative activity than that shown by phenol 3a.
Table 2.
Antiproliferative activity [μM] of variously modified 3′,4′-dimethoxybenzyl-substituted THIQs against DU-145 human prostate and MDA MB-231 human breast cancer cells in vitro.[a]

| ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | X | R2 | R5 | DU-145 | MDA MB-231 |
| 11j[b] | H | Me | OMe | Br | 0.4 | 0.3 |
| 12j[b] | SO2NH2 | Me | OMe | Br | 0.05 | 0.04 |
| 14 | H | H | Et | Br | 1.2 | 0.4 |
| 15 | SO2NH2 | H | Et | Br | 0.02 | 0.03 |
| 17a | Me | H | OMe | OMe | > 100 | 77.6 |
| 17b | Ac | H | OMe | OMe | 0.407 | 0.34 |
| 17c | SO2Me | H | OMe | OMe | 0.22 | 0.188 |
Results are GI50 values in μM and are the mean of three determinations.
Compounds 11j and 12j are racemic mixtures.
Next, we examined the effect of deletion of the C7 substituent that in the steroidomimetic series had proven essential for activity. In conjunction, we varied the C6 substituent and the linker at N2 and, in so doing, discovered an SAR strongly contrasting to that previously elucidated (Table 3). Here, the compound with a hydroxy group at C6, phenol 20b, is by far the most active (GI50=151 nm in DU-145 and GI50 =227 nm in MDA MB-231). Larger C6 substituents are universally less active, for example 20c (about 20- to 200-fold) or inactive (e.g., 20a and sulfamate 20d) in the concentration range tested. Modifying the 3′,4′,5′-trimethoxybenzyl motif at N2 also proves to have a dramatic effect on antiproliferative activity. The only compounds to show modest activity are the 3′,4′-di-methoxy-5′-halobenzyl THIQs 20e,f, albeit they are between 100- to 600-fold less active than 20b. Increasing the size of one or more groups in this motif similarly delivers inactive compounds such as 20g,h. Finally, exchanging the methylene linker between the 3′,4′,5′-trimethoxy aryl motif and N2 for a carbonyl group or extending it to ethylene proves fruitless (see compounds 20i,j). Although not shown here, it should be mentioned that compounds wherein the 3′,4′,5′-trimethoxybenzyl motif at N2 in 20b and 20d is replaced by alternate mono-, di- and other trimethoxybenzyl motifs, and the sulfamoylated derivatives of 20e–j show at best only modest activity (high micromolar GI50s).
Table 3.
Antiproliferative activity [μM] of C7 hydrogen-substituted THIQs against DU-145 human prostate and MDA MB-231 human breast cancer cells in vitro.[a]
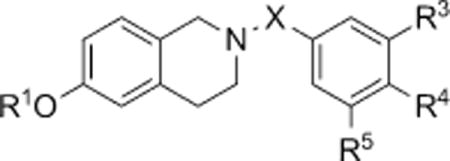
| |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | X | R3 | R4 | R5 | DU-145 | MDA MB-231 |
| 20a | Bn | CH2 | OMe | OMe | OMe | > 100 | > 100 |
| 20b | H | CH2 | OMe | OMe | OMe | 0.151 | 0.227 |
| 20c | Me | CH2 | OMe | OMe | OMe | 31 | 4.89 |
| 20d | SO2NH2 | CH2 | OMe | OMe | OMe | > 100 | > 100 |
| 20e | H | CH2 | OMe | OMe | Cl | 86 | 74 |
| 20f | H | CH2 | OMe | OMe | Br | 35 | 25 |
| 20g | H | CH2 | OMe | OEt | OMe | > 100 | > 100 |
| 20h | H | CH2 | OEt | OEt | OEt | > 100 | > 100 |
| 20i | H | CO | OMe | OMe | OMe | > 100 | > 100 |
| 20j | H | CH2CH2 | OMe | OMe | OMe | > 100 | > 100 |
Results are GI50 values in μM and are the mean of three determinations.
A selection of compounds was also tested at the US National Cancer Institute (NCI) in the full 60-cell-line assay (Table 4) that allows activity across a wide range of cancer types to be assessed. Data from six cell lines are presented along with the mean activity across the whole panel (MGM value). The data obtained in the assay are consistent with those obtained in the antiproliferative screens discussed above and confirm the potential of these compounds against a broad range of cancer phenotypes with, in particular, 12e,f and 20b proving highly active.
Table 4.
Antiproliferative activity [μM] of selected compounds against various cancer cell lines from the NCI-60 cell line panel.[a]
| Compd | Lung HOP-62 | Colon HCT-116 | CNS SF-539 | Melanoma UACC-62 | Ovarian OVCAR-3 | Renal SN12-C | MGM |
|---|---|---|---|---|---|---|---|
| 1a | 0.051 | 0.045 | 0.036 | <0.01 | <0.01 | 0.126 | 0.087 |
| 11e | 0.869 | 0.474 | 0.365 | 0.35 | 0.187 | 0.91 | 0.501 |
| 12e | 0.056 | 0.039 | 0.019 | 0.025 | 0.02 | 0.066 | 0.039 |
| 11f | 0.353 | 0.143 | 0.177 | 0.305 | 0.074 | 0.768 | 0.38 |
| 12f | 0.08 | 0.038 | 0.022 | 0.028 | 0.018 | 0.055 | 0.044 |
| 12i | 2.45 | 1.05 | 0.916 | 0.592 | 0.209 | 5.29 | 1.17 |
| 20b | 0.062 | 0.035 | 0.036 | 0.049 | 0.019 | 0.049 | 0.045 |
Results are GI50 values in μM and are the mean of three determinations. The MGM represents the mean concentration that caused 50% growth inhibition in all 60 cell lines. Data for 1a are taken from the literature [9].
We also wished to establish the microtubule disruptor activity in particular of 12e,f, 12j, 15 and 20b alongside the established potent disruptor combretastatin A-4 (CA-4) and the 3′,4′,5′-trimethoxybenzyl THIQ derivatives 3a and 4a,b (Table 5). The 3′,4′-dimethoxy-5′-halobenzyl THIQs are superior to the first generation chimeras 3a and 4a as inhibitors of tubulin assembly and approach the activity of CA-4, with 15 disrupting the polymerisation of tubulin with an IC50 value of 1.8±0.04 μM. In tubulin-based assays, the concentration needed far exceeds the antiproliferative dose. It presumably suffices to disrupt microtubule dynamics to arrest the cell cycle. Additionally, of course, the nominal compound concentration recorded is that of agent added to the culture medium, and is not the concentration within cells. We also determined that sulfamates 12e,f, 12j and 15 inhibit colchicine binding to tubulin, with 15 being the best, showing 78% inhibition at 5 μM, approaching again the activity of CA-4 (98%). Interestingly, deletion of the methoxy group at C7 in 3a leads to phenol 20b that shows slightly improved antiproliferative activity but also, somewhat surprisingly in comparison, excellent antitubulin data compared to both 3a and its sulfamate 4a. Moreover, 20b is markedly better as an antiproliferative agent than its respective sulfamate derivative 20d. The origin of these very considerable improvements remains to be determined, but might be due to steric effects leading to better accessibility of the unsubstituted phenolic hydroxy group and/or an alternate and stronger binding conformation at the tubulin binding site.
Table 5.
Activity of selected THIQs as inhibitors of tubulin polymerisation and [3H]colchicine binding to tubulin.
| Compd | Tubulin assembly IC50 [μM][a] |
Colchicine binding Inhibition at 5 μM inhibitor [%][a] |
|---|---|---|
| CA-4 | 1.2 ±0.1 | 98 ±0.7 |
| 3a | > 20 (no activity)[b] | 4.1 ±2 |
| 4a | > 20 (partial activity)[b] | 10±0.9 |
| 4b | 2.5 ±0.3 | 49 ±0.5 |
| 12e | 5.6 ±0.7 | 32 ±3 |
| 12f | 2.4 ±0.4 | 45 ±0.6 |
| 12j[c] | 2.4 ±0.2 | 50 ±4 |
| 15 | 1.8 ±0.04 | 78±2 |
| 20b | 1.6 ±0.1 | 73±3 |
Data are the mean ±SD of at least two determinations.
Compound 4a inhibits tubulin assembly at 20 μM while 3a is inactive at this concentration.
Compound 12j is a racemic mixture. Data for CA-4, 3a and 4a,b are taken from the literature [12].
It thus appears reasonable to suggest that the interaction of the novel THIQ derivatives can at least partially be ascribed to their ability to disrupt the normal dynamic polymerisation of tubulin by interaction at, or around, the colchicine binding site. On the strength of their in vitro antiproliferative activity, 11f and 12f were selected for in vivo evaluation at the NCI in the hollow fibre assay that involves assessment of activity against the proliferation of various cancer lines in sealed polyvinylidine fluoride fibres implanted i.p. or s.c. in mice.[18] A 50% net cell growth inhibition is awarded a score of 2 and over 48 fibres (12 cell lines ×2 sites ×2 dose levels) a maximum score of 96 is possible. The results obtained for these compounds showed the strong difference in activity between the phenol 11f and its corresponding sulfamate 12f. Dosing of 11f i.p. at 150 mgkg−1 resulted in 50% inhibition of cell growth in only one fibre and thus a score of only 2. In contrast, sulfamate 12f when dosed by the same route at 75 mgkg−1 delivered a score of 34 (16 for i.p. fibres and 18 for s.c. fibres). This demonstrates both good activity and tissue distribution for the sulfamoylated THIQs and augers well for further development of this class of compounds as in vivo agents, with the anticipated better in vivo performance of the sulfamoyl ester versus the phenol. Although the activity surpasses normal criteria (a score >20) for further investigations at the NCI, compound 12f was not selected for additional study. Despite the excellent in vitro and antitubulin data, and with in vivo data for the related phenol 11f in hand, phenol 20b, despite its clearly attractive antitubulin activities, is not very likely to show the same excellent in vivo and bioavailability properties as the sulfamoylated compound 12f. Therefore, no further studies were carried out so far with this compound class.
The antimitotic natural product agents colchicine, combretastatin A-4, podophyllotoxin and steganacin interact with the colchicine site on tubulin and all possess a trimethoxyaryl unit. This unit in colchicine is thought to be derived via the shikimate biosynthetic pathway and that for combretastatin A-4, currently in clinical trials, is probably derived similarly. This A-ring unit in colchicine has long been thought to make an additive contribution to the strength of binding to tubulin, possibly through hydrogen bonding, and to serve also as an anchor to maintain the whole molecule in the proper orientation within the binding site. Alteration of the oxygenation pattern from a trimethoxy motif on the A-ring of the combretastatins was noted to adversely affect their biological properties and indeed, until recently, it was thought that the trimethoxyaryl group is critical for efficient binding. A range of combretastatins was synthesised with the trimethoxy motif substituted by other functionalities. This study demonstrated interestingly that modifying this motif could significantly reduce cytotoxicity and enhance antitubulin properties.[19] Our initial series of chimeric ligands[12] demonstrated that excellent antiproliferative in vitro and in vivo activities of this new class of compounds could be obtained with a chimera possessing a trimethoxyaryl motif, although the properties of such compounds with respect to inhibition of tubulin polymerisation and colchicine binding are somewhat less impressive. While it is as yet unclear if members of our prototype chimeric microtubule disruptors possessing the trimethoxyaryl motif (e.g., 3–4) bind with this motif in a position comparable to that of colchicine, the present studies do generally support the previous observations that the trimethoxy motif is not critical[19] and demonstrate that molecules of a more optimised series possessing functionalities other than trimethoxy can interact potently with tubulin. Thus, in direct comparison, for example, the ca. 10-fold improvement in DU-145 antiproliferative activity of bromodimethoxyaryl-substituted 12f versus the parent trimethoxyaryl 4a, coupled with improvement in inhibition of tubulin assembly and colchicine binding, provide further evidence that excessive reverence for the trimethoxyaryl motif in optimisation of antimitotic properties should perhaps not remain. With antiproliferative activity maintained and enhanced in our more optimised series, together with the much enhanced antitubulin activity, members of the new series reported herein should yield even more impressive in vivo data in xenograft models of cancer.
Conclusions
A second generation class of tetrahydroisoquinoline (THIQ)-based chimeric microtubule disruptors with improved and excellent activity in vitro and in vivo, combined with a desirable drug-like profile, was identified. The best compounds possess antiproliferative activities in the 20–40 μm range, inhibit tubulin assembly, interfere with the colchicine binding site and possess a pendant N-aryl group without a trimethoxy motif. The sole sulfamoylated compound to be evaluated in vivo shows highly promising preliminary activity, validating the chimeric design and optimisation strategy for this new class of anticancer agents.
Experimental Section
Biology: In vitro studies
Cell Lines
DU-145 (brain metastasis carcinoma of the prostate) and MDA-MB-231 (metastatic pleural effusion of breast adenocarcinoma) established human cell lines were obtained from ATCC Global Bioresource Center. Cells were maintained in a 5% CO2 humidified atmosphere at 37 °C in RPMI-1640 medium, supplemented with 10% fetal bovine serum, penicillin (100 UmL−1), and streptomycin (0.1 mgmL−1).
Antiproliferative assays
DU-145 and MDA-MB-231 cells were seeded into 96-well microtiter plates (5000 cells/well) and treated with 10−9–10−4 M of compounds or with vehicle control. At 96 h post-treatment, live cell counts were determined by the WST-1 cell proliferation assay (Roche, Penzberg, Germany), as per the manufacturer’s instructions. Viability results were expressed as a percentage of mean control values resulting in the calculation of the 50% growth inhibition (GI50). All experiments were performed in triplicate.
Tubulin assays
Bovine brain tubulin, prepared as described previously,[20] was used in the studies presented here. Assembly IC50 values were determined as described in detail elsewhere.[21] Briefly, 1.0 mgmL−1 (10 μM) tubulin was preincubated without GTP with varying compound concentrations for 15 min at 30 °C. The reaction mixtures were placed on ice, and GTP (final concentration, 0.4 mM) was added. The reaction mixtures were transferred to cuvettes, held at 0 °C in a recording spectrophotometer. Baselines were established at 0 °C, and increase in turbidity was followed for 20 min following a rapid (<30 s) jump to 30 °C. Compound concentrations required to reduce the turbidity increase by 50% were determined. The method for measuring inhibition of the binding of [3H]colchicine to tubulin was described in detail previously.[22] Reaction mixtures contained 0.1 mgmL−1 (1.0 μM) tubulin, 5.0 μM [3H]colchicine, and potential inhibitor at 5.0 μM. Compounds were compared to CA-4, a particularly potent inhibitor of the binding of colchicine to tubulin.[23] Reaction mixtures were incubated 10 min at 37 °C, a time point at which the binding of colchicine in control reaction mixtures is generally 40–60% complete. A minimum of two experiments was performed for each compound.
Chemistry
All chemicals were either purchased from Aldrich Chemical Co. (Gillingham, UK) or Alfa Aesar (Heysham, UK). Organic solvents of A.R. grade were supplied by Fisher Scientific (Loughborough, UK) and used as supplied. The petroleum ether (PE) used for column chromatography was of fractions 40–60 °C. CH2Cl2, CHCl3, N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMA) and tetrahydrofuran (THF) were purchased from Aldrich and stored under a positive pressure of N2 after use. Sulfamoyl chloride was prepared by an adaptation of the method of Appel and Berger[24] and was stored in the refrigerator under a positive pressure of N2 as a solution in toluene as described by Woo et al.[25] An appropriate volume of this solution was freshly concentrated in vacuo immediately before use. Compounds 5, 6a,b, 8a–d, 11a–d, 12a–d, 13 and 18a–c were prepared according to literature procedures.[3, 12, 13, 16, 17] Reactions were carried out at room temperature (RT) unless stated otherwise. Thin-layer chromatography (TLC) was performed on precoated aluminium plates (Merck, silica gel 60 F254). Product spots were visualised either by UV irradiation at 254 nm or by staining with either alkaline KMnO4 solution or 5% dodecamolybdophosphoric acid in EtOH, followed by heating. Flash column chromatography was performed using gradient elution (solvents indicated in text) on either prepacked columns (Isolute) on a Flashmaster II system (Biotage, Uppsala, Sweden) or on a CombiFlash Rf Automated Flash Chromatography System (Teledyne Isco, Lincoln, NE, USA) with RediSep Rf disposable flash columns. 1H NMR and 13C NMR spectra were recorded with either a Delta JMN-GX 270 (Jeol, Peabody, MA, USA) at 270 and 67.5 MHz, respectively, or a Mercury VX 400 NMR spectrometer (Varian, Paolo Alto, CA, USA) at 400 and 100 MHz, respectively. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane (TMS) as internal standard. Coupling constants J are recorded to the nearest 0.1 Hz. Mass spectra were recorded at the Mass Spectrometry Service Centre, University of Bath, UK. FAB-MS was carried out using m-nitrobenzyl alcohol (NBA) as the matrix. Melting points were determined using a Stuart SMP3 or a Stanford research systems Optimelt MPA100 melting point apparatus (Stanford Research Systems, Sunnyvale, CA, USA) and are uncorrected. All compounds were ≥98% pure by reverse phase HPLC run with CH3CN/H2O or MeOH/H2O (Sunfire C18 reverse phase column, 4.6×150 mm, 3.5 μM pore size).
2-(3-Chloro-4,5-dimethoxybenzoyl)-7-methoxy-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (9a)
Compound 6a (504 mg, 1.5 mmol) and 3-chloro-4,5-dimethoxybenzoic acid (487 mg, 2.25 mmol) were placed in an oven-dried tube and dissolved in CH2Cl2 (3.0 mL) and THF (3.0 mL). N′-(3-Dimethylaminopropyl)-N-ethylcarbodiimide (EDCI; 573 mg, 3.0 mmol) was added, and the reaction mixture was stirred at RT for 18 h. HCl (2M, 30 mL) was then added, and the mixture was extracted with CH2Cl2 (2×50 mL). The combined organics were dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane→hexane/EtOAc 4:1→EtOAc) afforded compound 9a as a colourless oil (535 mg, 66%): 1H NMR (270 MHz, CDCl3): δ=1.04 (18 H, d, J=6.9 Hz), 1.11–1.30 (3 H, m), 2.73 (2 H, s, br), 3.59 and 3.69 (2 H, m), 3.73 (3 H, s, br), 3.84 (3 H, s, br), 3.85 (3 H, s), 4.49 and 4.73 (2 H, s, br), 6.37 and 6.58 (1 H, s), 6.61 (1 H, s), 6.92 (1 H, s), 7.03 ppm (1 H, d, J=1.9 Hz); HRMS (ES+): m/z [M+H]+calcd for C28H41ClNO5Si+: 534.2437, found: 534.2431.
2-(3,5-Dimethoxy-4-ethoxybenzoyl)-7-methoxy-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (9b)
Method as for 9a using compound 6a (503 mg, 1.5 mmol), 3,5-dimethoxy-4-ethoxybenzoic acid[26] (508 mg, 2.25 mmol) and EDCI (574 mg, 3.0 mmol) in CH2Cl2 (3.0 mL) and THF (3.0 mL) at RT for 18 h. Flash column chromatography (hexane→hexane/EtOAc 7:3→EtOAc) afforded compound 9b as a colourless oil (449 mg, 55%): 1H NMR (270 MHz, CDCl3): δ=1.04 (18 H, d, J=6.9 Hz), 1.11–1.28 (3 H, m), 1.32 (3 H, t, J=7.0 Hz), 2.73 and 2.80 (2 H, s, br), 3.60 and 3.69 (2 H, m), 3.73 (3 H, s, br), 3.80 (6 H, s), 4.03 (2 H, q, J=7.1 Hz), 4.48 and 4.74 (2 H, s, br), 6.35 and 6.60 (1 H, s), 6.60 (1 H, s), 6.64 ppm (2 H, s); HRMS (ES+): m/z [M+H]+ calcd for C30H46NO6Si+: 544.3089, found: 544.3093.
7-Methoxy-2-(3,4,5-triethoxybenzoyl)-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (9c)
Method as for 9a using compound 6a (503 mg, 1.5 mmol), 3,4,5-triethoxybenzoic acid (571 mg, 2.25 mmol) and EDCI (573 mg, 3.0 mmol) in CH2Cl2 (3.0 mL) and THF (3.0 mL) at RT for 18 h. Flash column chromatography (hexane→hexane/EtOAc 7:3→EtOAc) afforded compound 9c as a colourless oil (550 mg, 64%): 1H NMR (270 MHz, CDCl3): δ=1.06 (18 H, d, J=6.9 Hz), 1.14–1.29 (3 H, m), 1.33 (3 H, t, J=7.2 Hz), 1.39 (6 H, t, J=6.9 Hz), 2.72 and 2.80 (2 H, s, br), 3.60 and 3.69 (2 H, m), 3.75 (3 H, s, br), 4.03 (2 H, q, J=7.2 Hz), 4.06 (4 H, q, J=7.4 Hz), 4.48 and 4.74 (2 H, s, br), 6.35 and 6.62 (1 H, s), 6.62 ppm (3 H, s); HRMS (ES+): m/z [M+H]+ calcd for C32H50NO6Si+: 572.3402, found: 572.3405.
7-Methoxy-2-(3,4,5-triethylbenzoyl)-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (9d)
Method as for 9a using compound 6a (503 mg, 1.5 mmol) and 3,4,5-triethylbenzoic acid[27, 28] (483 mg, 2.25 mmol) and EDCI (572 mg, 3.0 mmol) in CH2Cl2 (3.0 mL) and THF (3.0 mL) at RT for 18 h. Flash column chromatography using (hexane→hexane/EtOAc 9:1→EtOAc) afforded compound 9d as a colourless oil (342 mg, 43%): 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.9 Hz), 1.10–1.31 (3 H, m), 1.12 (3 H, t, J=7.7 Hz), 1.21 (6 H, t, J=7.7 Hz), 2.58–2.76 (2 H, m), 2.66 (4 H, q, J=7.7 Hz), 2.68 (2 H, q, J=7.7 Hz), 3.61 and 3.68 (2 H, m), 3.76 and 3.93 (3 H, s, br), 4.51 and 4.77 (2 H, s, br), 6.35 and 6.62 (1 H, s), 6.62 (1 H, s), 7.10 ppm (2 H, s); HRMS (ES+): m/z [M+H]+ calcd for C32H50NO3Si+: 524.3555, found: 524.3562.
2-(3-Chloro-4,5-dimethoxybenzyl)-7-methoxy-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (10a)
LiAlH4 (103 mg, 2.7 mmol) was placed in an oven-dried tube and covered with THF (1.0 mL). 9a (481 mg, 0.9 mmol) was dissolved in THF (4.4 mL) and added dropwise via syringe. The reaction mixture was stirred at RT for 2 h. EtOAc (5 mL) was added carefully. The mixture was then diluted with EtOAc (100 mL) and left without stirring in a beaker for 0.5 h. The mixture was filtered through Celite that was then washed with EtOAc (4×10 mL), and the filtrate was concentrated in vacuo. Flash column chromatography (hexane/EtOAc 9:1→9:1 and 2% Et3N) afforded compound 10a as a pale yellow oil (328 mg, 70%): 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.9 Hz), 1.13–1.32 (3 H, m), 2.61–2.69 (2 H, m), 2.70–2.78 (2 H, m), 3.52 (2 H, s), 3.54 (2 H, s), 3.71 (3 H, s), 3.85 (6 H, s), 6.44 (1 H, s), 6.58 (1 H, s), 6.88 (1 H, d, J= 1.7 Hz), 6.96 ppm (1 H, d, J= 1.6 Hz); HRMS (ES+): m/z [M+H]+ calcd for C28H43ClNO4Si+: 520.2644, found: 520.2632.
2-(3-Bromo-4,5-dimethoxybenzyl)-7-methoxy-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (10b)
Compound 6a (369 mg, 1.1 mmol) was dissolved in DMF (3.3 mL), and diisopropylethylamine (DIPEA; 287 mg, 2.2 mmol) and 3-bromo-4,5-dimethoxybenzyl bromide[15] (335 mg, 1.2 mmol) were added. The mixture was stirred at 80 °C for 18 h, cooled to RT, diluted with EtOAc (100 mL) and washed with H2O (100 mL) and NH4Cl (sat., 5 mL). The aqueous layer was extracted with EtOAc (100 mL). The combined organics were dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 19:1→19:1 and 2% Et3N) afforded compound 10b as a yellow oil (272 mg, 43%): 1H NMR (270 MHz, CDCl3): δ= 1.06 (18 H, d, J=6.9 Hz), 1.13–1.32 (3 H, m), 2.61–2.69 (2 H, m), 2.70–2.78 (2 H, m), 3.52 (2 H, s), 3.55 (2 H, s), 3.71 (3 H, s), 3.83 (3 H, s), 3.84 (3 H, s), 6.44 (1 H, s), 6.58 (1 H, s), 6.93 (1 H, d, J= 1.7 Hz), 7.12 ppm (1 H, d, J= 1.4 Hz); HRMS (ES+): m/z [M+H]+ calcd for C28H43BrNO4Si+: 564.2139, found: 564.2132.
2-(3,5-Dimethoxy-4-ethoxybenzyl)-7-methoxy-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (10c)
Method as for 10a using compound 9b (435 mg, 0.8 mmol) and LiAlH4 (92 mg, 2.4 mmol) in THF (4.8 mL) at RT for 2 h. Flash column chromatography (hexane/EtOAc 9:1 →4:1→4:1 and 2% Et3N) afforded compound 10c as a viscous yellow oil (398 mg, 93%): 1H NMR (270 MHz, CDCl3): δ= 1.06 (18 H, d, J=6.9 Hz), 1.12–1.29 (3 H, m), 1.33 (3 H, t, J= 7.2 Hz), 2.60–2.68 (2 H, m) 2.69–2.77 (2 H, m), 3.53 (2 H, s), 3.56 (2 H, s), 3.69 (3 H, s), 3.80 (6 H, s), 4.02 (2 H, q, J=7.2 Hz), 6.44 (1 H, s), 6.57 (1 H, s), 6.59 ppm (2 H, s); HRMS (ES+): m/z [M+H]+ calcd for C30H48NO5Si+: 530.3296, found: 530.3303.
7-Methoxy-2-(3,4,5-triethoxybenzyl)-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (10d)
Method as for 10a using compound 9c (515 mg, 0.9 mmol) and LiAlH4 (104 mg, 2.7 mmol) in THF (5.4 mL) at RT for 2 h. Flash column chromatography (hexane/EtOAc 9:1→4:1→4:1 and 2% Et3N) afforded compound 10d as a viscous yellow oil (379 mg, 75%): 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.9 Hz), 1.14–1.30 (3 H, m), 1.34 (3 H, t, J= 7.2 Hz), 1.39 (6 H, t, J= 7.2 Hz), 2.59–2.68 (2 H, m) 2.69–2.78 (2 H, m), 3.53 (2 H, s), 3.55 (2 H, s), 3.71 (3 H, s), 4.05 (6 H, q, J=7.2 Hz), 6.44 (1 H, s), 6.59 ppm (3 H, s); HRMS (ES+): m/z [M+H]+ calcd for C32H52NO5Si+: 558.3609, found: 558.3616.
7-Methoxy-2-(3,4,5-triethylbenzyl)-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (10e)
Method as for 10a using compound 9d (340 mg, 0.65 mmol) and LiAlH4 (74 mg, 1.95 mmol) in THF (4.0 mL) at RT for 2 h. Flash column chromatography (hexane/EtOAc 19:1→19:1 and 2% Et3N) afforded compound 10e as a viscous yellow oil (289 mg, 87%): 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.9 Hz), 1.13–1.32 (3 H, m), 1.15 (3 H, t, J=7.6 Hz), 1.23 (6 H, t, J=7.6 Hz), 2.61–2.69 (2 H, m) 2.66 (4 H, q, J=7.5 Hz), 2.67 (2 H, q, J=7.7 Hz), 2.70–2.78 (2 H, m), 3.56 (2 H, s), 3.60 (2 H, s), 3.72 (3 H, s), 6.45 (1 H, s), 6.58 (1 H, s), 7.04 ppm (2 H, s); HRMS (ES+): m/z [M+H]+ calcd for C32H52NO2Si+: 510.3762, found: 510.3769.
(±)-2-(3-Bromo-4,5-dimethoxybenzyl)-7-methoxy-3-methyl-6-(triisopropylsilyloxy)-1,2,3,4-tetrahydroisoquinoline (10 f)
Method as for 10b using compound 6b (419 mg, 1.2 mmol), 3-bromo-4,5-dimethoxybenzyl bromide[15] (449 mg, 1.45 mmol) and DIPEA (310 mg, 2.4 mmol) in DMF (3.6 mL) at 80 °C for 18 h. Flash column chromatography (hexane/EtOAc 9:1→9:1 and 2% Et3N) afforded compound 10f as a yellow oil (598 mg, 86%): 1H NMR (270 MHz, CDCl3): δ=1.07 (18 H, d, J=6.6 Hz), 1.09 (3 H, d, J=5.0 Hz), 1.14–1.32 (3 H, m), 2.45 (1 H, dd, J= 16.1, 5.9 Hz), 2.85 (1 H, dd, J=16.0, 4.7 Hz), 3.04 (1 H, sext, J= 6.1 Hz), 3.40–3.73 (4 H, m), 3.71 (3 H, s), 3.83 (3 H, s), 3.84 (3 H, s), 6.41 (1 H, s), 6.57 (1 H, s), 6.89 (1 H, d, J= 1.9 Hz), 7.11 ppm (1 H, d, J=1.6 Hz); HRMS (ES+): m/z [M+H]+ calcd for C29H45BrNO4Si+: 578.2296, found: 578.2251.
2-(3-Chloro-4,5-dimethoxybenzyl)-6-hydroxy-7-methoxy-1,2,3,4-tetrahydroisoquinoline (11e)
Compound 10a (286 mg, 0.55 mmol) was dissolved in THF (2.5 mL). Tetra-n-butylammonium fluoride (TBAF; 0.66 mL, 1 M in THF, 0.66 mmol) was added dropwise via syringe, and the reaction mixture was stirred at 0 °C for 0.5 h. MeOH (5 mL) and CH2Cl2 (30 mL) were added, and the mixture was concentrated in vacuo. Flash column chromatography (CHCl3/acetone 9:1→9:1 and 2% MeOH) afforded compound 11e as a white solid (168 mg, 84%): mp: 193–195 °C; 1H NMR (270 MHz, CDCl3): δ= 2.61–2.68 (2 H, m), 2.69–2.76 (2 H, m), 3.46 (2 H, s), 3.51 (2 H, s), 3.75 (3 H, s), 3.80 (6 H, s), 6.42 (1 H, s), 6.57 (1 H, s), 6.88 (1 H, d, J=1.6 Hz), 6.90 ppm (1 H, d, J=1.6 Hz); 13C NMR (67.5 MHz, CDCl3): δ=28.0, 50.6, 55.5, 55.8, 56.0, 60.6, 62.0, 108.9, 111.5, 114.3, 122.2, 125.3, 126.4, 127.6, 134.6, 144.1, 145.1, 153.7 ppm; LC-MS (ES+): m/z 364.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C19H23ClNO4+: 364.1310, found: 364.1305.
2-(3-Bromo-4,5-dimethoxybenzyl)-6-hydroxy-7-methoxy-1,2,3,4-tetrahydroisoquinoline (11f)
Method as for 11e using compound 10b (254 mg, 0.45 mmol) and TBAF (0.54 mL, 1 M in THF, 0.54 mmol) in THF (2.7 mL) at 0 °C for 0.5 h. Flash column chromatography (CHCl3/acetone 9:1→9:1 and 2% MeOH) gave compound 11f as a pale yellow solid (148 mg, 80%): mp: 195–198 °C; 1H NMR (270 MHz, CDCl3): δ= 2.59–2.67 (2 H, m), 2.68–2.76 (2 H, m), 3.45 (2 H, s), 3.50 (2 H, s), 3.74 (3 H, s), 3.78 (3 H, s), 3.79 (3 H, s), 6.41 (1 H, s), 6.55 (1 H, s), 6.91 (1 H, d, J= 1.6 Hz), 7.05 ppm (1 H, d, J= 1.6 Hz); 13C NMR (67.5 MHz, CDCl3): δ= 27.9, 50.6, 55.5, 55.8, 55.9, 60.4, 61.9, 108.9, 112.3, 114.3, 117.0, 125.0, 125.2, 126.4, 135.2, 144.1, 145.2, 153.5 ppm; LC-MS (ES+): m/z 408.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C19H23BrNO4+: 408.0805, found: 408.0796.
2-(3,5-Dimethoxy-4-ethoxybenzyl)-6-hydroxy-7-methoxy-1,2,3,4-tetrahydroisoquinoline (11 g)
Method as for 11e using compound 10c (371 mg, 0.7 mmol) and TBAF (0.84 mL, 1 M in THF, 0.84 mmol) in THF (4.2 mL) at 0 °C for 0.5 h. Flash column chromatography (CHCl3/acetone 9:1→4:1→4:1 and 2% MeOH) afforded compound 11g as a yellow glass (194 mg, 74%): 1H NMR (270 MHz, CDCl3): δ=1.34 (3 H, t, J= 7.0 Hz), 2.65–2.72 (2 H, m), 2.72–2.79 (2 H, m), 3.53 (2 H, s), 3.58 (2 H, s), 3.78 (3 H, s), 3.80 (6 H, s), 4.03 (2 H, q, J=7.1 Hz), 5.52 (1 H, s, br), 6.45 (1 H, s), 6.60 (1 H, s), 6.61 ppm (2 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 15.5, 28.2, 50.6, 55.7, 55.9, 56.0, 62.8, 68.8, 105.7, 108.8, 114.3, 125.7, 126.8, 133.7, 135.8, 144.1, 145.0, 153.3 ppm; LC-MS (ES+): m/z 374.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C21H28NO5+: 374.1962, found: 374.1953.
6-Hydroxy-7-methoxy-2-(3,4,5-triethoxybenzyl)-1,2,3,4-tetra-hydroisoquinoline (11h)
Method as for 11e using compound 10d (336 mg, 0.6 mmol) and TBAF (0.72 mL, 1 M in THF, 0.72 mmol) in THF (3.6 mL) at 0 °C for 0.5 h. Flash column chromatography (CHCl3/acetone 9:1→9:1 and 2% MeOH) afforded compound 11h as a pale yellow solid (189 mg, 78%): mp: 119–121 °C; 1H NMR (270 MHz, CDCl3): δ=1.34 (3 H, t, J=63 Hz), 1.38 (6 H, t, J=6.9 Hz), 2.62–2.70 (2 H, m), 2.71–2.79 (2 H, m), 3.51 (2 H, s), 3.55 (2 H, s), 3.79 (3 H, s), 4.03 (6 H, q, J= 7.0 Hz), 5.63 (1 H, s, br), 6.45 (1 H, s), 6.58 (1 H, s), 6.62 ppm (2 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 15.0, 15.6, 28.4, 50.6, 55.8, 56.0, 62.8, 64.6, 68.7, 107.4, 108.8, 114.2, 126.1, 127.0, 133.6, 144.0, 144.9, 152.8 ppm; LC-MS (ES+): m/z 402.3 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C23H32NO5+: 402.2275, found: 402.2268.
6-Hydroxy-7-methoxy-2-(3,4,5-triethylbenzyl)-1,2,3,4-tetrahydroisoquinoline (11i)
Method as for 11e using compound 10e (255 mg, 0.5 mmol) and TBAF (0.6 mL, 1 M in THF, 0.6 mmol) in THF (3.0 mL) at 0 °C for 0.5 h. Flash column chromatography (CHCl3/acetone 9:1→9:1 and 2% MeOH) afforded compound 11i as a yellow glass (134 mg, 75%): 1H NMR (270 MHz, CDCl3): δ= 1.16 (3 H, t, J=7.6 Hz), 1.24 (6 H, t, J= 7.4 Hz), 2.67 (4 H, q, J= 7.4 Hz), 2.67–2.82 (6 H, m), 3.57 (2 H, s), 3.63 (2 H, s), 3.80 (3 H, s), 5.49 (1 H, s, br), 6.48 (1 H, s), 6.61 (1 H, s), 7.06 ppm (2 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 15.3, 15.8, 21.2, 25.7, 28.2, 50.5, 55.7, 55.9, 62.5, 108.9, 114.3, 126.0, 126.9, 127.1, 135.2, 138.2, 141.9, 144.0, 144.9 ppm; LC-MS (ES+): m/z 354.3 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C23H32NO2+: 354.2428, found: 354.2414.
(±)-2-(3-Bromo-4,5-dimethoxybenzyl)-6-hydroxy-7-methoxy-3-methyl-1,2,3,4-tetrahydroisoquinoline (11j)
Method as for 11e using compound 10f (520 mg, 0.9 mmol) and TBAF (1 M in THF, 1.08 mL, 1.08 mmol) in THF (4.5 mL) at 0 °C for 0.5 h. Flash column chromatography (CHCl3/acetone 9:1→9:1 and 2% MeOH) afforded compound 11j as a yellow solid (342 mg, 90%): mp: 111–114 °C; 1H NMR (270 MHz, CDCl3): δ= 1.11 (3 H, d, J= 6.3 Hz), 2.46 (1 H, dd, J= 16.2, 6.1 Hz), 2.87 (1 H, dd, J= 16.1, 4.8 Hz), 3.06 (1 H, sext, J= 6.1 Hz), 3.41–3.73 (4 H, m), 3.78 (3 H, s), 3.81 (3 H, s), 3.83 (3 H, s), 5.32 (1 H, s, br), 6.43 (1 H, s), 6.59 (1 H, s), 6.92 (1 H, d, J= 1.9 Hz), 7.10 ppm (1 H, d, J=1.7 Hz); 13C NMR (67.5 MHz, CDCl3): δ= 15.2, 34.4, 51.2, 52.2, 55.8, 56.0, 56.3, 60.5, 108.6, 112.0, 114.5, 117.1, 124.5, 125.0, 126.2, 136.7, 144.0, 144.9, 145.1, 153.6 ppm; LC-MS (ES+): m/z 422.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H25BrNO4S+: 422.0962, found: 422.0942.
2-(3-Chloro-4,5-dimethoxybenzyl)-7-methoxy-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (12e)
Compound 11e (146 mg, 0.4 mmol) was placed in an oven-dried 50 mL round-bottom flask and dissolved in DMA (2.0 mL). Sulfamoyl chloride (0.57 M in toluene, 2.1 mL, 1.2 mmol) was concentrated in vacuo and re-dissolved in DMA (1.0 mL). This solution was added dropwise via syringe at 0 °C. The reaction mixture was stirred at RT for 2 h. EtOAc (100 mL) was added, and the mixture was washed with NaHCO3 (saturated, 50 mL) and H2O (4×50 mL). The organic layer was dried (NaCl), filtered and concentrated in vacuo. The residue was stirred in CH2Cl2/Et2O/hexane (~1:2:2), filtered and dried to afford compound 12e as a white solid (85 mg, 48%): mp: 130–134 °C; 1H NMR (270 MHz, CDCl3): δ=2.62–2.70 (2 H, m), 2.73–2.81 (2 H, m), 3.50 (2 H, s), 3.52 (2 H, s), 3.76 (3 H, s), 3.81 (6 H, s), 5.98 (2 H, s, br), 6.56 (1 H, s), 6.83 (1 H, d, J=1.6 Hz), 6.92 (1 H, d, J=1.7 Hz), 7.04 ppm (1 H, s); 13C NMR (67.5 MHz, CDCl3): δ=28.0, 50.4, 55.5, 56.1, 56.1, 60.6, 61.8, 110.9, 111.2, 121.8, 123.8, 126.9, 127.8, 134.0, 134.8, 137.4, 144.3, 149.6, 153.7 ppm; LC-MS (ES+): m/z 443.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C19H24ClN2O6S+: 443.1038, found: 443.1034.
2-(3-Bromo-4,5-dimethoxybenzyl)-7-methoxy-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (12f)
Method as for 12e using compound 11f (122 mg, 0.3 mmol) and sulfamoyl chloride (0.9 mmol) in DMA (2.5 mL) at RT for 2 h. The residue was stirred in CH2Cl2/Et2O/hexane (~1:2:2), filtered and dried to afford compound 12f as a pale yellow solid (54 mg, 37%): mp: 132–136 °C; 1H NMR (270 MHz, CDCl3): δ=2.62–2.70 (2 H, m), 2.73–2.81 (2 H, m), 3.50 (2 H, s), 3.53 (2 H, s), 3.76 (3 H, s), 3.80 (3 H, s), 3.80 (3 H, s), 5.99 (2 H, s, br), 6.56 (1 H, s), 6.87 (1 H, d, J=1.6 Hz), 7.04 (1 H, s), 7.07 ppm (1 H, d, J=1.6 Hz); 13C NMR (67.5 MHz, CDCl3): δ=28.0, 50.4, 55.5, 56.0, 56.1, 60.4, 61.7, 110.9, 112.0, 117.2, 123.8, 124.6, 126.8, 134.0, 135.4, 137.4, 145.4, 149.6, 153.6 ppm; LC-MS (ES+): m/z 487.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C19H24BrN2O6S+: 487.0533, found: 487.0511.
2-(3,5-Dimethoxy-4-ethoxybenzyl)-7-methoxy-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (12g)
Method as for 12e using compound 11g (169 mg, 0.45 mmol) and sulfamoyl chloride (1.35 mmol) in DMA (4.0 mL) at RT for 2 h. The residue was stirred in CH2Cl2/Et2O (~1:4), filtered and dried to afford compound 12g as a pale yellow solid (125 mg, 61%): mp: 127–130 °C; 1H NMR (270 MHz, CDCl3): δ=1.34 (3 H, t, J=7.0 Hz), 2.66–2.75 (2 H, m), 2.76–2.85 (2 H, m), 3.57 (2 H, s), 3.59 (2 H, s), 3.79 (3 H, s), 3.82 (6 H, s), 4.03 (2 H, q, J=7.1 Hz), 5.05 (2 H, s, br), 6.59 (3 H, s), 7.06 ppm (1 H, s); 13C NMR (67.5 MHz, CDCl3): δ=15.5, 27.9, 50.2, 55.6, 56.1, 56.2, 62.6, 68.9, 105.8, 111.1, 124.0, 127.3, 133.3, 134.4, 136.0, 137.3, 149.4, 153.4 ppm; LC-MS (ES+): m/z 453.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C21H29N2O7S+: 453.1690, found: 453.1688.
7-Methoxy-6-sulfamoyloxy-2-(3,4,5-triethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (12h)
Method as for 12e using compound 11h (160 mg, 0.4 mmol) and sulfamoyl chloride (1.2 mmol) in DMA (3.0 mL) at RT for 2 h. The residue was stirred in CH2Cl2/Et2O/ hexane (~1:2:2), filtered and dried to afford compound 12h as a pale yellow solid (84 mg, 46%): mp: 133–134°C; 1H NMR (270 MHz, CDCl3): δ=1.33 (3 H, t, J=6.9 Hz), 1.39 (6 H, t, J=6.9 Hz), 2.64–2.72 (2 H, m), 2.75–2.83 (2 H, m), 3.54 (2 H, s), 3.55 (2 H, s), 3.79 (3 H, s), 4.03 (2 H, q, J=7.0 Hz), 4.04 (4 H, q, J=7.0 Hz), 5.07 (2 H, s, br), 6.57 (2 H, s), 6.59 (1 H, s), 7.05 ppm (1 H, s); 13C NMR (67.5 MHz, CDCl3): δ=14.9, 15.6, 28.0, 50.2, 55.6, 56.2, 62.6, 64.6, 68.8, 107.4, 111.1, 124.0, 127.5, 133.1, 134.6, 137.0, 137.3, 149.3, 152.8 ppm; LC-MS (ES+): m/z 481.3 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C23H33N2O7S+: 481.2003, found: 481.1989.
7-Methoxy-6-sulfamoyloxy-2-(3,4,5-triethylbenzyl)-1,2,3,4-tetra-hydroisoquinoline (12i)
Method as for 12e using compound 11i (106 mg, 0.3 mmol) and sulfamoyl chloride (0.9 mmol) in DMA (2.5 mL) at RT for 2 h. The residue was stirred in CH2Cl2/hexane (~1:4), filtered and dried to afford compound 12i as a pale yellow solid (65 mg, 50%): mp: 138–140 °C; 1H NMR (270 MHz, CDCl3): δ= 1.14 (3 H, t, J=7.4 Hz), 1.22 (6 H, t, J=7.6 Hz), 2.61–2.75 (4 H, m), 2.65 (4 H, q, J=7.6 Hz), 2.77–2.84 (2 H, m), 3.59 (2 H, s), 3.61 (2 H, s), 3.79 (3 H, s), 5.07 (2 H, s, br), 6.61 (1 H, s), 7.02 (2 H, s), 7.06 ppm (1 H, s); 13C NMR (67.5 MHz, CDCl3): δ=15.5, 27.9, 50.2, 55.6, 56.1, 56.2, 62.6, 68.9, 105.8, 111.1, 123.9, 127.3, 133.3, 134.4, 136.0, 137.3, 149.3, 153.4 ppm; LC-MS (ES+): m/z 433.3 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C23H33N2O4S+: 433.2156, found: 433.2147.
(±)-2-(3-Bromo-4,5-dimethoxybenzyl)-7-methoxy-3-methyl-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (12j)
Method as for 12e using compound 11j (212 mg, 0.5 mmol) and sulfamoyl chloride (1.5 mmol) in DMA (3.0 mL) at RT for 2 h. The residue was stirred in CH2Cl2, filtered and dried to afford compound 12j as a pale yellow solid (241 mg, 96%): mp: 155–158 °C; 1H NMR (270 MHz, CDCl3): δ= 0.98 (3 H, d, J= 6.6 Hz), 2.39 (1 H, dd, J= 16.6, 5.6 Hz), 2.81 (1 H, dd, J= 16.2, 4.4 Hz), 2.95 (1 H, sext, J= 5.6 Hz), 3.30–3.62 (4 H, m), 3.66 (3 H, d, J= 2.2 Hz), 3.69 (3 H, d, J= 2.2 Hz), 3.71 (3 H, d, J= 2.2 Hz), 6.45 (2 H, s), 6.50 (1 H, s), 6.76 (1 H, s), 6.94 (1 H, s), 6.97 ppm (1 H, d, J=1.7 Hz); LC-MS (ES+): m/z 501.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H26BrN2O6S+: 501.0617, found: 501.0644;.
2-(3-Bromo-4,5-dimethoxybenzyl)-7-ethyl-6-hydroxy-1,2,3,4-tetrahydroisoquinoline (14)
Method as for 10b using compound 13[3] (266 mg, 1.5 mmol), DIPEA (581 mg, 4.5 mmol) and 3-bromo-4,5-dimethoxybenzyl bromide[15] (511 mg, 1.65 mmol) in DMF (5 mL) at 80 °C for 20 h. Flash column chromatography (CHCl3/acetone 4:1) afforded compound 14 as a tan solid (246 mg, 40%): mp: 156–160 °C; 1H NMR (270 MHz, CDCl3): δ= 1.12 (3 H, t, J= 7.4 Hz), 2.50 (2 H, q, J= 7.4 Hz), 2.60 (2 H, d, J= 5.2 Hz), 2.67 (2 H, d, J= 5.2 Hz), 3.51 (2 H, s), 3.57 (2 H, s), 3.75 (3 H, s), 3.84 (3 H, s), 6.12 (1 H, s), 6.70 (1 H, s), 6.98 (1 H, s), 7.12 ppm (1 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 14.1, 22.7, 28.1, 50.7, 55.4, 55.9, 60.5, 62.1, 112.6, 114.8, 117.1, 125.3, 127.0, 128.5, 132.1, 134.8, 145.5, 145.5, 152.5, 153.6 ppm; LC-MS (ES+): m/z 406.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H25BrNO3+: 406.1012, found: 406.1005.
2-(3-Bromo-4,5-dimethoxybenzyl)-7-ethyl-6-sulfamoyloxy-1,2,3,4-tetrahydroisoquinoline (15)
Method as for 12e using compound 14 (150 mg, 0.37 mmol) and sulfamoyl chloride (1.48 mmol) in DMA (1.0 mL) at RT for 20 h. Flash column chromatography (CHCl3/acetone 4:1) afforded compound 15 as a pale yellow foam (85 mg, 47%): 1H NMR (270 MHz, CDCl3): δ= 1.16 (3 H, t, J= 7.6), 2.64 (2 H, q, J= 7.6 Hz), 2.70 (2 H, t, J= 5.5 Hz), 2.84 (2 H, t, J= 5.5 Hz), 3.56–3.58 (4 H, m), 3.84 (6 H, s), 6.88–6.93 (2 H, m), 7.08–7.13 ppm (2 H, m); 13C NMR (67.5 MHz, CDCl3): δ= 14.3, 22.7, 28.4, 50.1, 55.2, 56.1, 60.6, 61.7, 112.3, 117.3, 121.4, 125.0, 127.9, 133.1, 133.2, 134.4, 135.0, 145.6, 146.7, 153.7 ppm; LC-MS (ES+): m/z 485.2 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H26BrN2O5S+: 485.0740, found: 485.0736.
6,7-Dimethoxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (17a)
Compound 16 (0.23 g, 1.0 mmol), 3,4,5-trimethoxybenzyl chloride (0.26 g, 1.2 mmol) and Et3N (0.5 mL, 3.6 mmol) were placed in a 10 mL microwave vessel and dissolved in EtOH (2.5 mL). The mixture was then irradiated at 130 °C for 1 h in the microwave oven. After cooling to RT, the mixture was poured into H2O and extracted with EtOAc. The organic layer was washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 6:1→1:1) gave a white solid that was stirred in Et2O, filtered and dried to afford compound 17a as a white powder (230 mg, 62%): mp: 118–119 °C; 1H NMR (270 MHz, CDCl3): δ= 2.70 (2 H, t, J= 5.6 Hz), 2.81 (2 H, t, J= 5.6 Hz), 3.55 (2 H, s), 3.59 (2 H, s), 3.81 (3 H, s), 3.83 (3 H, s), 3.84 (3 H, s), 3.85 (6 H, s), 6.50 (1 H, s), 6.60 (1 H, s), 6.62 ppm (2 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 28.8, 50.7, 55.9, 56.0, 56.2, 61.0, 63.1, 105.6, 109.5, 111.4, 126.3, 126.8, 134.5, 136.9, 147.3, 147.6, 153.2 ppm; LC-MS (ES+): m/z 374.27 [M+H]+; Anal. calcd for C21H27NO5: C 67.54, H 3.75, N 7.29, found: C 67.5, H 3.76, N 7.10.
6-Acetoxy-7-methoxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (17b)
Compound 3a (108 mg, 0.3 mmol), Et3N (0.21 mL, 1.6 mmol) and Ac2O (0.16 mL, 1.6 mmol) were stirred in CHCl3 (10 mL) at RT for 24 h. The reaction mixture was diluted with CHCl3 (30 mL) and washed with H2O (4×30 mL) and brine, dried (MgSO4), filtered and concentrated in vacuo. The residue was stirred in Et2O, filtered and dried in vacuo to afford compound 17b as a white powder (95 mg, 79%): mp: 143–144 °C; 1H NMR (270 MHz, CDCl3): δ= 2.28 (3 H, s), 2.69–2.83 (4 H, m), 3.60 (4 H, s), 3.75 (3 H, s), 3.84 (3 H, s), 3.85 (6 H, s), 6.58 (1 H, s), 6.62 (2 H, s), 6.77 ppm (1 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 20.7, 28.3, 50.5, 55.9, 56.0, 56.2, 60.9, 62.8, 105.7, 110.5, 122.7, 124.1, 126.5, 133.3, 136.9, 138.1, 149.1, 153.3, 169.4 ppm; LC-MS (ES+): m/z 402.24 [M+H]+; Anal. calcd for C22H27NO6: C 65.82, H 6.78, N 3.49, found: C 65.8, H 6.81, N 3.49.
7-Methoxy-6-methanesulfonyloxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (17c)
Compound 3a (80 mg, 0.22 mmol) was dissolved in pyridine (1.0 mL) and cooled to 0 °C. Methanesulfonyl chloride (20 μL, 0.26 mmol) was added, and the reaction mixture was stirred at 0 °C for 2 h and then at RT for 4 h. The mixture was diluted with EtOAc, washed with H2O and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane/EtOAc 1:1) gave a solid that was stirred in Et2O, filtered and dried to afford compound 17c as a white solid (70 mg, 73%): mp: 134–135 °C; 1H NMR (270 MHz, CDCl3): δ= 2.68–2.84 (4 H, m), 3.15 (3 H, s), 3.57 (2 H, s), 3.59 (2 H, s), 3.81 (3 H, s), 3.83 (3 H, s), 3.85 (6 H, s), 6.61 (3 H, s), 7.04 ppm (1 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 28.3, 38.2, 50.4, 55.9, 56.1, 56.2, 61.0, 62.9, 105.6, 110.9, 124.5, 127.4, 134.2, 135.0, 136.7, 137, 149.3, 153.3 ppm; LC-MS (ES+): m/z 438.14 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C21H28NO7S+: 438.1581, found: 438.1569.
2-(3-Chloro-4,5-dimethoxybenzoyl)-6-hydroxy-1,2,3,4-tetrahydroisoquinoline (19a)
Method as for 9a using compound 18b (345 mg, 1.5 mmol), 3-chloro-4,5-dimethoxybenzoic acid (357 mg, 1.65 mmol), EDCI (575 mg, 3.0 mmol) and Et3N (0.25 mL, 1.8 mmol) in CH2Cl2 (10 mL) at RT for 18 h. The mixture was diluted with CH2Cl2, washed with H2O (10 mL), citric acid (10%, 10 mL), Na2CO3 (sat.) and brine, dried (MgSO4), filtered and concentrated in vacuo. Flash column chromatography (hexane→hexane/EtOAc 1:1) afforded compound 19a as an off-white solid (313 mg, 60%): mp: 151–164 °C; 1H NMR (270 MHz, CDCl3): δ= 2.75–2.82 (2 H, m), 3.61 (1 H, s, br), 3.81 (3 H, s, br), 3.86 (3 H, s), 3.85–3.92 (1 H, m), 4.49 (1 H, s, br), 4.74 (1 H, s, br), 6.56–6.90 (4 H, m), 7.02 (1 H, s), 7.80 ppm (1 H, s, br); LC-MS (ES+): m/z 348.4 [M+H]+; HRMS (ES+): m/z [M+H] + calcd for C18H19ClNO4+: 348.0997, found: 348.0987.
2-(3,5-Dimethoxy-4-ethoxybenzoyl)-6-hydroxy-1,2,3,4-tetrahydroisoquinoline (19b)
Method as for 19a using compound 18b (345 mg, 1.5 mmol), 4-ethoxy-3,5-dimethoxybenzoic acid[26] (373 mg, 1.65 mmol), EDCI (575 mg, 3.0 mmol) and Et3N (0.25 mL, 1.8 mmol) in CH2Cl2 (10 mL) at RT for 18 h. Flash column chromatography (hexane→hexane/EtOAc 1:4) afforded compound 19b as a white foam (296 mg, 55%): 1H NMR (270 MHz, CDCl3): δ=1.34 (3 H, t, J=6.9 Hz), 2.75–2.86 (2 H, m), 3.60–3.67 (1 H, m), 3.81 (6 H, s), 3.81–3.90 (1 H, m), 4.06 (2 H, q, J= 6.9 Hz), 4.51 (1 H, s, br), 4.76 (1 H, s, br), 6.60–6.97 ppm (5 H, m); LC-MS (ES+): m/z 358.1 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H24NO5+: 358.1649, found: 358.1637.
6-Hydroxy-2-(3,4,5-triethoxybenzoyl)-1,2,3,4-tetrahydroisoquinoline (19c)
Method as for 19a using compound 18b (690 mg, 3.0 mmol), 3,4,5-triethoxybenzoic acid (1.14 g, 4.5 mmol), EDCI (1.15 g, 6.0 mmol) and Et3N (0.5 mL, 3.6 mmol) in CH2Cl2 (10 mL) and THF (5 mL) at RT for 18 h. Flash column chromatography (hexane→hexane/EtOAc 3:2) afforded compound 19c as a white solid (731 mg, 63%): mp: 157–158 °C; 1H NMR (270 MHz, CDCl3): δ= 1.32–1.42 (9 H, m), 2.80 (2 H, s, br), 3.62 and 3.92 (2 H, s, br), 3.98–4.11 (6 H, m), 4.51 and 4.75 (2 H, s, br), 5.97 (1 H, s, br), 6.62–6.66 (4 H, m), 6.67 and 7.00 ppm (1 H, s, br); LC-MS (APCI+): m/z 386.5 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C22H28NO5+: 386.1962, found: 386.1960.
6-Hydroxy-2-(3,4,5-trimethoxyphenacetyl)-1,2,3,4-tetrahydroisoquinoline (19d)
Method as for 19a using compound 18b (345 mg, 1.5 mmol) and 3,4,5-trimethoxyphenylacetic acid (373 mg, 1.65 mmol), EDCI (575 mg, 3.0 mmol) and Et3N (0.25 mL, 1.8 mmol) in CH2Cl2 (10 mL) at RT for 18 h. Flash column chromatography (hexane→EtOAc) afforded compound 19d as a gummy foam (470 mg, 88%): 1H NMR (270 MHz, CDCl3): δ= 2.66 and 2.78 (2 H, t, J= 6.2 Hz), 3.64 and 3.81 (2 H, t, J= 6.2 Hz), 3.70–3.82 (11 H, m), 4.55 and 4.67 (2 H, s), 5.50 (1 H, s, br), 6.39 and 6.46 (2 H, s), 6.56–6.68 (2 H, m), 6.83 and 6.97 ppm (1 H, d, J= 8.1 Hz); LC-MS (APCI): m/z 358.3 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H24NO5+: 358.1649, found: 358.1643.
6-Benzyloxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (20a)
Method as for 17a using compound 18c (431 mg, 1.8 mmol), 3,4,5-trimethoxybenzyl chloride (433 mg, 2 mmol) and Et3N (0.5 mL, 3.6 mmol) in EtOH (2.5 mL) at 130 °C for 1.5 h in the microwave oven. Flash column chromatography (hexane/EtOAc 3:1→1:1) gave a solid that was stirred in Et2O, filtered and dried to afford compound 20a as a yellow powder (450 mg, 60%): mp: 103–104 °C; 1H NMR (270 MHz, CDCl3): δ= 2.69 (2 H, t, J= 5.8 Hz), 2.86 (2 H, t, J= 5.8 Hz), 3.58 (2 H, s), 3.60 (2 H, s), 3.84 (3 H, s), 3.85 (6 H, s), 5.03 (2 H, s), 6.62 (2 H, s), 6.74–6.78 (2 H, m), 6.92 (1 H, d, J= 8.0 Hz), 7.26–7.43 ppm (5 H, s); 13C NMR (67.5 MHz, CDCl3): δ= 29.4, 50.3, 55.7, 56.1, 60.8, 63.0, 69.9, 105.5, 112.8, 114.3, 127.4, 127.5, 127.9, 128.5, 134.4, 135.6, 136.7, 137.1, 153.1, 157.1 ppm; LC-MS (ES+): m/z 420.25 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C26H30NO4+: 420.2169, found: 420.2158; Anal. calcd for C26H29NO4: C 74.44, H 6.97, N 3.34, found: C 74.3, H 6.91, N 3.30.
6-Hydroxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (20b)
Compound 20a (300 mg, 0.72 mmol) was treated with Pd/C (10%, 40 mg) in THF (20 mL) and MeOH (20 mL) under H2 at RT for 1.5 h. The reaction mixture was then filtered through Celite and washed with MeOH (4×10 mL). The combined filtrates were concentrated in vacuo, and the residue was stirred in EtOAc, filtered and dried to afford compound 20b as a white powder (175 mg, 74%): mp: 197–199 °C; 1H NMR (270 MHz, [D6]DMSO): δ = 2.60 (2 H, t, J= 5.4 Hz), 2.72 (2 H, t, J= 5.4 Hz), 3.45 (2 H, s), 3.54 (2 H, s), 3.64 (3 H, s), 3.75 (6 H, s), 6.48–6.53 (2 H, m), 6.65 (2 H, s), 6.81 (1 H, d, J= 8.0 Hz), 9.11 ppm (1 H, s); 13C NMR (67.5 MHz, [D6]DMSO): δ= 28.8, 50.2, 55.2, 55.8, 60.0, 62.1, 105.5, 113.0, 114.6, 125.2, 127.3, 134.3, 135.1, 136.2, 152.8, 155.4 ppm; LC-MS (ES−): m/z 328.16 [M—H] −; HRMS (ES+): m/z [M+H]+ calcd for C19H24NO4+: 330.1700, found: 330.1689; Anal. calcd for C19H23NO4: C 69.28, H 7.04, N 4.25, found: C 69.1, H 7.33, N 4.56.
6-Methoxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (20c)
Method as for 17a using compound 18a (300 mg, 1.5 mmol), 3,4,5-trimethoxybenzyl chloride (325 mg, 1.5 mmol) and Et3N (0.5 mL, 3.6 mmol) in EtOH (5 mL) at 120 °C for 1 h in the microwave oven. Flash column chromatography (hexane→hexane/EtOAc 1:1) afforded compound 20c as a white solid (81 mg, 16%): mp: 102–103 °C; 1H NMR (270 MHz, CDCl3): δ= 2.69 (2 H, t, J= 7.8 Hz), 2.87 (2 H, t, J= 7.8 Hz), 3.58 (2 H, s), 3.59 (2 H, s), 3.77 (3 H, s), 3.84 (3 H, s), 3.85 (6 H, s), 6.62–6.71 (4 H, m), 6.92 ppm (1 H, d, J= 8.2 Hz); 13C NMR (67.5 MHz, [D6]DMSO): δ=15.5, 28.5, 50.5, 55.4, 56.0, 63.0, 68.9, 106.3, 113.7, 115.1, 125.4, 127.5, 132.8, 135.0, 135.9, 153.3, 154.9 ppm; LRMS (ES+): m/z 344.4 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H26NO4+: 344.1856, found: 344.1842.
6-Sulfamoyloxy-2-(3,4,5-trimethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (20d)
Method as for 12e using compound 20b (100 mg, 0.3 mmol) and sulfamoyl chloride (0.6 mmol) in DMA (1.0 mL) at RT for 24 h. Flash column chromatography (hexane/EtOAc 3:1→EtOAc) afforded compound 20d as a white powder (110 mg, 88%): mp: 173–174 °C; 1H NMR (270 MHz, [D6]DMSO): δ= 2.65 (2 H, t, J=5.5 Hz), 2.84 (2 H, t, J=5.5 Hz), 3.56 (2 H, s), 3.58 (2 H, s), 3.64 (3 H, s), 3.75 (6 H, s), 6.65 (2 H, s), 6.98–7.02 (2 H, m), 7.12 (1 H, d, J=8.0 Hz), 7.91 ppm (2 H, br); 13C NMR (67.5 MHz, [D6]DMSO): δ=28.8, 49.7, 55.1, 55.8, 60.0, 61.8, 105.4, 119.5, 121.8, 127.7, 133.3, 134.2, 135.8, 136.3, 148.3, 152.8 ppm; HRMS (ES+): m/z [M+H]+ calcd for C19H25N2O6S+: 409.1428, found: 409.1428; Anal. calcd for C19H24N2O6S: C 55.87, H 5.92, N 6.86, found: C 55.6, H 6.02, N 6.51.
2-(3-Chloro-4,5-dimethoxybenzyl)-6-hydroxy-1,2,3,4-tetrahydroisoquinoline (20e)
Method as for 10a using compound 19a (203 mg, 0.58 mmol) and LiAlH4 (111 mg, 2.9 mmol) in THF (8 mL) at RT for 18 h. The reaction mixture was cooled to 0 °C, and H2O (0.15 mL) was added slowly followed by aq NaOH (15 %, 0.15 mL) and then H2O (0.45 mL). The mixture was stirred at RT for 0.25 h, then diluted with EtOAc (100 mL) and stirred another 0.25 h, filtered, dried (MgSO4) and concentrated in vacuo. Flash column chromatography (hexane→hexane/EtOAc 2:3) afforded compound 20e as a colourless solid (134 mg, 69%): mp: 155–157 °C; 1H NMR (270 MHz, CDCl3): δ=2.69 (4 H, s), 3.51 (2 H, s), 3.56 (2 H, s), 3.73 (3 H, s), 3.84 (3 H, s), 6.25 (1 H, d, J=2.3 Hz), 6.45 (1 H, dd, J=8.3, 2.3 Hz), 6.76 (1 H, d, J=8.3 Hz), 6.93 ppm (2 H, s); 13C NMR (67.5 MHz, CDCl3): δ=28.6, 50.5, 55.4, 56.0, 60.6, 62.2, 111.8, 113.6, 115.1, 122.5, 125.5, 127.6, 127.7, 134.1, 135.1, 144.4, 153.8, 154.6 ppm; LC-MS (ES+): m/z 333.9 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C18H21ClNO3+: 334.1204, found: 334.1197; Anal. calcd for C18H20ClNO3: C 64.77, H 6.04, N 4.20, found: C 64.7, H 6.03, N 4.19.
2-(3-Bromo-4,5-dimethoxybenzyl)-6-hydroxy-1,2,3,4-tetrahydroisoquinoline (20f)
Method as for 10b using compound 18b (200 mg, 0.87 mmol), 3-bromo-4,5-dimethoxybenzyl bromide[15] (296 mg, 0.96 mmol), DIPEA (790 mg, 6.1 mmol) in DMF (5.0 mL) at 80 °C for 60 h. Flash column chromatography (hexane→hexane/EtOAc 2:3) afforded compound 20f as a white solid (246 mg, 85%): mp: 161–165 °C; 1H NMR (270 MHz, CDCl3): δ=2.68–2.72 (4 H, m), 3.52 (2 H, s), 3.56 (2 H, s), 3.76 (3 H, s), 3.83 (3 H, s), 6.33 (1 H, d, J=2.2 Hz), 6.49 (1 H, dd, J=8.1, 2.2 Hz), 6.78 (1 H, d, J= 8.1 Hz), 6.96 (1 H, d, J=1.5 Hz), 7.09 ppm (1 H, d, J=1.5 Hz); 13C NMR (67.5 MHz, CDCl3): δ=28.7, 50.5, 55.4, 56.0, 60.6, 62.1, 112.4, 113.5, 115.0, 117.2, 125.2, 125.8, 127.6, 135.0, 135.2, 145.5, 153.7, 154.5 ppm; LC-MS (ES+): m/z 377.7 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C18H21BrNO3+: 378.0699, found: 378.0685; Anal. calcd for C18H20BrNO3: C 57.15, H 5.33, N 3.70, found: C 57.0, H 5.39, N 3.61.
2-(3,5-Dimethoxy-4-ethoxybenzyl)-6-hydroxy-1,2,3,4-tetrahydroisoquinoline (20g)
Method as for 20e using compound 19b (280 mg, 0.78 mmol) and LiAlH4 (149 mg, 3.9 mmol) in THF (8 mL) at RT for 18 h. Flash column chromatography (hexane→EtOAc) afforded compound 20g as a colourless solid (179 mg, 67%): mp: 154–157 °C; 1H NMR (270 MHz, CDCl3): δ=1.33 (3 H, t, J=7.2 Hz), 2.68 (4 H, s), 3.52 (2 H, s), 3.59 (2 H, s), 3.75 (6 H, s), 4.02 (2 H, q, J= 7.2 Hz), 6.23 (1 H, d, J=1.9 Hz), 6.44 (1 H, dd, J=8.3, 1.9 Hz), 6.61 (2 H, s), 6.74 ppm (1 H, d, J=8.3 Hz); LC-MS (ES+): m/z 344.0 [M+ H]+; HRMS (ES+): m/z calcd for C20H26NO4+: 344.1856, found: 344.1842; Anal. calcd for C20H25NO4: C 69.95, H 7.34, N 4.08, found: C 69.60, H 7.35, N 4.00.
6-Hydroxy-2-(3,4,5-triethoxybenzyl)-1,2,3,4-tetrahydroisoquinoline (20h)
Method as for 20e using compound 19c (258 mg, 0.67 mmol) and LiAlH4 (127 mg, 3.4 mmol) in THF (8 mL) at RT for 18 h. Flash column chromatography (hexane→hexane/EtOAc 1:4) afforded compound 20h as a white solid (75 mg, 38%): mp: 140–142 °C; 1H NMR (400 MHz, CDCl3): δ=1.34 (3 H, t, J=7.2 Hz), 1.35 (6 H, t, J=6.8 Hz), 2.68 (4 H, s), 3.52 (2 H, s), 3.57 (2 H, s), 3.97 (4 H, q, J=6.8 Hz), 4.04 (2 H, q, J=7.2 Hz), 6.27 (1 H, d, J=2.4 Hz), 6.45 (1 H, dd, J=8.4, 2.4 Hz), 6.59 (2 H, s), 6.75 ppm (1 H, d, J=8.4 Hz); 13C NMR (100 MHz, CDCl3): δ=14.9, 15.6, 28.6, 50.5, 55.4, 63.0, 64.5, 68.8, 107.7, 113.6, 115.1, 125.5, 127.5, 132.6, 135.1, 136.8, 152.7, 154.7 ppm; LC-MS (ES+): m/z 372.4 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C22H30NO4+: 372.2169, found: 372.2156; Anal. calcd. for C22H29NO4: C 71.13, H 7.87, N 3.77, found: C 70.9, H 7.87, N 3.77.
6-Hydroxy-2-(3,4,5-trimethoxybenzoyl)-1,2,3,4-tetrahydroisoquinoline (20i)
Compound 18b (200 mg, 0.87 mmol) and 3,4,5-trimethoxybenzoyl chloride (200 mg, 0.87 mmol) were dissolved in CH2Cl2 (10 mL) and cooled to 0 °C. Et3N (0.36 mL, 2.6 mmol) was added. The reaction mixture was stirred at RT for 18 h, then diluted with CH2Cl2, washed with H2O (10 mL) and brine, dried (MgSO4) and concentrated in vacuo. Flash column chromatography (hexane→hexane/EtOAc 1:4) afforded compound 20i as an off-white solid (200 mg, 67%): mp: 175–176 °C; 1H NMR (270 MHz, CDCl3): δ=2.78–2.86 (2 H, m), 3.62 (1 H, s, br), 3.82–3.88 (10 H, m), 4.51 (1 H, s, br), 4.76 (1 H, s, br), 6.60–6.70 ppm (5 H, m); LC-MS (ES+): m/z 344.0 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C19H22NO5+: 344.1492, found: 344.1480.
6-Hydroxy-2-(3,4,5-trimethoxyphenethyl)-1,2,3,4-tetrahydroisoquinoline (20j)
Method as for 20e using compound 19d (200 mg, 0.56 mmol) and LiAlH4 (106 mg, 2.79 mmol) in THF (8 mL) at RT for 18 h. Flash column chromatography (hexane→EtOAc) afforded compound 20j as a colourless solid (120 mg, 63%): mp: 180–186 °C; 1H NMR (270 MHz, CDCl3): δ=2.72–2.87 (8 H, m), 3.64 (2 H, s), 3.82 (3 H, s), 3.84 (6 H, s), 5.02 (1 H, s, br), 6.44 (2 H, s), 6.54 (1 H, d, J=2.5 Hz), 6.59 (1 H, dd, J=8.3, 2.5 Hz), 6.89 ppm (1 H, d, J=8.3 Hz); 13C NMR (67.5 MHz, CDCl3): δ=28.7, 34.0, 51.0, 55.4, 56.1, 60.2, 60.8, 105.6, 113.8, 115.2, 125.4, 127.6, 135.1, 135.8, 136.3, 153.1, 154.9 ppm; LC-MS (ES+): m/z 344.3 [M+H]+; HRMS (ES+): m/z [M+H]+ calcd for C20H26NO4+: 344.1856, found: 344.1840; Anal. calcd for C20H25NO4·0.25H2O: C 69.04, H 7.39, N 4.03, found: C 69.1, H 7.31, N 3.91.
Acknowledgments
This work was supported by Sterix Ltd., a member of the IPSEN Group. We thank Ms Alison Smith (University of Bath) for technical support and the NCI DTP for providing in vitro and in vivo screening resources.
References
- 1.a) Nowell PC, Hungerford DA. Science. 1960;132:1497. [Google Scholar]; b) Nowell PC. J Clin Invest. 2007;117:2033–2035. doi: 10.1172/JCI31771. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M. Ann Intern Med. 2003;138:819–830. doi: 10.7326/0003-4819-138-10-200305200-00010. [DOI] [PubMed] [Google Scholar]
- 2.Faderl S, Talpaz M, Estrov Z, Kantarjian HM. Ann Intern Med. 1999;131:207–219. doi: 10.7326/0003-4819-131-3-199908030-00008. [DOI] [PubMed] [Google Scholar]
- 3.Leese MP, Jourdan F, Kimberley MR, Cozier GE, Thiyagarajan N, Stengel C, Regis-Lydi S, Foster PA, Newman SP, Acharya KR, Ferrandis E, Purohit A, Reed MJ, Potter BVL. Chem Commun. 2010;46:2907–2909. doi: 10.1039/c002558e. [DOI] [PubMed] [Google Scholar]
- 4.Bubert C, Leese MP, Mahon MF, Ferrandis E, Regis-Lydi S, Kasprzyk PG, Newman SP, Ho YT, Purohit A, Reed MJ, Potter BVL. J Med Chem. 2007;50:4431–4443. doi: 10.1021/jm070405v. [DOI] [PubMed] [Google Scholar]
- 5.Jourdan F, Leese MP, Dohle W, Ferrandis E, Newman SP, Chander S, Purohit A, Potter BVL. J Med Chem. 2011;54:4863–4879. doi: 10.1021/jm200483x. [DOI] [PubMed] [Google Scholar]
- 6.Jourdan F, Leese MP, Dohle W, Hamel E, Ferrandis E, Newman SP, Purohit A, Reed MJ, Potter BVL. J Med Chem. 2010;53:2942–2951. doi: 10.1021/jm9018806. [DOI] [PubMed] [Google Scholar]
- 7.Leese MP, Hejaz HAM, Mahon MF, Newman SP, Purohit A, Reed MJ, Potter BVL. J Med Chem. 2005;48:5243–5256. doi: 10.1021/jm050066a. [DOI] [PubMed] [Google Scholar]
- 8.Leese MP, Jourdan FL, Gaukroger K, Mahon MF, Newman SP, Foster PA, Stengel C, Regis-Lydi S, Ferrandis E, Di Fiore A, De Simone G, Supuran CT, Purohit A, Reed MJ, Potter BVL. J Med Chem. 2008;51:1295–1308. doi: 10.1021/jm701319c. [DOI] [PubMed] [Google Scholar]
- 9.Leese MP, Leblond B, Smith A, Newman SP, Di Fiore A, De Simone G, Supuran CT, Purohit A, Reed MJ, Potter BVL. J Med Chem. 2006;49:7683–7696. doi: 10.1021/jm060705x. [DOI] [PubMed] [Google Scholar]
- 10.Leese MP, Jourdan F, Dohle W, Kimberley MR, Thomas MP, Bai R, Hamel E, Ferrandis E, Potter BVL. ACS Med Chem Lett. 2012;3:5–9. doi: 10.1021/ml200232c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pettit GR, Minardi MD, Rosenberg HJ, Hamel E, Bibby MC, Martin SW, Jung MK, Pettit RK, Cuthbertson TJ, Chapuis JC. J Nat Prod. 2005;68:1450–1458. doi: 10.1021/np058038i. [DOI] [PubMed] [Google Scholar]
- 12.Leese MP, Jourdan FL, Major MR, Dohle W, Hamel E, Ferrandis E, Fiore A, Kasprzyk PG, Potter BVL. ChemMedChem. 2014;9:85–108. doi: 10.1002/cmdc.201300261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dohle W, Leese MP, Jourdan FL, Major MR, Bai R, Hamel E, Ferrandis E, Kasprzyk PG, Fiore A, Newman SP, Purohit A, Potter BVL. ChemMedChem. 2014;9:350–370. doi: 10.1002/cmdc.201300412. [DOI] [PubMed] [Google Scholar]
- 14.Okada M, Iwashita S, Koizumi N. Tetrahedron Lett. 2000;41:7047–7051. [Google Scholar]
- 15.a) Fürstner A, Stelzer F, Rumbo A, Krause H. Chem Eur J. 2002;8:1856–1871. doi: 10.1002/1521-3765(20020415)8:8<1856::AID-CHEM1856>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]; b) Takenaka Y, Tanahashi T, Nagakura N, Hamada N. Chem Pharm Bull. 2003;51:794–797. doi: 10.1248/cpb.51.794. [DOI] [PubMed] [Google Scholar]; c) Al-Farhan E, Falana OM, Keehn PM, Stevenson R. Tetrahedron Lett. 1992;33:5885–5886. [Google Scholar]; d) Wang Y-C, Georghiou PE. Org Lett. 2002;4:2675–2678. doi: 10.1021/ol0261635. [DOI] [PubMed] [Google Scholar]; e) Akbaba Y, Balaydin HT, Goeksu S, Sahin E, Menzek A. Helv Chim Acta. 2010;93:1127–1135. [Google Scholar]
- 16.a) Zhong HM, Villani FJ, Marzouq R. Org Process Res Dev. 2007;11:463–465. [Google Scholar]; b) Sall DJ, Grunewald GL. J Med Chem. 1987;30:2208–2216. doi: 10.1021/jm00395a006. [DOI] [PubMed] [Google Scholar]
- 17.Yang SH, Song CH, Van HTM, Park E, Khadka DB, Gong E-Y, Lee K, Cho WJ. J Med Chem. 2013;56:3414–3418. doi: 10.1021/jm3014103. [DOI] [PubMed] [Google Scholar]
- 18.Hollingshead M, Alley MC, Camalier RF, Abbott BJ, Mayo JG, Malspeis L, Grever MR. Life Sci. 1995;57:131–141. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
- 19.Gaukroger K, Hadfield JA, Lawrence NJ, Nolan S, McGown AT. Org Biomol Chem. 2003;1:3033–3037. doi: 10.1039/b306878a. [DOI] [PubMed] [Google Scholar]
- 20.Hamel E, Lin CM. Biochemistry. 1984;23:4173–4184. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 21.Hamel E. Cell Biochem Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 22.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 23.Lin CM, Ho HH, Pettit GR, Hamel E. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 24.Appel R, Berger G. Chem Ber. 1958;91:1339–1341. [Google Scholar]
- 25.Woo LWL, Lightowler M, Purohit A, Reed MJ, Potter BVL. J Steroid Biochem Mol Biol. 1996;57:79–88. doi: 10.1016/0960-0760(95)00244-8. [DOI] [PubMed] [Google Scholar]
- 26.Tetrahydroisoquinolines as Tumour Growth Inhibitors,; Jourdan F, Kimberley M, Leese M, Potter BVL, Purohit A, Reed MJ. Sterix Ltd; Slough, UK: WO 2008/117061. Int PCT Pat Appl. 2008 Oct;A2
- 27.Roth B, Aig E. J Med Chem. 1987;30:1998–2004. doi: 10.1021/jm00394a012. [DOI] [PubMed] [Google Scholar]
- 28.McNulty PJ, Pearson DE. J Am Chem Soc. 1959;81:612–618. [Google Scholar]