

Summary
A multi-disciplinary scientific conference focused on diffuse and interstitial lung diseases in children was held in La Jolla, CA in June 2012. The conference brought together clinicians (including Pediatric and Adult Pulmonologists, Neonatologists, Pathologists, and Radiologists), clinical researchers, basic scientists, government agency representatives, patient advocates, as well as children affected by diffuse lung disease (DLD) and their families, to review recent scientific developments and emerging concepts in the pathophysiology of childhood DLD. Invited speakers discussed translational approaches, including genetics and proteomics, epigenetics and epigenomics, models of DLD, including animal models and induced pluripotent stem cells, and regenerative medicine approaches. The presentations of the invited speakers are summarized here.
Keywords: diffuse lung disease, childhood interstitial lung disease, genetics, epigenetics, stem cell
INTRODUCTION
In June of 2012, a scientific conference on diffuse lung disease (DLD) in Children was convened in La Jolla, California, supported by the National Heart, Lung and Blood Institute (NHLBI, see Acknowledgements, for additional supporters). The conference brought together clinicians (including Pediatric and Adult Pulmonologists, Neonatologists, Pathologists, and Radiologists), clinical researchers, basic scientists, government agency representatives, patient advocates, as well as children affected by DLD and their families, from five continents, to review recent scientific developments and emerging concepts in the pathophysiology of childhood DLD. This conference had several important antecedents. In recognition of the unique features of pediatric interstitial lung disease (ILD) and the lack of knowledge and understanding of these disorders, the NHLBI convened workshops in 1988 and 1989. The participants advocated a multi-disciplinary approach including pediatric pulmonary clinicians, pathologists, and radiologists, formation of a patient data registry, and development of standard protocols for diagnosis. Unfortunately, funding was not available to achieve these goals. In 2004, with support from the NIH Rare Lung Diseases Consortium (RLDC) an interdisciplinary group of clinicians and investigators from multiple institutions, subsequently named the ChILD Research Network (ChILDRN), was convened to begin to codify the clinical spectrum of childhood DLD and to establish a research agenda. Importantly, families of affected patients were invited to attend the initial conference and they formed the Children’s Interstitial and Diffuse Lung Disease (chILD) Foundation, which has held annual meetings in conjunction with members of the ChILDRN. The scientific conference which is summarized here was scheduled immediately preceding the annual meeting of the chILD Foundation and the ChILDRN, in order to maximize interaction of families and clinicians with the speakers and attendees of the conference. Eminent investigators from fields which represent emerging paradigms in lung biology relevant to lung development, repair and regeneration were invited to speak, and abstracts of their presentations comprise the sections of this document (the initials following the section headings refer to the authors, who are listed as the authors of the overall document). Others who contributed to the content of each section are listed in the Acknowledgements section. In addition, selected abstracts submitted for the conference were chosen for oral presentation, and these will be published in a separate document. Family members presented brief narratives of their own experiences relevant to the content of the scientific sessions. It is hoped that the interactions among the attendees will promote novel collaborations which will move this field forward and offer new hope for children affected by DLD and their families.
INHERITED DISORDERS OF SURFACTANT METABOLISM (AH)
Mutations in critical molecules in the surfactant metabolic pathway result in acute neonatal respiratory failure or chronic ILD that presents in early infancy and beyond. In the acute neonatal presentation, term newborns develop severe respiratory failure shortly after birth that may be transiently responsive to intensive care interventions, including mechanical ventilation, surfactant replacement, inhaled nitric oxide, or corticosteroids. This acute form is typical for infants with recessive mutations in the genes encoding surfactant protein-B (SFTPB) or the ATP Binding Cassette member A3 (ABCA3), or dominant mutations in the thyroid transcription factor gene (NKX2-1), but has also been seen in infants with dominant mutations in gene encoding surfactant protein-C (SFTPC).1–5 Typically, infants with this severe neonatal presentation succumb to intractable respiratory failure or are considered for lung transplantation, although some infants with partial deficiencies can survive beyond infancy. Later onset, less severe, and more variable disease is associated with mutations in SFTPC, NKX2-1, and ABCA3. Some children present in the newborn period with mild respiratory dysfunction, but most present in the first several months with gradual onset of respiratory insufficiency, failure to gain weight, and ILD on chest radiographs.6–9 Absence of a consistent history of an associated viral illness at disease onset and the identification of asymptomatic carriers of dominant mutations in SFTPC suggest that other genetic or environmental modifiers influence the presentation of disease associated with mutations in all these genes. This variability in severity and course of the disease is not mutation specific and precludes definitive prediction of outcome.
SP-B deficiency is a rare recessive disorder with a disease frequency of approximately 1 per million live births in populations of European descent, though isolated cases have been identified in other ethnic groups.10,11 The frequency of SP-C associated disease is unknown but is probably very rare; the population-based frequency of mutations in SFTPC is significantly less than 0.1%.12 The frequency of mutations and disease associated with NKX2-1 is unknown. Children with NKX2-1 mutations may present with isolated pulmonary disease but the majority of the children will present with any combination of neurologic abnormalities, hypothyroidism, and respiratory disease, the so-called “Brain-Thyroid-Lung Syndrome.”13 The frequency of ABCA3-associated disease in the population is unknown, but the frequency of mutations in ABCA3 in the general population may be as high as 3–5%, suggesting that ABCA3 deficiency is the most common of these disorders of surfactant homeostasis.14 Several individuals with DLD and only a single recessive mutation in ABCA3 have also come to clinical attention. Explanations include the possibility that these individuals are one of the 3–5% of the general population who carry an ABCA3 mutation and their lung disease is unrelated to ABCA3, or perhaps a mutation in another gene or an environmental “2nd hit” is interacting with the ABCA3 mutation to elicit disease.15 The recent identification of an intronic variant 98 nucleotides away from exon 26 (IVS25-98) in ABCA3 that alters splicing and the structure and function of ABCA3 demonstrates that there may be variants elsewhere in ABCA3 that are not captured by standard clinical sequencing techniques.16
Mutations in SFTPC and ABCA3 have also been identified in adults with idiopathic pulmonary fibrosis (IPF), suggesting that diffuse ILD in children and IPF in adults may be part of the same spectrum of disease. Mutations in the genes encoding surfactant protein-A2 (SFTPA2) and telomerases (TERT and TERC) have been associated with familial IPF and lung cancer in adulthood, but whether or not mutations in these genes contribute to childhood ILD is unknown.17,18
Studies investigating the mechanisms that result in variable expression of disease associated with mutations in surfactant associated genes are ongoing and will ultimately provide the basis for development of mechanism-specific interventions.
BRONCHOALVEOLAR LAVAGE PROTEOMICS AND INSIGHTS INTO LUNG DISEASE (RD)
The field of Systems Biology, at times referred to as “omics,” often involves the investigation of the genome (DNA), transcriptome (mRNA), proteome (protein), and metabolome (metabolites).19 Genomics and more recently transcriptomics have yielded improved understanding of disease. Though less commonly employed, proteomics, the large-scale study of the expression, structure or function of proteins in a biological system, can provide valuable information in concert with genomics. Proteomic analysis has the advantage of studying: (1) networks of proteins that provide “real-time” status of disease state, (2) modulation of protein function by diseases and drugs, (3) gene activity, (4) pathogenesis of disease, and (5) the prediction of new therapeutic approaches.20–22 However, the application of proteomics has been limited by the complexity of studying over 20,000 protein-coding genes that can all undergo post-translational modification and the lack of inexpensive, timely, high-throughput systems.
Discovery and validation of proteomic signatures holds great promise for children with interstitial and diffuse lung disease (chILD). Despite successes in recognizing clinical phenotypes and genetic etiologies in chILD, significant limitations remain in diagnostics, understanding mechanisms of disease and developing evidence based therapies.23 Initial work in chILD has been done by Fan and colleagues to show that Krebs von den Lungen-6 (KL-6) and SPD are serum biomarkers that can identify children with ILD and that KL-6 may specifically distinguish surfactant dysfunction mutations from Neuroendocrine Cell Hyperplasia of Infancy (NEHI).24,25 Though approved and used as a biomarker for adult ILDs in Japan, KL-6 (a submolecule of human MUC1 mucin protein) is not clinically available in the U.S. Popler and colleagues from Colorado have investigated cytokine and chemokine profiles from bronchoalveolar lavage fluid (BAL) from a small cohort of children with chILD (NEHI and follicular bronchitis [FB]) compared to disease controls. They report that NEHI had lower levels of IL-1β, MIP-1β, and IL-8 and FB had higher levels of IL-1ra, G-CSF, IL-6, and VEGF compared to all groups.26 These data reinforce the idea that NEHI is not a classic inflammatory disease state, which is also supported by the lack of inflammation seen in lung histology.27 Using new SOMAmer (slow off-rate modified aptamer) proteomic techniques, we investigated whether specific proteomic signatures could be defined for 811 proteins and molecular biologic pathways identified in BAL from subjects with NEHI and Surfactant dysfunction mutations. SOMAmers are unique chemically synthesized protein binding agents designed by SOMALogic, Inc. (Boulder, CO) from DNA derivatives.22 Proteins can be quantified to less than pg/ml concentrations in 25 μl of fluid with SOMAmer technology. BAL from NEHI and Surfactant Dysfunction Mutation subjects had specific and distinguishing SOMAmer protein signatures compared to each other and other disease controls.28 Interestingly, distinguishing proteins for Surfactant Dysfunction Mutations, such as CCL2, CCL18, and CCL22, have also been reported as predictors of poor outcome in IPF in adults.29,30 Using the PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System,31 a resource that classifies genes/proteins by their functions, We analyzed proteomic signatures for NEHI and Surfactant Dysfunction Mutations to predict pathways involved in molecular and biologic function.
These preliminary studies provide proof of concept for the power new molecular proteomic technologies may hold for chILD. Follow-up studies are now underway to increase the sample size and type of chILD diseases studied to further validate these novel protein signatures. Expanding SOMAmer technology to define signatures in serum and tissue and increase the number of proteins evaluated to over 6,000 in the next few years has the potential to extend our understanding even further. The involvement of the Children’s Interstitial and Diffuse Lung Disease Research Network (chILDRN) and chILD Foundation will be critical for the success of this work. In summary, SOMAmer proteomics holds great promise to improve diagnostic evaluations, better define pathogenesis, and suggest new therapeutic approaches for chILD.
EMERGING CONCEPTS AND THERAPEUTICS FOR EARLY ONSET LUNG DISEASE (WEB)
While the genome functions as a storage device that dictates an individual’s unique hereditary make-up, it is the proteins encoded by the genome that dictate the ability of each person to respond to the environment on a daily basis.
There is increasing awareness that proteins are highly malleable and that human health is actively managed by an active protein folding program referred to as protein homeostasis or proteostasis—a conserved and ancient biological language base directing the function of the protein fold and managing the overall health of the human proteome.
The cell continually exploits the emergent properties of the proteostasis system which includes >2,500 protective chaperones and degradative pathways that remove defective proteins. The proteostasis language base generates a “cloud” of management capacity around each protein in the proteome to promote proteome health as well as remove proteins that are defective (Fig. 1) as often occurs in disease. The proteostasis program responds to numerous folding stress signaling pathways of high clinical relevance to the neonate, childhood development and adult liver and lung pathophysiology. These include environmentally triggered inflammatory diseases (COPD, emphysema, asthma) and a multitude of inherited diseases (i.e., α1-antitrypsin (A1AT) deficiency (ATD), cystic fibrosis (CF) (cystic fibrosis transmembrane conductance regulator (CFTR)) as well as DLD) (Fig. 1).
Fig. 1.

The proteostasis cloud protecting the protein fold in health and disease. Illustrated are two examples where deficiencies in protein folding due to hereditary changes (mutations), including ATD and CF, require the activity of the proteostasis language base to manage the protein fold to protect the neonate and child from the severe consequences of protein misfolding. We are learning that the proteostasis “cloud” (cloud icons) can be bolstered therapeutically with small molecules to provide an improved folding environment to restore protein function and provide benefit to human disease.
Management by the proteostasis language is very dynamic-protecting and coordinating proteome health in diverse cell, tissue and organ environments. Using the same pathways that the cell uses to optimize proteostasis for protection on a daily basis, and that often fail in inherited and sporadic complex diseases, we are learning to manipulate the proteostasis folding language by altering the composition and function of the proteostasis network. In both cystic fibrosis and A1AT we have conducted high throughput genomic and proteomic screens as well as small molecule “corrector” screens to understand the basic principles responsible for these diseases, the changes in the proteostasis program that are responsible for a loss of protein function, and through use of small molecule correctors, how to alter the proteostasis language base to correct the diseased state. In each case we are discovering a common framework based on changes in the proteostasis network to therapeutics that can bring about substantial change in the folding efficiency of mutant proteins and that are anticipated to provide substantial benefit in terms of beginning to restore normal activity in the clinic. By merging our advanced genomic, proteomic and small molecule HTS approaches with bioinformatic tools we are beginning to build a multi-layered, system-wide view of the language of proteostasis to develop therapeutics that should provide benefit to the many diseases affecting human healthspan (Fig. 1).32–37
GENOME-WIDE DNA METHYLATION PATTERNS IN INTERSTITIAL LUNG DISEASE (ILD) AND CHRONIC OBSTRUCTIVE LUNG DISEASE (COPD) (DAS)
Epigenetic mechanisms are likely to be involved in the control of gene expression in COPD and ILD, especially given the association of these diseases with cigarette smoking and the relationship between cigarette smoke and changes in epigenetic marks.
Using the comprehensive high-throughput arrays for relative methylation (CHARM) method, we collected genome-wide DNA methylation profiles on lung tissue from 179 subjects with COPD, 159 subjects with ILD, and 79 non-disease controls from the LTRC. Percent methylation estimates from the normalized and scaled dataset were fit to disease status, age, gender and smoking status in a linear regression model. We correlated DNA methylation changes with changes in gene expression collected on the Agilent platform. We also performed similar analyses to identify differentially methylated regions (DMRs) associated with cigarette smoke, decline in lung function, and %emphysema in COPD.
We identified 3737 unique genes with a significant DMR (Bonferroni-adjusted P < 0.05) located ≤2 kb from the gene associated with ILD and 5,409 associated with COPD, after controlling for age, gender, and smoking status. Fifteen percent of these DMRs are associated with changes in gene expression, including a number of genes known to be associated with the development of chronic lung disease (chemokine receptors, ADAM family genes, integrins, collagens, coagulation genes).
Our genome-wide analysis of DNA methylation patterns identified DMRs associated with COPD and ILD that may be involved in regulating gene expression in these diseases. Understanding epigenetic regulation of biological processes in the lung may lead to the development of novel diagnostic and therapeutic approaches for chronic lung disease.
VENTILATION AFFECTS HISTONE MARKS IN LUNG AND BRAIN OF PRETERM LAMBS (KHA)
Mechanical ventilation (MV) injures the lung and brain of chronically ventilated preterm neonates. Our studies using chronically ventilated preterm lambs show that both organs have genome-wide hypoacetylation of histones when managed by mechanical ventilation. Lung and brain injury is reduced and genome-wide hyperacetylation of histones occurs with a gentler ventilation mode: nasal high-frequency ventilation (HFV; similar to nasal CPAP).
Dichotomy of genome-wide acetylation state of histones between ventilation modes (MV vs. nasal HFV) suggests that epigenetic mechanisms may participate in the pathogenesis of multiple-organ injury that typifies neonatal chronic lung disease. We hypothesized that treating preterm lambs with histone deacetylase (HDAC) inhibitors during MV would lead to histone hyperacetylation and reduce injury to the lung and brain.
Preterm lambs (~132 days gestation; term ~150 days), treated with antenatal steroids and postnatal surfactant, were managed by MV, MV + valproic acid (VPA; non-specific HDAC inhibitor), MV + trichostatin A (TSA; specific HDAC inhibitor), or nasal HFV (n = 4/group). Each inhibitor was given once/d, intravenously. At the end of 3 days, lung parenchyma and temporal lobe white matter were analyzed by immunoblot for acetylated (ac) H3K14, H3K27ac, H3K18ac, trimethylated (me3) H3K36, and histone deacetylase. Immunoblot results were normalized for total H3. Structural measurements were made of alveolar formation in the lung and apoptosis/proliferation of neurons and glia in the brain.
Histone marks H3K14ac, H3K27ac, H3K18ac, H3K36me3 were significantly lower (P < 0.05) in the lung and temporal lobe of the brain of preterm lambs that were supported by MV than nasal HFV. HDAC protein abundance was significantly higher in the MV group. During MV, inhibition of histone deacetylation, with either VPA or TSA, significantly increased H3K14ac, H3K27ac, H3K18ac, H3K36me3 in the lung, and white and gray matter of the brain. HDAC protein abundance was significantly decreased in both organs. Structurally, the lung of both groups of treated preterm lambs had significantly greater morphometric indices of alveolar formation in the lung. Markers of apoptosis (cleaved caspase 3) and proliferation (proliferating nuclear cell antigen) were significantly lower in white matter of the temporal lobe of the brain.
In summary, treating preterm lambs with HDAC inhibitors during MV leads to histone hyperacetylation and reduces injury to the lung and brain. These molecular and structural outcomes are similar to the outcomes following support of preterm lamb with nasal HFV. These results suggest an epigenetic mechanism for the pathogenesis of neonatal chronic lung disease. We speculate that the lung epigenetic and structural effects are triggered by ventilator-induced stretch. We further speculate that the brain epigenetic and structural effects result from release of pro-inflammatory cytokines and/or chemokines from the injured lung that triggers local inflammatory responses in the brain.
MODELING LUNG REMODELING IN TRANSGENIC MOUSE MODELS (JAW)
Transgenic mice have been used widely for the study of lung diseases for more than two decades, providing the ability to delete, mutate or express genes of interest in pulmonary cells. The molecular pathways regulating lung morphogenesis in function are highly conserved among vertebrate species. While the structure of the murine lung differs in size, extent of branching, and ontogeny from that in humans, the cellular and molecular features of the mouse and human lung are remarkably similar. Thus, studies in transgenic mice have been highly useful in identifying and assessing the pathological basis of lung diseases.
Cell and tissue specificity, conditionality of the control of gene expression using cell-specific gene regulatory elements have been developed and refined. These mouse models permit lineage analysis, cell specific ablation, deletion or mutation, and the ability to manipulate the mouse genome to create models relevant for the study of lung disease and repair.38
Alveolar Injury in the Pathogenesis of ILD
Genetic studies in children and families, as well as genetic engineering in complementary transgenic mouse models, strongly implicate injury of the alveolar epithelial cell in the pathogenesis of ILD, lung fibrosis, and other lung disorders related to surfactant dysfunction. Mutations in genes encoding the proteins critical for the surfactant system are expressed in alveolar type II cells, for example, SP-A, SP-B, SP-C, and ABCA3, all cause epithelial cell dysfunction and/or surfactant deficiency that underlie epithelial cell injury, inflammation and remodeling that are associated with respiratory failure and ILD in children and adults. Deletion or mutation of SP-B, ABCA3, or SP-C caused similar pulmonary abnormalities in humans and transgenic mice (Fig. 2). Mutations in telomerases, genes that are critical for maintenance of telomeres and cell survival, are associated with ILD in adult patients. Taken together, experimental and genetic studies support the concept that alveolar cell injury is an important mechanism underlying chronic lung disease (see Ref.39 for review).
Fig. 2.
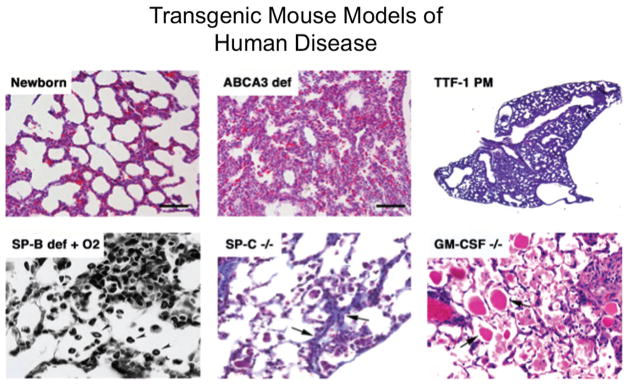
Representative histopathology of diseases caused by gene variants in surfactant-associated genes in humans which have been modeled in transgenic mice. See the Inherited Disorders of Surfactant Metabolism (AH) Section for a description of these disorders.
Direct proof that epithelial cell injury causes lung remodeling and fibrosis was provided by experiments in which a floxed diphtheria toxin A gene was conditionally activated in pulmonary epithelial cells under control of doxycycline-dependent elements expressing Cre-recombinase in type II alveolar cells or conducting airway cells. Repeated injury to the airway epithelium caused epithelial cell specific apoptosis, causing lung fibrosis, creating a model of epithelial cell injury and repair in the mouse, useful for elucidating the mechanisms involved in lung fibrosis and remodeling.40
Growth Factor and Cytokine Mediated Lung Injury
Chronic lung injury and abnormal repair are associated with dysregulation of the immune system, leading to chronic inflammation as mediated, at least in part, by a number of growth factors and cytokines, for example TGF-β, TGF-α, CTGF, IL-13, IL-1β, and others. Expression of these factors in respiratory epithelial cells of conducting or peripheral airway epithelial cells is readily achieved in transgenic mouse models. Expression or deletion of genes mediating inflammation and repair provide models of lung disease that are useful in understanding the pathogenesis of lung disease. For example, conditional expression of IL-13 in the mouse lung using doxycycline-regulated strategies induces mucus metaplasia, lung inflammation, and remodeling with features of asthma. Conditional expression of TGF-α in the respiratory epithelium causes extensive lung fibrosis. Many of the physiological and structural changes induced by these signaling molecules are reversible upon withdrawal of doxycycline.41 Thus, these transgenic models provide insight into the processes involved in both pathogenesis and resolution of lung pathology. Transgenic mouse models of human disease related genes, for example caused by mutations in CFTR, ABCA3, SFTPC, and SFTPB, share features with those caused by mutations in the genes in patients, providing models useful for understanding the molecular physiological and structural consequences of hereditable lung diseases that are useful for identifying and testing new therapeutic strategies for treatment of lung disease.
REGULATION OF PROGENITOR CELL FATE IN THE AIRWAY EPITHELIUM (WVC)
In the mature lung, airways are lined by a well-balanced population of ciliated, secretory and neuroendocrine (NE) cells, which perform functions as diverse as air humidification, detoxification and clearance of environmental particles. How these different cell types emerge and these fates are maintained in airways is still little understood. Aberrant patterns of differentiation with increase in specific cell populations are seen in a number of diseases, such as asthma, COPD, pulmonary NE cell hyperplasias, among others. Our studies in the embryonic lung show a critical role of Notch in formation of secretory cells and restriction of the ciliated and NE cell phenotypes during development.42,43 This role has been confirmed by both loss and gain of function approaches.42–44 Notch signaling was also found to be required during neonatal life to repress a goblet cell program in Clara cells.45 Disruption of Notch signaling during neonatal life resulted in aberrant formation of goblet cells in proximal airways. Moreover, we provide evidence that during development distinct subpopulations of secretory precursors cells are defined at a very early stage in airways. We show that when airways are forming these subpopulations differ in gene expression, regional distribution and in their association with NE cells. We identified a novel subpopulation of secretory precursors juxtaposed to presumptive NE bodies (NEB) distinguished by their strong secretoglobin Scgb3a2 and Uroplakin3A signals, and reduced Clara Cell Secretory Protein (Ccsp or Scgb1a1) expression. Disrupting expression of the transcription factor Ascl1 abolished NEBs and prevented formation of these secretory precursors in mutant mice.46 Thus, NEB seemed to influence dramatically the features of gene expression and, potentially, the behavior of these cells. Analysis of the Notch pathway reveals a differential distribution of Jagged and Delta ligands throughout the airways, suggesting that distinct subpopulations of secretory cells may arise from Notch activation by a selective ligand. We conclude that Notch is a major signal regulating cell fate in airways likely playing a role in conditions that result in unbalance of cell phenotypes and faulty adaptation responses of the lung to environmental agents.
INDUCED PLURIPOTENT STEM CELLS FOR THE STUDY AND TREATMENT OF ChILD (DNK)
Recent advances in reprogramming technologies have led to the derivation of pluripotent stem cells from the somatic cells of humans, including those suffering from lung diseases. What are the implications of these newly described stem cells, termed induced pluripotent stem (iPS) cells, for individuals with childhood interstitial lung diseases (ChILD)? Pluripotent stem cells, such as embryonic stem (ES) cells, can be differentiated in vitro into many lineages, including endodermal epithelia, such as lung airway and alveolar cells. However, until recently it was not possible to derive pluripotent stem cells from individual patients. In 2006, Dr. Shinya Yamanaka and colleagues published a reprogramming methodology able to induce pluripotency in somatic cells, such as dermal fibroblasts.47,48 The pluripotent stem cells derived by exogenous expression of Oct4, Klf4, Sox2, and CMyc closely resembled ES cells derived from blastocyst embryos, and these newly derived cells were named iPS cells indicating their derivation did not require any embryonic or fetal tissue, in contrast to ES cells. Since 2006, Dr. Yamanaka’s approach has been adapted and optimized resulting in many methods for generating iPS cells from a variety of somatic tissues, including human skin biopsy-derived fibroblasts or aliquots of peripheral blood cells.49 There are now numerous reprogramming methodologies; for example, the use of an excisable lentiviral vector has been employed to generate the first lung disease-specific iPS cell lines from adults with inherited lung diseases.50 To date numerous lung disease-specific iPSC clones have been banked, the majority generated from humans suffering from epithelial and vascular lung diseases, such as cystic fibrosis, emphysema, alpha-1 antitrypsin deficiency, scleroderma, sickle cell disease, and pediatric lung diseases due to mutations in NKX2.1, SFTPB, SFTPC, or ABCA3. Until recently there was little to do with this bank as the protocols had not yet been developed to differentiate iPSCs into lung lineages. The recent publication of protocols for the derivation of Nkx2.1+ lung epithelial lineages from pluripotent stem cells51–53 now allows unprecedented opportunities to utilize disease-specific iPS cell to model lung disease pathogenesis in vitro. These in vitro models should enable the study of the earliest moments of human lung development as well as examination of the cell fate decisions and epigenetic mechanisms involved in specifying lung epithelia. Ultimately, drug screens, individualized therapeutic tests, and tissue engineering approaches may be developed for each patient using this new in vitro model system. Since iPS cell-derived lung epithelia from each patient contain each individual’s own genetic background, these cells potentially provide individualized models of disease pathogenesis and individualized trials of drug efficacy testing. iPS cells derived from individuals carrying mutations causing ChILD (children’s interstitial lung disease) provide a particularly powerful platform to study disease pathogenesis of diseases whose onset in the developing lung epithelium occurs at a fetal stage that is otherwise difficult to study in vivo.
Stem Cells as Therapy for chILD Lung Disease (SK)
Bronchopulmonary dysplasia (BPD) is a complex chronic lung disease with multifactorial etiology, characterized by the arrest of alveolar and vascular growth associated with inflammation and parenchymal fibrosis. Historically, oxygen toxicity and ventilator-induced injury have been the prerequisites for BPD in premature infants born at less than 28–32 weeks gestation with respiratory distress syndrome, but BPD may also occur in immature infants with few signs of initial lung injury.
The saccular stage of murine lung development is completed after two weeks of postnatal alveolarization. Hence, the developmental stage of the mouse lung at birth resembles that in the human preterm neonate between 24 and 28 weeks gestation, making the newborn mouse an excellent model to study human developmental lung injury. Hyperoxia-induced lung damage in neonatal mice is characterized by rarefication and simplification of alveoli and thickened alveolar septa, inflammation and parenchymal fibrosis, a pattern which is similar to human BPD.
Mesenchymal stem cells, also referred to as multi-potent stromal cells (MSCs), have attracted significant attention as potential cell-based therapy for BPD and other severe lung diseases because these multipotent cells exhibit beneficial effects in related animal models through anti-inflammatory, immunomodulatory, pro-survival (endothelial, epithelial), and anti-fibrotic mechanisms. We have previously demonstrated that intravenous injection of bone marrow-derived MSCs in newborn mice conferred significant vascular and immunological protection from hyperoxia-induced injury but had limited effect in preserving alveolar architecture. Concentrated MSC-conditioned media (MSC-CM), however, prevented both vascular and alveolar hyperoxic injury resulting in normal alveolar number and thin septa, comparable to controls in room air.54 These findings suggested that MSCs have important cytoprotective effects in the hyperoxia mouse model of BPD via paracrine mechanisms including the release of immunomodulatory and vasoprotective mediators. Given that clinically it’s more relevant to reverse BPD as prevention cannot be readily achieved, we tested the ability of MSC-CM to rescue injury in this animal model. Indeed, a single intravenous dose of MSC-CM reversed—to a significant degree—hyperoxia-induced BPD and pulmonary vascular disease versus mouse lung fibroblast-CM (MLF-CM) control: MSC-CM-treatment (1) reversed the hyperoxia-induced parenchymal fibrosis and peripheral pulmonary artery (PA) devascularization (PA pruning), (2) partially reversed alveolar injury, (3) normalized lung function (airway hyperresponsiveness, dynamic lung compliance), (4) fully reversed the moderate pulmonary hypertension (PH), and (5) attenuated peripheral PA muscularization associated with hyperoxia-induced BPD.55
Factors secreted in MSC-CM promote signaling pathways of lung repair and include inhibitors of inflammation that are linked to the development of PH and pulmonary fibrosis. An attractive speculation is that the beneficial effect of MSC-CM may be, at least in part, due to activation of endogenous lung stem cells. To explore this hypothesis, we investigated bronchioalveolar stem cells (BASCs), an adult lung stem cell population capable of self-renewal and differentiation in culture, and proliferation in response to bronchiolar and alveolar lung injury in vivo. We demonstrated that MSC-CM treatment led to a significant increase in BASCs in the lung compared to untreated controls. Treatment of BASCs with MSC-CM in culture resulted in an increase in growth efficiency, suggesting a paracrine effect of MSCs on BASCs. Lineage tracing in bleomycin-treated adult mice showed that CCSP-expressing cells, including BASCs, are capable of contributing to alveolar repair after lung injury. Thus, MSC-derived factors likely stimulate BASCs and/or other endogenous lung stem cells to contribute to the restoration of distal lung cell epithelia in BPD.
In preliminary proteomic analysis to identify the active moiety within MSC-CM, we noted the presence of proteins, including CD63, CD81, moesin, and hsp70, reported to be associated with intracellular vesicles known as exosomes. Exosomes are small microvesicles, 30–100 nm in diameter, that are stored within multi-vesicular bodies and released into the environment by fusion with the cell membrane. Microvesicles are now considered to be important mediators of intercellular communication by transferring their contents including proteins, mRNAs and miRNAs, to recipient cells. The physiological relevance of MSC-derived exosomes has not been evaluated in lung disease in vivo, even though their cellular source and factors they secrete have promising therapeutic potential on lung injury.
Using the adult hypoxic murine model of PH, we demonstrated that intravenous delivery of MSC-derived exosomes (MEX) inhibited hypoxic lung inflammation, vascular remodeling and PH, whereas MEX-depleted media or fibroblast-derived exosomes (FEX) had no effect.56 MEX suppressed the hypoxic activation of signal transducer and activator of transcription 3 (STAT3) and the upregulation of the miR-17 superfamily of microRNA clusters, whereas it increased lung levels of miR-204, a key microRNA, the expression of which is decreased in human pulmonary hypertension. MEX produced by human umbilical cord mesenchymal stromal cells inhibited STAT3 signaling in isolated human pulmonary artery endothelial cells, demonstrating a direct effect of MEX on hypoxic vascular cells. Ongoing work will evaluate the mechanisms and signaling pathways by which MEX modulate lung injury, inhibit lung inflammation and reverse lung diseases such as BPD.
Acknowledgments
The conference was funded in large part by the NHLBI (R13 HL114359-01, to J.S.H.). Additional support was received from the UCSD Department of Pediatrics, Rady Children’s Specialists of San Diego, A Medical Foundation, and an educational grant from InterMune. In-kind support and travel awards were sponsored by the chILD Foundation, the chILD Lung Foundation (U.K.), and the Mauli Ola Foundation. Additional funding relevant to the data presented in the sections of the document was from the following grants: RC2-HL101715 (to D.A.S.); and HL62875, HL11002, HL56401, and HD41075 (to K.A.). Special thanks are due to the scientific advisory committee (Lawrence Nogee, MD and Lisa Young, MD), as well as to Mrs. Elizabeth Boyd and Mrs. Maureen Helinski. Additional contributors to the individual sections were: Emerging Concepts and Therapeutics for Early Onset Lung Disease: M. Bouchareilh, D. Hutt, D. Roth, S. Pankow, S. Beckers, Hui Gei, Peter Reinhart, and J. Yates III. Genome-Wide DNA Methylation Patterns in Interstitial Lung Disease (ILD) and Chronic Obstructive Lung Disease (COPD): Ivana V. Yang, Brent Pedersen, Corinne Hennessy, Elizabeth Davidson, Megan Bonney, Sonia Leach, Joseph Brown, Julia Turner, Brenda Juan Guardela, John Tedrow, Mick Correll, Mark Geraci, Naftali Kaminski, John Quackenbush, Frank Sciurba, and Avrum Spira. Ventilation Affects Histone Marks in Lung and Brain of Preterm Lambs: Jeremy Alvord, Brady Houston, Matthew McCoy, Li Dong, Mar Janna Dahl, Christopher Callaway, Robert McKnight, Donald Null, Bradley Yoder, Robert DiGeronimo, and Robert Lane. Stem Cells as Therapy for chILD Lung Disease: Georg Hansmann, Angeles Fernandez-Gonzalez, Changjin Lee, Muhammad Aslam, Kostis Sdrimas, and Alex Mitsialis.
Footnotes
Conflict of interest: None.
References
- 1.Garmany T, Moxley MA, White FV, Dean M, Nogee LM, Hamvas A. Surfactant composition and function in patients with ABCA3 mutations. Pediatr Res. 2006;59:801–805. doi: 10.1203/01.pdr.0000219311.14291.df. [DOI] [PubMed] [Google Scholar]
- 2.Guillot L, Carre A, Szinnai G, Castanet M, Tron E, Jaubert F, Broutin I, Counil F, Feldmann D, Clement A, Polak M, Epaud R. NKX2-1 mutations leading to surfactant protein promoter dysregulation cause interstitial lung disease in “Brain-Lung-Thyroid Syndrome”. Hum Mutat. 2010;31:E1146–E1162. doi: 10.1002/humu.21183. [DOI] [PubMed] [Google Scholar]
- 3.Nogee L, Wert SE, Proffit SA, Hull WM, Whitsett JA. Allelic heterogeneity in hereditary surfactant protein B (SP-B) deficiency. Am J Respir Crit Care Med. 2000;161:973–981. doi: 10.1164/ajrccm.161.3.9903153. [DOI] [PubMed] [Google Scholar]
- 4.Nogee L, Dunbar AE, III, Wert S, Askin F, Hamvas A, Whitsett JA. Mutations in the surfactant protein C gene associated with interstitial lung disease. Chest. 2002;121:20S–21S. doi: 10.1378/chest.121.3_suppl.20s. [DOI] [PubMed] [Google Scholar]
- 5.Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350:1296–1303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 6.Brasch F, Griese M, Tredano M, Johnen G, Ochjs M, Rieger C, Mulugeta S, Muller KM, Bahuau M, Beers MF. Interstitial lung disease in a baby with a de novo mutation in the SFTPC gene. Eur Respir J. 2004;24:30–39. doi: 10.1183/09031936.04.00000104. [DOI] [PubMed] [Google Scholar]
- 7.Bullard J, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Resp Crit Care Med. 2005;172:1026–1031. doi: 10.1164/rccm.200503-504OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cameron H, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE, Deutsch G, Nogee LM. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr. 2005;146:370–375. doi: 10.1016/j.jpeds.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 9.Krude H, Schutz B, Biebermann H, von Moers A, Schnabel D, Neitzel H, Tonnies H, Weise D, Lafferty A, Schwarz S, DeFelice M, von Deimling A, van Landeghem F, DiLauro R, Gruters A. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-haploinsufficiency. J Clin Invest. 2002;109:475–480. doi: 10.1172/JCI14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamvas A, Trusgnich M, Brice H, Baumgartner J, Hong Y, Nogee LM, Cole FS. Population-based screening for rare mutations: high throughput DNA extraction and amplification from Guthrie cards. Pediatr Res. 2001;50:666–668. doi: 10.1203/00006450-200111000-00021. [DOI] [PubMed] [Google Scholar]
- 11.Tredano M, Cooper DN, Stuhrmann M, Christodoulou J, Chuzhanova NA, Roudot-Thoraval F, Boelle PY, Elion J, Jeanpierre M, Feingold J, couderc R, Bahuau M. Origin of the prevalent SFTPB indel g. 1549C>GAA (121ins2) mutation causing surfactant protein B (SP-B) deficiency. Am J Med Genet A. 2006;140:62–69. doi: 10.1002/ajmg.a.31050. [DOI] [PubMed] [Google Scholar]
- 12.Garmany TH, Wambach JA, Heins HB, Watkins-Torry J, Wegner DJ, Bennet K, An P, Land G, Saugstad OD, Henderson H, Nogee LM, Cole FS, Hamvas A. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res. 2008;63:645–649. doi: 10.1203/PDR.0b013e31816fdbeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carre A, Szinnai G, Castanet M, Sura-Trueba S, Tron E, Broutin-L’Hermite I, Barat P, Goizet C, Lacombe D, Moutard ML, Raybaud C, Raynaud-Ravni C, Romana S, Ythier H, Leger J, Polak M. Five new TTF1/NKX2. 1 mutations in brain-lung-thyroid syndrome: rescue by PAX8 synergism in one case. Hum Mol Genet. 2009;18:2266–2276. doi: 10.1093/hmg/ddp162. [DOI] [PubMed] [Google Scholar]
- 14.Wambach JA, Wegner DJ, Depass K, Heins H, Druley TE, Mitra RD, An P, Zhang Q, Nogee LM, Cole FS, Hamvas A. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics. 2012;130:e1575–e1582. doi: 10.1542/peds.2012-0918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bullard J, Nogee LM. Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein-C gene (SFTPC) mutation. Pediatr Res. 2007;62:176–179. doi: 10.1203/PDR.0b013e3180a72588. [DOI] [PubMed] [Google Scholar]
- 16.Agrawal A, Hamvas A, Cole FS, Wambach JA, Wegner D, Coghill C, Harrison K, Nogee LM. An intronic ABCA3 mutation that is responsible for respiratory disease. Pediatr Res. 2012;71:633–637. doi: 10.1038/pr.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ge H, Walhout AJ, Vidal M. Integrating ‘omic’ information: a bridge between genomics and systems biology. Trends Genet. 2003;19:551–560. doi: 10.1016/j.tig.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Hirsch J, Hansen KC, Burlingame AL, Matthay MA. Proteomics: current techniques and potential applications to lung disease. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1–L23. doi: 10.1152/ajplung.00301.2003. [DOI] [PubMed] [Google Scholar]
- 21.Zichi D, Eaton B, Singer B, Gold L. Proteomics and diagnostics: let’s get specific, again. Curr Opin Chem Biol. 2008;12:78–85. doi: 10.1016/j.cbpa.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic N, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE. 2010;5:e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deterding RR. Infants and young children with children’s interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 2010;23:25–31. doi: 10.1089/ped.2010.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Salmi QA, Walter JN, Colasurdo GN, Sockrider MM, Smith EO, Takahashi H, Fan LL. Serum KL-6 and surfactant proteins A and D in pediatric interstitial lung disease. Chest. 2005;127:403–407. doi: 10.1378/chest.127.1.403. [DOI] [PubMed] [Google Scholar]
- 25.Doan ML, Elidemir O, Dishop MK, Zhang H, Smith EO, Black PG, Deterding RR, Roberts DM, Al-Salmi QA, Fan LL. Serum KL-6 differentiates neuroendocrine cell hyperplasia of infancy from the inborn errors of surfactant metabolism. Thorax. 2009;64:677–681. doi: 10.1136/thx.2008.107979. [DOI] [PubMed] [Google Scholar]
- 26.Popler J, Wagner BD, Accurso FJ, Deterding RR. Airway cytokine profiles in children’s interstitial lung diseases. Am J Respir Crit Care Med. 2010;181:A6733. [Google Scholar]
- 27.Dishop M. Diagnostic pathology of diffuse lung disease in children. Pediatr Allergy Immunol Pulmonol. 2010;23:69–85. doi: 10.1089/ped.2010.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deterding RR, Wolfson A, Harris J, Walker JJ, Accurso FJ. Aptamer proteomic analysis of bronchoalveolar lavage fluid yields different protein signatures from children with Children’s Interstitial Lung Disease, Cystic Fibrosis and disease controls. Am J Respir Crit Care Med. 2010;181:A6722. [Google Scholar]
- 29.Deterding RR, Wolfson A, Harris JK, Walker J, Wagner BD, Accurso FJ. Bronchoalveolar lavage protein biomarkers in children with surfactant dysfunction mutations; an aptamer proteomics approach (Aspen Lung Conference) Proc Am Thorac Soc. 2011;8:210. [Google Scholar]
- 30.Shinoda H, Tasaka S, Fujishima S, Yamasawa W, Miyamoto K, Nakano Y, Kamata H, Hasegawa N, Ishizaka A. Elevated CC chemokine level in bronchoalveolar lavage fluid is predictive of a poor outcome of idiopathic pulmonary fibrosis. Respiration. 2009;78:285–292. doi: 10.1159/000207617. [DOI] [PubMed] [Google Scholar]
- 31.Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitzky-Lazareva B, Muruganujan A, Rabkin S, Vandergriff JA, Doremieux O. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balch WE, Roth DM, Hutt DM. Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harbor Perspect Biol. 2011;3:a004499. doi: 10.1101/cshperspect. pii: a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 34.Bouchecareilh M, Balch WE. Proteostasis, an emerging therapeutic paradigm for managing inflammatory airway stress disease. Curr Mol Med. 2012;12:815–826. doi: 10.2174/156652412801318782. [DOI] [PubMed] [Google Scholar]
- 35.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 36.Hutt D, Balch WE. Cell biology. The proteome in balance Science. 2010;329:766–767. doi: 10.1126/science.1194160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powers ET, Balch WE. Protein folding: protection from the outside. Nature. 2011;471:42–43. doi: 10.1038/471042a. [DOI] [PubMed] [Google Scholar]
- 38.Perl AK, Zhang L, Whitsett JA. Conditional expression of genes in the respiratory epithelium in transgenic mice: cautionary notes and toward building a better mouse trap. Am J Respir Cell Mol Biol. 2009;40:1–3. doi: 10.1165/rcmb.2008-0011ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med. 2002;347:2141–2148. doi: 10.1056/NEJMra022387. [DOI] [PubMed] [Google Scholar]
- 40.Perl AK, Riethmacher D, Whitsett JA. Conditional depletion of airway progenitor cells induces peribronchiolar fibrosis. Am J Respir Crit Care Med. 2011;183:511–521. doi: 10.1164/rccm.201005-0744OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L741–L749. doi: 10.1152/ajplung.00208.2003. [DOI] [PubMed] [Google Scholar]
- 42.Guseh JS, Bores SA, Stanger BZ, Zhou Q, Anderson WJ, Melton DA, Rajagopal J. Notch signaling promotes airway mucous metaplasia and inhibits alveolar development. Development. 2009;136:1751–1759. doi: 10.1242/dev.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development. 2009;136:2297–2307. doi: 10.1242/dev.034884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morimoto M, Liu Z, Cheng HT, Winters N, Bader D, Kopan R. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci. 2010;123:213–224. doi: 10.1242/jcs.058669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsao PN, Wei SC, Wu MF, Huang MT, Lin HY, Lee MC, Lin KM, Wang IJ, Kaartinen V, Yang LT, Cardoso WV. Notch signaling prevents mucous metaplasia in mouse conducting airways during postnatal development. Development. 2011;138:3533–3543. doi: 10.1242/dev.063727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guha A, Vasconcelos M, Cai Y, Yoneda M, Hinds A, Qian J, Li G, Dickel L, Johnson JE, Kimura S, Guo J, McMahon J, McMahon AP, Cardoso WV. Neuroepithelial body microenvironment is a niche for a distinct subset of Clara-like precursors in the developing airways. Proc Natl Acad Sci USA. 2012;109:12592–12597. doi: 10.1073/pnas.1204710109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 49.Stadtfeld M, Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24:2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Somers A, Jean JC, Sommer CA, Omari A, Ford CC, Mills JA, Ying L, Sommer AG, Jean JM, Smith BW, Lafyatis RA, Demierre MF, Weiss DJ, French DL, Gadue P, Murphy GJ, Mostoslavsky G, Kotton DN. Generation of transgene-free lung disease-specific human iPS cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Longmire TA, Ikonomou L, Hawkins F, Christodoulou C, Cao Y, Jean JC, Kwok LW, Mou H, Rajagopal J, Shen SS, Dowton AA, Serra M, Weiss DJ, Green MD, Snoeck HW, Ramirez MI, Kotton DN. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell. 2012;10:398–411. doi: 10.1016/j.stem.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mou H, Zhao R, Sherwood R, Ahfeldt T, Lapey A, Wain J, Sicilian L, Izvolsky K, Musunuru K, Cowan C, Rajagopal J. Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell Stem Cell. 2012;10:385–397. doi: 10.1016/j.stem.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Green MD, Chen A, Nostro MC, d’Souza SL, Schaniel C, Lemischka IR, Gouon-Evans V, Keller G, Snoeck HW. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat Biotechnol. 2011;29:267–272. doi: 10.1038/nbt.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, Kourembanas S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med. 2009;180:1122–1130. doi: 10.1164/rccm.200902-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansmann G, Fernandez-Gonzalez A, Aslam M, Vitali SH, Martin T, Mitsialis SA, Kourembanas S. Mesenchymal stem cell-mediated reversal of bronchopulmonary dysplasia and associated pulmonary hypertension. Pulm Circ. 2012;2:170–181. doi: 10.4103/2045-8932.97603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, Sdrimas K, Fernandez-Gonzalez A, Kourembanas S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation. 2012;126:2601–2611. doi: 10.1161/CIRCULATIONAHA.112.114173. [DOI] [PMC free article] [PubMed] [Google Scholar]