

Abstract
In this study, PC12 cells were induced to differentiate into neuron-like cells using nerve growth factor, and were subjected to oxygen-glucose deprivation. Cells were treated with 0, 10, 20, 30, 50, 100 ng/mL exogenous Activin A. The 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide assay and Hoechst 33324 staining showed that the survival percentage of PC12 cells significantly decreased and the rate of apoptosis significantly increased after oxygen-glucose deprivation. Exogenous Activin A significantly increased the survival percentage of PC12 cells in a dose-dependent manner. Reverse transcription-PCR results revealed a significant increase in Activin receptor IIA, Smad3 and Smad4 mRNA levels, which are key sites in the Activin A/Smads signaling pathway, in neuron-like cells subjected to oxygen-glucose deprivation, while mRNA expression of the apoptosis-regulation gene caspase-3 decreased. Our experimental findings indicate that exogenous Activin A plays an anti-apoptotic role and protects neurons by means of activating the Activin A/Smads signaling pathway.
Keywords: neural regeneration, brain injury, biological factor, oxygen-glucose deprivation, Activin A, Activin A/Smads signaling pathway, caspase-3, apoptosis, grants-supported paper, neuroregeneration
Research Highlights
(1) Activin A-mediated signal transduction mainly relies on the Activin A/Smads pathway, which has multiple functional sites and related regulatory factors. However, little is known about the changes in pathway expression and target genes, and the mechanism of action after ischemic brain injury.
(2) To date, most studies on Activin A have focused on its neuroprotective effects in in vivo experiments. Here, we used in vitro conditions and aimed to explore the role of Activin A in the Activin A/Smads pathway. We found that exogenous Activin A had a neuroprotective effect following oxygen-glucose deprivation-induced neuronal injury.
(3) Exogenous Activin A plays a neuroprotective role by preventing apoptosis, and protecting neurons from injury through activation of the Activin A/Smads signal transduction pathway.
INTRODUCTION
According to the definition of the World Health Organization, ischemic stroke is one of the leading causes of death and adult disability worldwide[1,2]. Ischemic stroke occurs when blood supply to the brain is obstructed. Accumulating evidence suggests that the cell death observed during the first few hours of cerebral ischemia is a result of apoptosis as opposed to necrosis, which was considered the predominant form of neuron damage generated by ischemia[3,4,5]. The ischemic damage of nerve cells leads to the disruption of a series of complex signaling pathways that produces an effect on corresponding biological functions and thus affects brain function[6,7,8]. A better understanding of signal transduction mechanisms that are involved in the process of brain ischemic injury could identify key targets for neuroprotective substances.
Activin A, a member of the transforming growth factor-β superfamily, was initially isolated from gonads and served as a modulator of follicle-stimulating hormone secretion[9,10,11,12,13,14]. In the Activin A/Smads signaling pathway, Activin A interacts with two types of transmembrane receptors (Activin receptors IA and IIA), and Activin receptor IIA is a key component in the pathway that can bind Activin A and initiate the signal[15,16]. After ligand binding, Activin receptor IIA phosphorylates and thereby activates Activin receptor IA, which conducts the signal to specific intracellular receptors, Smads (Smad2/Smad3), and forms heterodimers with Smad4 (Smad2/4 or Smad3/4). The heterodimers translocate to the nucleus and recruit transcriptional co-activators or co-repressors, regulating transforming growth factor-β target genes[17,18,19]. Activin A is an important factor in modulating tissue and cellular function, participating in the regulation of cellular development in a variety of tissues[20]. Activin A is also expressed in the central nervous system. In vivo studies have shown that Activin A expression is up-regulated in asphyxiated full-term newborns with hypoxic-ischemic encephalopathy[21] and Activin A may be essential for neurogenesis following neurodegeneration[22]. Recently, interest in the role of Activin A in central nervous system diseases has emerged[23,24]. A number of in vivo studies have shown that Activin A may have a protective effect on neurons in some degenerative diseases such as Huntington's disease[25]. In mouse models of neurological deficits, Activin A plays a neuroprotective role in combination with basic fibroblast growth factor[26]. However, direct evidence of the neuroprotective effect of Activin A is poorly documented. In addition, the interaction of Activin A with downstream Smads remains undefined in in vitro models of ischemic neuronal injury.
PC12 cells are developed from the rat adrenal pheochromocytomas, and have been the object of intense neurobiological study for the investigation of signal transduction mechanisms[27,28] and studies on cell differentiation, survival[29,30], apoptosis[31], and Huntington's disease[32]. The oxygen-glucose deprivation-damage model is most commonly used in studies addressing cerebral ischemia. The principle of the oxygen-glucose deprivation model is that Na2S2O4 quickly clears the oxygen in the culture matrix, does not damage the cell membrane, and is better able to simulate the hypoxic environment when compared with in vivo models[33]. In this study, we used nerve growth factor to induce the differentiation of PC12 cells into neuron-like cells. PC12 cells were cultured in different oxygen conditions, followed by oxygen-glucose deprivation to establish a cerebral hypoxia-ischemia model. In addition, we measured mRNA expression of major factors in the Activin A/Smads signaling pathway after pretreatment with exogenous Activin A and oxygen-glucose deprivation treatment, in a broader attempt to clarify possible mechanisms involved in the neuroprotective effects of exogenous Activin A in a model of hypoxic-ischemic brain disease.
RESULTS
Effect of exogenous Activin A on the survival of PC12 cells subjected to oxygen-glucose deprivation
The 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT) assay showed that the survival of PC12 cells in the 6-hour oxygen-glucose deprivation group was significantly decreased compared with the control group (P < 0.05). The survival of PC12 cells in the exogenous Activin A plus oxygen-glucose deprivation 6-hour group was much higher than that in the 6-hour oxygen-glucose deprivation group (P < 0.05), and the effect was dose-dependent. Because there was no significant difference between the survival rates of the 50 and 100 ng/mL exogenous Activin A-treated cells (P > 0.05), 50 ng/mL Activin A was used in all subsequent experiments (Figure 1).
Figure 1.
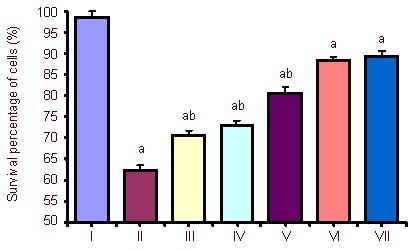
Effect of exogenous Activin A on the survival percentage of PC12 cells subjected to oxygen-glucose deprivation (OGD) for 6 hours.
The 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide assay was used to assess cell viability. PC12 cells at a density of 5 × 104 cells/well were treated with 10, 20, 30, 50 and 100 ng/mL exogenous Activin A in a 96-well plate for 24 hours. The survival percentage of PC12 cells in the exogenous Activin A plus OGD group was much higher than that in the OGD and control groups. Cell viability percentage = (absorbance of experimental group/ absorbance of control group) × 100%. Data are expressed as mean ± SD of three independent experiments. One-way analysis of variance was used to compare the mean values. aP < 0.05, vs. control group; bP < 0.05, vs. OGD 6 h and 50 ng Activin A + OGD 6 h groups.
I: Control; II: OGD 6 h; III: Activin A 10 ng + OGD 6 h; IV: Activin A 20 ng + OGD 6 h; V: Activin A 30 ng + OGD 6 h; VI: Activin A 50 ng + OGD 6 h; VII: Activin A 100 ng+ OGD 6 h; h: hours.
Hoechst 33342 fluorescence staining was carried out on normal activated PC12 cells, which were subjected to oxygen-glucose deprivation for 6 hours with or without different concentrations of exogenous Activin A for 24 hours to observe the morphological changes of apoptotic cells and the rate of apoptosis in different groups. In the control group, the nuclei of cells were ellipse in shape and stained light blue, with little apoptotic cells. After 6 hours of oxygen-glucose deprivation, apoptotic cells were classically shrunken and hyperchromatic. In the exogenous Activin A plus oxygen-glucose deprivation group, the number of apoptotic cells was decreased compared with the oxygen-glucose deprivation group (Figure 2).
Figure 2.
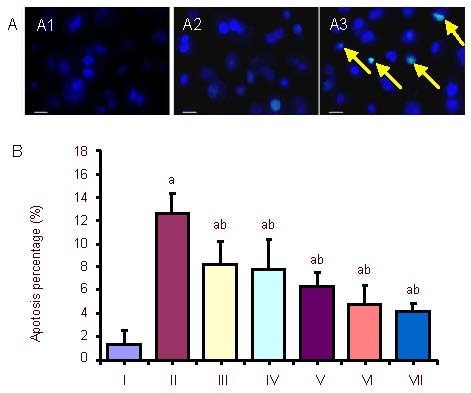
Morphological changes and rate of apoptosis in PC12 cells after oxygen-glucose deprivation (OGD) and exogenous Activin A treatment at different concentrations.
(A) Morphological changes in PC12 cells treated with exogenous Activin A plus OGD. Morphologic changes of condensed or fragmented nuclei were considered to be markers of apoptosis. The nuclei of cells in the control group (A1) were ellipse in shape and stained light blue, with little apoptosis. In the 6-hour OGD group (A2), apoptotic cells were observed to be shrunken and hyperchromatic. In the exogenous Activin A + OGD group, apoptotic cells (arrows) decreased compared with the OGD group (Hoechst 33342 fluorescence staining). Scale bars: 50 μm.
(B) Rate of apoptosis in PC12 cells. The rate of cell apoptosis in the OGD group was significantly higher than that in the control group. Treatment with exogenous Activin A for 24 hours resulted in reduced damage of cells compared with the OGD group. The percentage of apoptosis following Hoechst 33342 staining was inversely correlated to the concentration of exogenous Activin A. Five fields were chosen randomly, and 200 cells were counted in each field to obtain the rate of apoptosis. Data are expressed as mean ± SD. One-way analysis of variance was used to compare mean values. aP < 0.05, vs. control group; bP < 0.05, vs. OGD 6 h group.
I: Control; II: OGD 6 h; III: Activin A 10 ng + OGD 6 h; IV: Activin A 20 ng + OGD 6 h; V: Activin A 30 ng + OGD 6 h; VI: Activin A 50 ng + OGD 6 h; VII: Activin A 100 ng+ OGD 6 h; h: hours.
Exogenous Activin A activated the Activin A/Smads pathway of PC12 cells subjected to oxygen-glucose deprivation
Effect of exogenous Activin A on Activin receptor IIA mRNA expression in PC12 cells
The expression of Activin receptor IIA mRNA increased significantly after 6-hour oxygen-glucose deprivation compared with the control group, and was dramatically up-regulated in the exogenous Activin A plus oxygen- glucose deprivation group compared with the 6-hour oxygen-glucose deprivation group (Figure 3).
Figure 3.
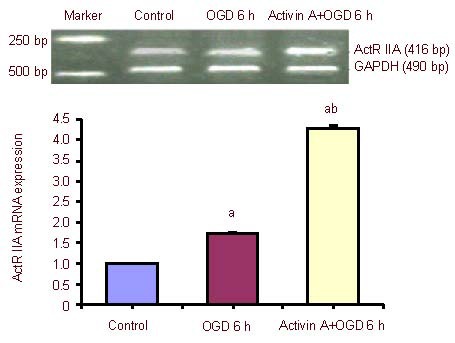
Effect of exogenous Activin A on Activin receptor IIA (ActR IIA) mRNA expression in PC12 cells subjected to oxygen-glucose deprivation (OGD).
ActR IIA mRNA levels were analyzed by reverse transcription-PCR and expressed as the absorbance ratio of ActR II A to GAPDH. Data are expressed as mean ± SD of three independent experiments. One-way analysis of variance was used to compare mean values. aP < 0.05, vs. control group; bP < 0.05, vs. OGD 6 h group. h: Hours.
Effect of exogenous Activin A on Smad3 mRNA expression in PC12 cells
The expression of Smad3 mRNA increased significantly after 6-hour oxygen-glucose deprivation compared with the control group, and was dramatically up-regulated in the exogenous Activin A plus oxygen-glucose deprivation group compared with the 6-hour oxygen-glucose deprivation group (Figure 4).
Figure 4.
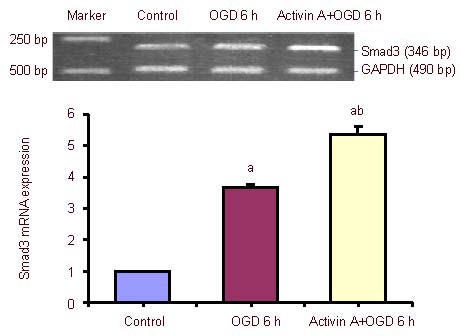
Effect of exogenous Activin A on Smad3 mRNA expression in PC12 cells subjected to oxygen-glucose deprivation (OGD).
Smad3 mRNA levels were analyzed by reverse transcription-PCR and expressed as the absorbance ratio of Smad3 to GAPDH. Data are expressed as mean ± SD of three independent experiments. One-way analysis of variance was used to compare mean values. aP < 0.05, vs. control group; bP < 0.05, vs. OGD 6 h group. h: Hours.
Effect of exogenous Activin A on Smad4 mRNA expression in PC12 cells
The expression of Smad4 mRNA increased significantly after 6-hour oxygen-glucose deprivation compared with the control group, and was dramatically up-regulated in the exogenous Activin A plus oxygen-glucose deprivation group compared with the 6-hour oxygen-glucose deprivation group (Figure 5).
Figure 5.
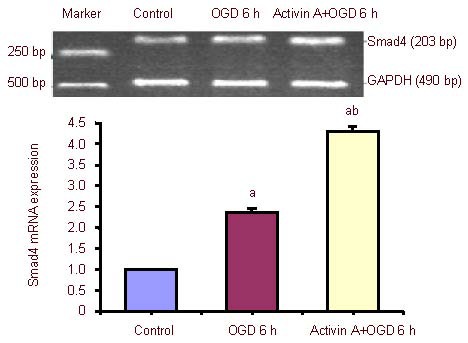
Effect of exogenous Activin A on Smad4 mRNA expression in PC12 cells subjected to oxygen-glucose deprivation (OGD).
Smad4 mRNA levels were analyzed by reverse transcription-PCR and expressed as the absorbance ratio of Smad4 to GAPDH. Data are expressed as mean ± SD of three independent experiments. One-way analysis of variance was used to compare mean values. aP < 0.05, vs. control group; bP < 0.05, vs. OGD 6 h group. h: Hours.
Effect of exogenous Activin A on caspase-3 mRNA expression in PC12 cells
The expression of caspase-3 mRNA increased significantly after 6-hour oxygen-glucose deprivation compared with the control group, and was dramatically up-regulated in the exogenous Activin A plus oxygen-glucose deprivation group compared with the 6-hour oxygen-glucose deprivation group (Figure 6).
Figure 6.
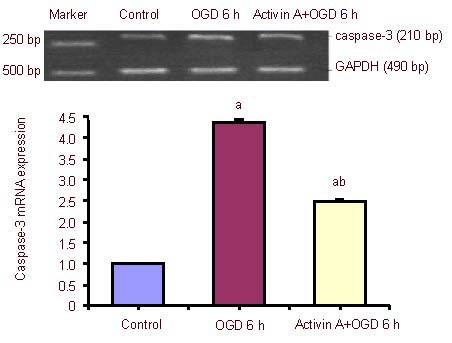
Effect of exogenous Activin A on caspase-3 mRNA expression in PC12 cells subjected to oxygen-glucose deprivation (OGD).
Caspase-3 mRNA levels were analyzed by reverse transcription-PCR and expressed as the absorbance ratio of Smad4 to GAPDH. Data are expressed as mean ± SD of three independent experiments. One-way analysis of variance was used to compare mean values. aP < 0.05, vs. control group; bP < 0.05, vs. OGD 6 h group. h: Hours.
DISCUSSION
To gain further insights into the Activin A/Smads pathway following ischemic brain injury and determine the signal transduction mechanism and protective substances in the Activin A/Smads pathway, a stable in vitro neuronal ischemia model was established[34]. Existing animal models are affected by many factors; therefore, a stable cell model of cerebral ischemia has clear advantages as it allows the researcher to control the experimental conditions, and requires smaller amounts of samples and short experimental periods. In this study, nerve growth factor stimulated PC12 cells combined with oxygen- glucose deprivation were adopted to establish an in vitro ischemia model[34]. The MTT assay and Hoechst 33324 staining were both used to detect the survival of PC12 cells exposed to hypoxic-ischemic conditions. Results showed that the survival percentage of PC12 cells in the oxygen-glucose deprivation group decreased compared with the control group, while the rate of apoptosis increased. This evidence was similar to previous reports and further confirmed establishment of an ischemic injury model (oxygen-glucose deprivation model)[35,36,37,38].
To determine the effect of Activin A and the downstream signaling of Smads on ischemic brain injury, exogenous Activin A was introduced to neuron-like cells subjected to oxygen-glucose deprivation. We found that the survival percentage of cells in the presence of exogenous Activin A following 6 hours of oxygen-glucose deprivation was much higher than that of the oxygen-glucose deprivation group without Activin A treatment. In addition, the rate of cell apoptosis significantly dropped following treatment with exogenous Activin A. Our results indicated that exogenous Activin A maintained the survival of neurons, preventing them from damage following ischemic and hypoxic injury.
Activin A regulates cellular growth and differentiation, and controls morphogenesis, angiogenesis and repair processes involved in wound healing and brain injury[39,40,41,42]. The transmembrane Activin receptor combines with Activin A to subsequently propagate downstream signaling by phosphorylating specific receptor-regulated Smad proteins[41]. Activin A may combine with Activin receptor II to activate Smad2 and Smad3, and promote the dimerization of Smad4 and Smad2/3, which enter the nucleus to influence gene transcription and activation of a series of biological functions[43,44,45].
To determine the signal transduction mechanism of the protective effect induced by exogenous Activin A, gene expression of the main factors in the Activin A/Smads pathway was detected. Compared with the control group, the expression of Activin receptor IIA, Smad3 and Smad4 mRNA significantly increased in the oxygen- glucose deprivation group, and were further up-regulated after treatment with exogenous Activin A compared with the oxygen-glucose deprivation group. Our results prove the existence of the Activin A/Smads pathway in ischemia and hypoxia injuries, which can stimulate the activation of this pathway. Because the Activin A/Smads pathway was also activated and provided a protective function under ischemic/hypoxic stimulation[46,47], we deduce that Activin A is an extracellular activator of the Activin A/Smads signal transduction pathway, and plays an important role in protecting neurons against apoptosis induced by ischemia.
Activin A can regulate the growth and apoptosis of liver cells through influencing the synthesis of DNA induced by mitogens, and there is a dose-dependent relationship between the quantity of apoptotic cells and Activin A[48]. However, in brain ischemic injury, the intranuclear target genes of the Activin A/Smads pathway remain undefined. The caspase-3 gene is the convergence point of various apoptotic pathways as well as the final approach for implementing apoptosis. It is an important effector that results in the apoptosis of ischemic neurons, which can lead to the death of cells directly[49,50,51,52]. In this study, the expression of caspase-3 mRNA in the oxygen-glucose deprivation group was markedly increased compared with the control group. This result confirmed that oxygen-glucose deprivation injury can up-regulate caspase-3 expression. We also observed down- regulation of caspase-3 gene expression following exogenous Activin A treatment, which correlated with a reduction in the rate of apoptosis after treatment with exogenous Activin A plus oxygen-glucose deprivation. Our experimental findings showed that exogenous Activin A can restrain neuronal apoptosis by activating the Activin A/Smads pathway, which may down-regulate caspase-3 expression.
In summary, ischemic injury could stimulate the activation of the Activin A/Smads pathway; exogenous Activin A can play an anti-apoptotic role and protect neuron-like PC12 cells from oxygen-glucose deprivation damage through further activation of the pathway and down-regulation of caspase-3 gene expression. Further research is required to fully understand the beneficial role of Activin A in ischemic injury, which may eventually lead to clinical interventions in ischemic cerebrovascular disease.
MATERIALS AND METHODS
Design
A controlled observational cell study.
Time and setting
This study was performed at the Central Laboratory, China-Japan Union Hospital of Jilin University, China from January 2010 to December 2012.
Materials
PC12 cells were purchased from the Cell Bank of the Chinese Academy of Sciences, China.
Methods
PC12 cell culture
The PC12 cell lines, at a density of 1 × 108 cells/L, were maintained in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum, 5% (v/v) fetal bovine serum (Gibco, New York, NY, USA), 100 IU/mL streptomycin and 100 IU/mL penicillin (pH 7.0). The cells were detached using 0.25% (w/v) trypsin (Sigma, New York, NY, USA). PC12 cells were grown at 37°C in 5% (v/v) CO2[34].
Differentiation of PC12 cells
PC12 cells, at a density of 1 × 108 cells/L, were treated with 100 ng/mL nerve growth factor (Promega, Madison, WI, USA). Twenty-four hours later, the cells were transfected and cultured for an additional 5 days in a medium containing nerve growth factor at a final concentration of 50 ng/mL[34]. PC12 cells were treated with nerve growth factor for 6 days.
Preparation of oxygen-glucose deprivation model
Cells at a density of 1 × 108 cells/L were rinsed three times with Dulbecco's modified Eagle's medium, and cultured with glucose-free Dulbecco's modified Eagle's medium containing 1 mM NaS2O4 in hypoxic conditions (37°C, 5% (v/v) CO2 and 95% (v/v) N2) for 6 hours[34]. The control group was neuron-like cells transformed from PC12 cells without any treatment.
Pretreatment with exogenous Activin A
The experimental group was made up of cells (at a density of 1 × 108 cells/L) pretreated with different concentrations of exogenous Activin A for 24 hours in vitro and then subjected to oxygen-glucose deprivation for 6 hours. The experimental group was divided into five subgroups according to different concentrations of exogenous Activin A (10, 20, 30, 50 and 100 ng/mL)[53]. According to the results, we chose a concentration of 50 ng/mL Activin A for use in the exogenous Activin A plus oxygen-glucose deprivation group.
Cell viability detected by the MTT assay
The MTT assay was used to assess cell viability[54]. The cells were grown at a density of 5 × 104 cells/well and then treated with 10, 20, 30, 50 and 100 ng/mL exogenous Activin A in a 96-well plate for 24 hours. At the end of the treatment, the Activin A-containing medium was carefully removed and the cells were subjected to oxygen-glucose deprivation for 6 hours. The culture medium was removed and 200 μL of medium containing MTT (20 μL, 5 mg/mL in PBS; Sigma) was added to each well. After cells were incubated for 4 hours at 37°C, the medium was removed and dimethyl sulfoxide (100 μL) was added to each well. The absorbance of each well was read at 490 nm. Cell viability percentage was calculated as follows: (absorbance of experimental group/absorbance of control group) × 100%[55].
Cell apoptosis detected by Hoechst 33342 fluorescence staining
PC12 cells were cultured at the same density (5 000 cells/well) in 12-well plates with Coverglass for Growth (Fisher Scientific, Pittsburgh, PA, USA) placed in advance[56]. After treatment as mentioned above, cell medium was removed, and cells in each well were fixed with 0.5 mL stationary liquid, which was removed after 10 minutes, and rinsed with PBS twice for 3 minutes each. Slides were incubated in 0.5 mL Hoechst 33342 (Sigma) at room temperature for 5 minutes, and rinsed with PBS before observation using the confocal microscopy (Olympus, Tokyo, Japan)[57]. Morphologic changes such as condensed or fragmented nuclei were considered to be apoptotic. Five fields were chosen randomly, and 200 cells were counted in each field to obtain the rate of apoptosis.
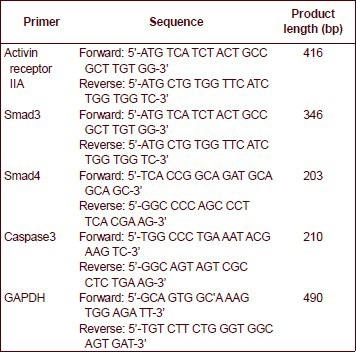
Activin receptor IIA, Smad3, Smad4 and caspase-3 mRNA levels analyzed by reverse transcription-PCR
Total RNA was extracted from cells using the RNAEasy kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer's recommendation. cDNA was synthesized using the retrotranscriptase enzyme according to the manufacturer's specification, and then stored at −20°C. GAPDH was used as an internal control. Reverse transcription-PCR was performed with 35 amplification cycles.
The products of reverse transcription-PCR were electrophoresed on a 1.2% (w/v) agarose gel, and the image was scanned using an image analysis system. The Bandscan system (Media Cybernetics, Bethesda, MD, USA) was used to analyze the absorbance of each band compared with GAPDH in cells to determine the mRNA expression of the genes.
Primer sequences are as follows:
Statistical analysis
Data were expressed as mean ± SD from three to six independent experiments. SPSS 10.0 software (SPSS, Chicago, IL, USA) was used for statistical analysis. One-way analysis of variance was used to compare mean values. A P-value less than 0.05 indicated statistically significant differences.
Acknowledgments
We would like to thank the Central Laboratory of China-Japan Union Hospital of Jilin University, China for providing the laboratory where the experiments were conducted.
Footnotes
Funding: The study is supported by the Natural Science Foundation of Jilin Province, China, No. 201015181; Jilin Province Science and Technology Development Projects, No. 20120723.
Conflicts of interest: None declared.
(Reviewed by Diwakarls S, Pack M, Wu M, Zhang JM)
(Edited by Yu J, Yang Y, Li CH, Song LP)
REFERENCES
- [1].Warlow C, Sudlow C, Dennis M, et al. Stroke. Lancet. 2003;362(9391):1211–1224. doi: 10.1016/S0140-6736(03)14544-8. [DOI] [PubMed] [Google Scholar]
- [2].Edvinsson LI, Povlsen GK. Vascular plasticity in cerebrovascular disorders. J Cereb Blood Flow Metab. 2011;31(7):1554–1571. doi: 10.1038/jcbfm.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Paeme S, Moorhead KT, Chase JG, et al. Mathematical multi-scale model of the cardiovascular system including mitral valve dynamics. Application to ischemic mitral insufficiency. Biomed Eng Online. 2011;10:86. doi: 10.1186/1475-925X-10-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hansen-Schwartz J, Svensson CL, Xu CB, et al. Protein kinase mediated upregulation of endothelin A, endothelin B and 5-hydroxytryptamine 1B/1D receptors during organ culture in rat basilar artery. Br J Pharmacol. 2002;137(1):118–126. doi: 10.1038/sj.bjp.0704838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Henriksson M, Stenman E, Edvinsson L. Intracellular pathways involved in upregulation of vascular endothelin type B receptors in cerebral arteries of the rat. Stroke. 2003;34(6):1479–1483. doi: 10.1161/01.STR.0000072984.79136.79. [DOI] [PubMed] [Google Scholar]
- [6].Barbarash OL, Zykov MV, Kashtalap VV, et al. Prevalence and clinical significance of multifocal atherosclerosis in patients with ischemic heart disease. Kardiologiia. 2011;51(8):66–71. [PubMed] [Google Scholar]
- [7].Meschia JF, Nalls M, Matarin M, et al. Siblings with ischemic stroke study: results of a genome-wide scan for stroke loci. Stroke. 2011;42(10):2726–2732. doi: 10.1161/STROKEAHA.111.620484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Foster GP, Westerdahl DE, Foster LA, et al. Ischemic preconditioning of the lower extremity attenuates the normal hypoxic increase in pulmonary artery systolic pressure. Respir Physiol Neurobiol. 2011;179(2-3):248–253. doi: 10.1016/j.resp.2011.09.001. [DOI] [PubMed] [Google Scholar]
- [9].Ageta H, Tsuchida K. Multifunctional roles of activins in the brain. Vitam Horm. 2011;85:185–206. doi: 10.1016/B978-0-12-385961-7.00009-3. [DOI] [PubMed] [Google Scholar]
- [10].Breit S, Ashman K, Wilting J, et al. The N-myc oncogene in human neuroblastoma cells: down-regulation of an angiogenesis inhibitor identified as activin A. Cancer Res. 2000;60(16):4596–4601. [PubMed] [Google Scholar]
- [11].Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- [12].Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121(5):671–674. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- [13].Le DA, Wu Y, Huang Z, et al. Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A. 2002;99(23):15188–15193. doi: 10.1073/pnas.232473399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bossenmeyer C, Chihab R, Muller S, et al. Hypoxia/reoxygenation induces apoptosis through biphasic induction of protein synthesis in cultured rat brain neurons. Brain Res. 1998;787(1):107–116. doi: 10.1016/s0006-8993(97)01527-8. [DOI] [PubMed] [Google Scholar]
- [15].Tsuchida K, Nakatani M, Uezumi A, et al. Signal transduction pathway through activin receptors as a therapeutic target of musculoskeletal diseases and cancer. Endocr J. 2008;55(1):11–21. doi: 10.1507/endocrj.kr-110. [DOI] [PubMed] [Google Scholar]
- [16].Attisano L, Cárcamo J, Ventura F, et al. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell. 1993;75(4):671–680. doi: 10.1016/0092-8674(93)90488-c. [DOI] [PubMed] [Google Scholar]
- [17].Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114(Pt 24):4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- [18].Harrison CA, Gray PC, Vale WW, et al. Antagonists of activin signaling: mechanisms and potential biological applications. Trends Endocrinol Metab. 2005;16(2):73–78. doi: 10.1016/j.tem.2005.01.003. [DOI] [PubMed] [Google Scholar]
- [19].Xu L, Chen YG, Massagué J. The nuclear import function of Smad2 is masked by SARA and unmasked by TGFbeta-dependent phosphorylation. Nat Cell Biol. 2000;2(8):559–562. doi: 10.1038/35019649. [DOI] [PubMed] [Google Scholar]
- [20].Cruise BA, Xu P, Hall AK. Wounds increase activin in skin and a vasoactive neuropeptide in sensory ganglia. Dev Biol. 2004;271(1):1–10. doi: 10.1016/j.ydbio.2004.04.003. [DOI] [PubMed] [Google Scholar]
- [21].Florio P, Frigiola A, Battista R, et al. Activin A in asphyxiated full-term newborns with hypoxic ischemic encephalopathy. Front Biosci (Elite Ed) 2010;2:36–42. doi: 10.2741/e62. [DOI] [PubMed] [Google Scholar]
- [22].Abdipranoto-Cowley A, Park JS, Croucher D, et al. Activin A is essential for neurogenesis following neurodegeneration. Stem Cells. 2009;27(6):1330–1346. doi: 10.1002/stem.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Florio P, Gazzolo D, Luisi S, et al. Activin A in brain injury. Adv Clin Chem. 2007;43:117–130. doi: 10.1016/s0065-2423(06)43004-3. [DOI] [PubMed] [Google Scholar]
- [24].Mukerji SS, Rainey RN, Rhodes JL, et al. Delayed activin A administration attenuates tissue death after transient focal cerebral ischemia and is associated with decreased stress-responsive kinase activation. J Neurochem. 2009;111(5):1138–1148. doi: 10.1111/j.1471-4159.2009.06406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hughes PE, Alexi T, Williams CE, et al. Administration of recombinant human Activin-A has powerful neurotrophic effects on select striatal phenotypes in the quinolinic acid lesion model of Huntington's disease. Neuroscience. 1999;92(1):197–209. doi: 10.1016/s0306-4522(98)00724-6. [DOI] [PubMed] [Google Scholar]
- [26].Venezia V, Nizzari M, Carlo P, et al. Amyloid precursor protein and presenilin involvement in cell signaling. Neurodegener Dis. 2007;4(2-3):101–111. doi: 10.1159/000101834. [DOI] [PubMed] [Google Scholar]
- [27].Tischler AS. Chromaffin cells as models of endocrine cells and neurons. Ann N Y Acad Sci. 2002;971:366–370. doi: 10.1111/j.1749-6632.2002.tb04498.x. [DOI] [PubMed] [Google Scholar]
- [28].Liu WB, Zhou J, Qu Y, et al. Neuroprotective effect of osthole on MPP+-induced cytotoxicity in PC12 cells via inhibition of mitochondrial dysfunction and ROS production. Neurochem Int. 2010;57(3):206–215. doi: 10.1016/j.neuint.2010.05.011. [DOI] [PubMed] [Google Scholar]
- [29].Agell N, Bachs O, Rocamora N, et al. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal. 2002;14(8):649–654. doi: 10.1016/s0898-6568(02)00007-4. [DOI] [PubMed] [Google Scholar]
- [30].Hatanaka M, Shibata N, Shintani N, et al. 15d-prostaglandin J2 enhancement of nerve growth factor-induced neurite outgrowth is blocked by the chemoattractant receptor-homologous molecule expressed on T-helper type 2 cells (CRTH2) antagonist CAY10471 in PC12 cells. J Pharmacol Sci. 2010;113(1):89–93. doi: 10.1254/jphs.10001sc. [DOI] [PubMed] [Google Scholar]
- [31].Macdonald NJ, Delderfield SM, Zhang W, et al. Tumour necrosis factor-alpha- vs. growth factor deprivation- promoted cell death: distinct converging pathways. Aging Cell. 2003;2(5):245–256. doi: 10.1046/j.1474-9728.2003.00059.x. [DOI] [PubMed] [Google Scholar]
- [32].Ryu EJ, Harding HP, Angelastro JM, et al. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22(24):10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Park D, Joo SS, Lee HJ, et al. Microtubule-associated protein 2, an early blood marker of ischemic brain injury. J Neurosci Res. 2012;90(2):461–467. doi: 10.1002/jnr.22769. [DOI] [PubMed] [Google Scholar]
- [34].Mei CL, He JT, Mang J, et al. NGF combined with OGD induces neural ischemia tolerance in PC12 cells. Afr J Biochem Res. 2011;5(10):315–320. [Google Scholar]
- [35].Müller MR, Zheng F, Werner S, et al. Transgenic mice expressing dominant-negative activin receptor IB in forebrain neurons reveal novel functions of activin at glutamatergic synapses. J Biol Chem. 2006;281(39):29076–29084. doi: 10.1074/jbc.M604959200. [DOI] [PubMed] [Google Scholar]
- [36].Bossenmeyer C, Chihab R, Muller S, et al. Hypoxia/reoxygenation induces apoptosis through biphasic induction of protein synthesis in cultured rat brain neurons. Brain Res. 1998;787(1):107–116. doi: 10.1016/s0006-8993(97)01527-8. [DOI] [PubMed] [Google Scholar]
- [37].Jones PA, May GR, McLuckie JA, et al. Apoptosis is not an invariable component of in vitro models of cortical cerebral ischaemia. Cell Res. 2004;14(3):241–250. doi: 10.1038/sj.cr.7290225. [DOI] [PubMed] [Google Scholar]
- [38].Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28(9):2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Munz B, Tretter YP, Hertel M, et al. The roles of activins in repair processes of the skin and the brain. Mol Cell Endocrinol. 2001;180(1-2):169–177. doi: 10.1016/s0303-7207(01)00514-7. [DOI] [PubMed] [Google Scholar]
- [40].Xu P, Liu J, Derynck R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012;586(14):1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Luukko K, Ylikorkala A, Mäkelä TP. Developmentally regulated expression of Smad3, Smad4, Smad6, and Smad7 involved in TGF-beta signaling. Mech Dev. 2001;101(1-2):209–212. doi: 10.1016/s0925-4773(00)00556-6. [DOI] [PubMed] [Google Scholar]
- [42].Flanders KC, Kim ES, Roberts AB. Immunohistochemical expression of Smads 1-6 in the 15-day gestation mouse embryo: signaling by BMPs and TGF-betas. Dev Dyn. 2001;220(2):141–154. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1096>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- [43].Tong JL, Nie F, Ran ZH, et al. Epigallocatechin gallate induces apoptosis in human hepatocellular carcinoma HepG2 cells via TGF/Smad signaling pathway. Zhonghua Zhong Liu Za Zhi. 2009;31(9):646–650. [PubMed] [Google Scholar]
- [44].Hata A, Lo RS, Wotton D, et al. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. 1997;388(6637):82–87. doi: 10.1038/40424. [DOI] [PubMed] [Google Scholar]
- [45].Yagi K, Goto D, Hamamoto T, et al. Alternatively spliced variant of Smad2 lacking exon 3. Comparison with wild-type Smad2 and Smad3. J Biol Chem. 1999;274(2):703–709. doi: 10.1074/jbc.274.2.703. [DOI] [PubMed] [Google Scholar]
- [46].Chen W, Woodruff TK, Mayo KE. Activin A-induced HepG2 liver cell apoptosis: involvement of activin receptors and smad proteins. Endocrinology. 2000;141(3):1263–1272. doi: 10.1210/endo.141.3.7361. [DOI] [PubMed] [Google Scholar]
- [47].Wu DD, Lai M, Hughes PE, et al. Expression of the activin axis and neuronal rescue effects of recombinant activin A following hypoxic-ischemic brain injury in the infant rat. Brain Res. 1999;835(2):369–378. doi: 10.1016/s0006-8993(99)01638-8. [DOI] [PubMed] [Google Scholar]
- [48].Yasuda H, Mine T, Shibata H, et al. Activin A: an autocrine inhibitor of initiation of DNA synthesis in rat hepatocytes. J Clin Invest. 1993;92(3):1491–1496. doi: 10.1172/JCI116727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407(6805):802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- [50].Gurney ME, Tomasselli AG, Heinrikson RL. Neurobiology. Stay the executioner's hand. Science. 2000;288(5464):283–284. doi: 10.1126/science.288.5464.283. [DOI] [PubMed] [Google Scholar]
- [51].Lee JM, Grabb MC, Zipfel GJ, et al. Brain tissue responses to ischemia. J Clin Invest. 2000;106(6):723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Condorelli G, Roncarati R, Ross J, Jr, et al. Heart-targeted overexpression of caspase3 in mice increases infarct size and depresses cardiac function. Proc Natl Acad Sci U S A. 2001;98(17):9977–9982. doi: 10.1073/pnas.161120198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lai M, Gluckman P, Dragunow M, et al. Focal brain injury increases activin betaA mRNA expression in hippocampal neurons. Neuroreport. 1997;8(12):2691–2694. doi: 10.1097/00001756-199708180-00011. [DOI] [PubMed] [Google Scholar]
- [54].Longxi P, Buwu F, Yuan W, et al. Expression of p53 in the effects of artesunate on induction of apoptosis and inhibition of proliferation in rat primary hepatic stellate cells. PLoS One. 2011;6(10):e26500. doi: 10.1371/journal.pone.0026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rao V, Balachandran B, Shen H, et al. In vitro and in vivo antioxidant properties of the plant-based supplement greens+™. Int J Mol Sci. 2011;12(8):4896–4908. doi: 10.3390/ijms12084896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mukerji SS, Katsman EA, Wilber C, et al. Activin is a neuronal survival factor that is rapidly increased after transient cerebral ischemia and hypoxia in mice. J Cereb Blood Flow Metab. 2007;27(6):1161–1172. doi: 10.1038/sj.jcbfm.9600423. [DOI] [PubMed] [Google Scholar]
- [57].Li XQ, Verge VM, Johnston JM, et al. CGRP peptide and regenerating sensory axons. J Neuropathol Exp Neurol. 2004;63(10):1092–1103. doi: 10.1093/jnen/63.10.1092. [DOI] [PubMed] [Google Scholar]