

Abstract
Alzheimer's disease is a neurodegenerative disorder characterized by progressive cognitive impairment and neuropathology. Recent preclinical and epidemiological studies proposed statins as a possible therapeutic drug for Alzheimer's disease, but the exact mechanisms of action are still unknown. Biliverdin reductase-A is a pleiotropic enzyme involved in cellular stress responses. It not only transforms biliverdin-IX alpha into the antioxidant bilirubin-IX alpha but its serine/threonine/tyrosine kinase activity is able to modulate cell signaling networks. We previously reported the beneficial effects of atorvastatin treatment on biliverdin reductase-A and heme oxygenase-1 in the brains of a well characterized pre-clinical model of Alzheimer's disease, aged beagles, together with observed improvement in cognition. Here we extend our knowledge of the effects of atorvastatin on inducible nitric oxide synthase in parietal cortex, cerebellum and liver of the same animals. We demonstrated that atorvastatin treatment (80 mg/day for 14.5 months) to aged beagles selectively increased inducible nitric oxide synthase in the parietal cortex but not in the cerebellum. In contrast, inducible nitric oxide synthase protein levels were significantly decreased in the liver. Significant positive correlations were found between biliverdin reductase-A and inducible nitric oxide synthase as well as heme oxygenase-1 protein levels in the parietal cortex. The opposite was observed in the liver. Inducible nitric oxide synthase up-regulation in the parietal cortex was positively associated with improved biliverdin reductase-A functions, whereas the oxidative-induced impairment of biliverdin reductase-A in the liver negatively affected inducible nitric oxide synthase expression, thus suggesting a role for biliverdin reductase-A in atorvastatin-dependent inducible nitric oxide synthase changes. Interestingly, increased inducible nitric oxide synthase levels in the parietal cortex were not associated with higher oxidative/nitrosative stress levels. We hypothesize that biliverdin reductase-A-dependent inducible nitric oxide synthase regulation strongly contributes to the cognitive improvement observed following atorvastatin treatment.
Keywords: neural regeneration, age, Alzheimer's disease, atorvastatin, biliverdin reductase-A, cell stress-response, cognitive function, 4-hydroxy-2-nonenal, heme oxygenase-1, inducible nitric oxide synthase, oxidative stress, neuroregeneration
Research Highlights
-
(1)
Atorvastatin increases inducible nitric oxide synthase protein levels in the parietal cortex whereas decreases inducible nitric oxide synthase protein levels in the liver of aged beagles
(2) Up-regulation of inducible nitric oxide synthase and heme oxygenase-1 are positively associated with biliverdin reductase-A protein levels and activity.
-
(3)
Down-regulation of inducible nitric oxide synthase and heme oxygenase-1 are negatively associated with biliverdin reductase-A oxidation
INTRODUCTION
Alzheimer's disease is the most common form of dementia among the elderly and is characterized by progressive loss of memory and cognition. Amyloid-β-peptide plaques and neurofibrillary tangles composed of hyperphosphorylated tau, are two major hallmarks of Alzheimer's disease neuropathology. Both Amyloid-β-peptide and tau promote the formation of reactive oxygen and nitrogen species, induce calcium-dependent excitotoxicity and impairment of cellular respiration[1]. Alzheimer's disease neuropathology is observed in the hippocampus and underlying cortex, as well as in neocortex but with the cerebellum remaining relatively spared until late in the disease[2,3]. Treatments currently available primarily target the symptoms of dementia but do not appear to modify disease processes. Thus, identifying new approaches to preventing or slowing disease progression in patients with Alzheimer's disease is critically important.
Statins, a class of hypolipidemic drugs, have been proposed as potential agents for the treatment or prevention of Alzheimer's disease[4,5,6]. Data from animal models studies suggest possible mechanisms underlying the beneficial role of atorvastatin in preventing Alzheimer's disease, including the reduction of amyloid-β-peptide[7], β-secretase protein levels[8] and oxidative stress[9]. In addition, atorvastatin induces the activation of the heme oxygenase-1/biliverdin reductase-A system, which was proposed to have neuroprotective effects in the brain[10,11]. Interestingly, previous studies showed that statins enhance neurogenesis, synaptogenesis, and angiogenesis, and significantly improves neurological outcome after stroke[12] as well as after traumatic brain injuries[13]. However, the importance of statin treatment in Alzheimer's disease is still under debate, given that randomized clinical trials did not show any significant benefit on cognition[6,14]. In previous studies from our group, atorvastatin promoted beneficial effects in the brain of aged beagles, potentially through the modulation of the heme oxygenase-1/biliverdin reductase-A system[9,10,11] (Table 1).
Table 1.
Main effects observed during previous studies by our group about oxidative stress levels and heme oxygenase-1/biliverdin reductase-A system in the brain and liver of aged beagles following atorvastatin treatment
Aged beagles naturally develop learning and memory impairments in association with the accumulation of amyloid-β-peptide of the same amino acid sequence as humans representing a valuable model of early Alzheimer's disease pathology[15].
We hypothesized that the observed increase of biliverdin reductase-A protein levels and activity[10] could trigger a cell stress response improvingcognition by the following mechanisms: (i) Interaction with members of the MAPK family, such as ERK1/2-Mek-Elk1, through which biliverdin reductase-A regulates important metabolic pathway as well as the expression of oxidative-stress-responsive genes such as heme oxygenase-1 or inducible nitric oxide synthase[16,17,18,19]; (ii) activation of both conventional and atypical protein kinase C isoforms[16] whose involvement in memory function is now well established[20]; (iii) production of the powerful antioxidant BR as result of its reductase activity. In this context, since the phosphorylation of biliverdin reductase-A on Tyr residues is required to interact with ERK-Mek-Elk1[17], the increase of phospho-tyrosine on biliverdin reductase-A in the parietal cortex following atorvastatin treatment[10], coupled with the negative correlation between phospho-tyrosine on biliverdin reductase-A and size discrimination error scores[10], could suggest an activation of the MAPK-related signal transduction pathways that in turn promote a robust cell stress response[16].
At the same time, the significant correlations found between biliverdin reductase activity and decreased total protein carbonyls and 3-nitrotyrosine levels[10] suggest, consistently with prior studies[21,22,23,24], a crucial antioxidant role for BR. Considering the broad functions of biliverdin reductase-A in the cell, increased biliverdin reductase-A phosphorylation likely would have consequence beyond immediate changes in its scaffolding and reductase activities. In order to analyze this aspect in depth, this study aimed to measure the expression levels of inducible nitric oxide synthase and the possible correlations with biliverdin reductase-A levels and activities following of atorvastatin treatment in aged beagles. We hypothesized that the improved function of biliverdin reductase-A is associated with increased inducible nitric oxide synthase levels and that biliverdin reductase-A could contribute to the maintenance of physiological levels of nitric oxide in order to avoid its neurotoxic effects. In addition, we provide new results about the association between biliverdin reductase-A and heme oxygenase-1, the latter another main target of biliverdin reductase-A activity during cell stress-response, as noted above.
RESULTS
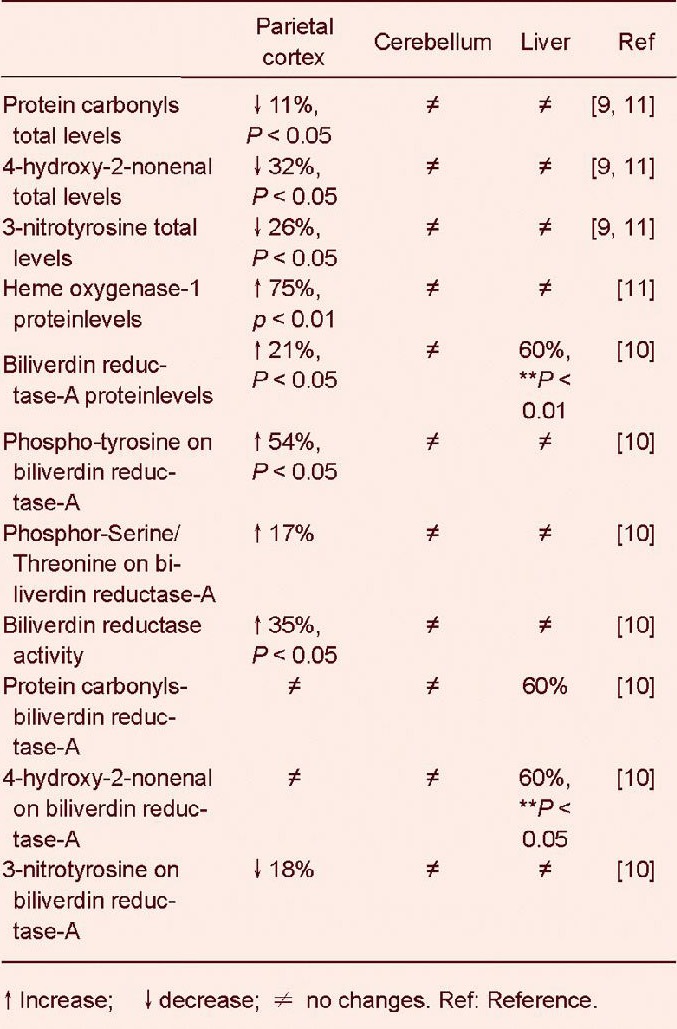
Biliverdin reductase-A protein levels increased with age in the parietal cortex of aged beagles
Age-associated changes of biliverdin reductase-A expression were previously reported in rat brain[25]. Here we provide novel data of an age-dependent modulation of biliverdin reductase-A protein in dogs. We observed a significant 2.4-fold increase of biliverdin reductase-A protein levels in 12 year old beagles (t(6) = 2.68, P < 0.05) compared to 4 years old (Figure 1).
Figure 1.
Age-dependent changes of biliverdin reductase-A (BVR-A) protein levels in parietal cortex of beagles.
Representative gel is shown. Data are expressed as mean ± SD (n = 3 replicates of the same sample for each time point). P < 0.05, vs. 4.49 years old.
Furthermore, a 1.6-fold decrease was observed in the parietal cortex of 14-year-old dogs, compared to 12 years old, although this value did not reach statistical significance.
Effect of atorvastatin treatment on inducible nitric oxide synthase protein levels in the parietal cortex, the cerebellum and liver of aged beagle
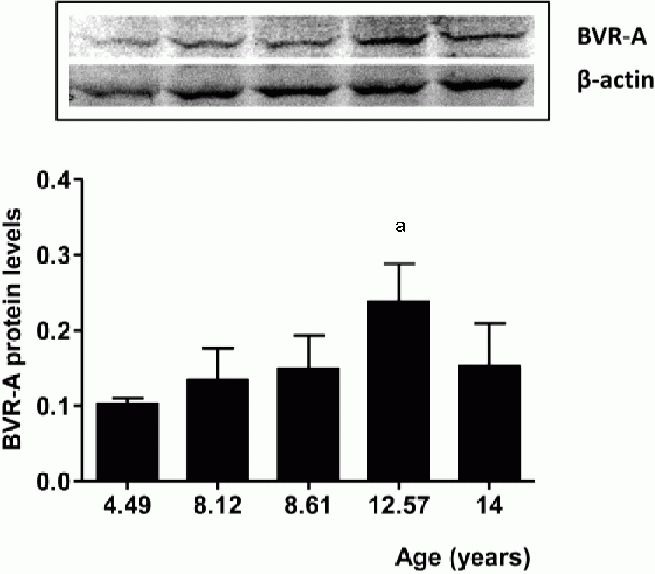
We previously reported differential effects of atorvastatin on biliverdin reductase-A protein levels and activities in the parietal cortex, cerebellum and liver of aged beagles[10] (Table 1). The current aging study shows that biliverdin reductase-A decreases as animals move from 12–14 years of age. In the canine atorvastatin study, beagles ranging in age from 8.9–13.2 received treatment for 14.5 months and a significant increase of biliverdin reductase-A protein levels in the parietal cortex following atorvastatin treatment was observed[10], perhaps suggesting it protects against further age-associated decreases of biliverdin reductase-A. Given tissue-specific changes in the levels of biliverdin reductase-A Table 1,[10]), and due to its role in the regulation of inducible nitric oxide synthase[16,26,27], we predicted that inducible nitric oxide synthase would be modified in response to atorvastatin treatment. Consistent with this hypothesis, atorvastatin (80 mg/kg per day for 14.5 months) increased inducible nitric oxide synthase protein levels by approximately 35% (t(6) = 1.57, P = 0.16) in the parietal cortex (Figure 2A). However, the increased ininducible nitric oxide synthase expression was not statistically significant compared to controls. There was no effect of treatment on inducible nitric oxide synthase protein levels in the cerebellum (t (6) = 0.14, P = 0.88) (Figure 2B). Conversely, a significant down-regulation of inducible nitric oxide synthase by 30% (t(6) = 3.17, P < 0.05) was observed in the liver (Figure 2C).
Figure 2.
Inducible nitric oxide synthase (iNOS) protein levels in the parietal cortex, the cerebellum and liver of aged beagles treated with atorvastatin.
iNOS protein levels in the (A) parietal cortex, (B) cerebellum and (C) liver of aged beagles. Representative gels are shown. Data are expressed as mean ± SD (n = 4 animals per group). P < 0.05, vs. control.
Atorvastatin-induced changes on biliverdin reductase-A protein levels, phosphorylation and oxidation were associated with changes of inducible nitric oxide synthaseand heme oxygenase-1 protein levels
Changes observed for inducible nitric oxide synthase protein levels were hypothesized to be associated with atorvastatin-induced alteration of biliverdin reductase-A. As our group previously reported, atorvastatin administration to aged beagles not only increased biliverdin reductase-A protein levels in the parietal cortex, but also promoted positive changes on the post-translational modifications affecting the protein (Table 1,[10].
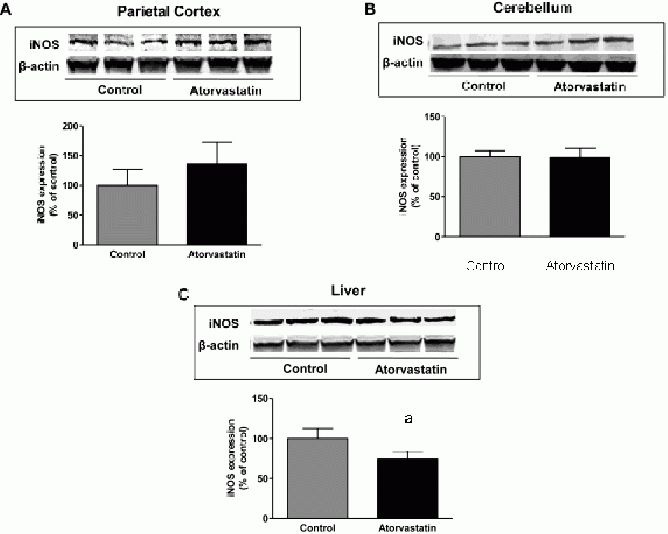
In particular, we found an increase of biliverdin reductase-A phosphorylation (phospho-tyrosine on biliverdin reductase-A, phosphor-Serine/Threonine on biliverdin reductase-A) and a decrease of biliverdin reductase-A nitration (3-nitrotyrosine on biliverdin reductase) which leaded to an increased activation of biliverdin reductase-A in the parietal cortex (Table 1[10]. Furthermore, an opposite situation characterized by an increase of biliverdin reductase-A protein levels together with an increase of its oxidation (4-hydroxy-2-nonenal on biliverdin reductase-A) was found in the liver (Table 1[10]. These latter results, obtained on samples coming from the same animals to which we refer in the current study, were used to analyze any possible association between inducible nitric oxide synthase protein levels and (i) biliverdin reductase-A protein levels, (ii) phospho-tyrosine on biliverdin reductase-A levels and (iii) phosphor-Serine/Threonine on biliverdin reductase-A levels in the parietal cortex (Figure 3A–C) and liver (Figure 3D–F).
Figure 3.
Inducible nitric oxide synthase (iNOS) protein levels are associated with biliverdin reductase-A (BVR-A) protein levels and phosphorylation in the parietal cortex and liver of aged beagles treated with atorvastatin.
Positive correlations were found between iNOS protein levels and (A) BVR-A protein levels (r = 0.77; P < 0.05), (B) phospho-tyrosine (pTyr) on BVR-A (r = 0.53, P < 0.05) and (C) phosphor-Serine/Threonine (pSer/Thr) on BVR-A (r = 0.78, P < 0.05) in the parietal cortex.
Negative correlations were found between iNOS protein levels and (D) BVR-A protein levels (r = −0.60, P = 0.11), (E) pTyr on BVR-A (r = −0.47, P = 0.24) and (F) pTyr/Thr on BVR-A (r = −0.31, P = 0.46) in the liver.
Due to the fact that no changes occurred in cerebellum, a further analysis of this brain region was not addressed. Biliverdin reductase-A protein levels positively and significantly correlated with inducible nitric oxide synthase protein levels (Pearson r = 0.78, P < 0.05; Figure 3A) only in the parietal cortex. In addition, the extent of biliverdin reductase-A phosphorylation positively correlated with inducible nitric oxide synthase protein levels in parietal cortex (phospho-tyrosine on biliverdin reductase-A (Pearson r = 0.53) and phosphor-Serine/Threonine on biliverdin reductase-A (Pearson r = 0.77; P < 0.05)] (Figure 3B, C, respectively), thus supporting the idea that an increase of biliverdin reductase-A protein levels together with its activation (increased phosphorylation) could play a main role in the regulation of inducible nitric oxide synthase expression in the brain. Very interestingly, we found the opposite in the liver. Indeed, after atorvastatin treatment, inducible nitric oxide synthase protein levels were significantly decreased in treated animals compared to controls despite a significant increase of biliverdin reductase-A protein levels[10] (Table 1). Furthermore, in the liver, correlation analysis demonstrated, negative associations between inducible nitric oxide synthase protein levels and (i) biliverdin reductase-A protein levels (Pearson r = −0.60, P = 0.11; Figure 3D) or (ii) phospshorylation[phospho-tyrosine on biliverdin reductase-A, (Spearman r = −0.47, P = 0.24) and phosphor-Serine/Threonine on biliverdin reductase-A (Spearman r = −0.31, p = 0.46)] (Figure 3E, F, respectively). In order to strengthen the hypothesis with regard to the role of biliverdin reductase-A on the effects produced by atorvastatin treatment in aged beagles, a correlation analysis between biliverdin reductase-A and heme oxygenase-1 protein levels was performed, since heme oxygenase-1 is another protein tightly regulated by the activation of biliverdin reductase-A[16,18,28,29,30]. We previously demonstrated a selective increase of heme oxygenase-1 in the parietal cortex of the same animals used for this study, following atorvastatin administration, whereas no changes were observed in the liver[11] (Table 1). Here a positive correlation between biliverdin reductase-A and heme oxygenase-1 in parietal cortex, was found.
In particular, biliverdin reductase-A and heme oxygenase-1 protein levels positively correlated in the parietal cortex (Spearman r = 0.69), and this association is close to statistical significance (P = 0.06) (Figure 4A).
Figure 4.
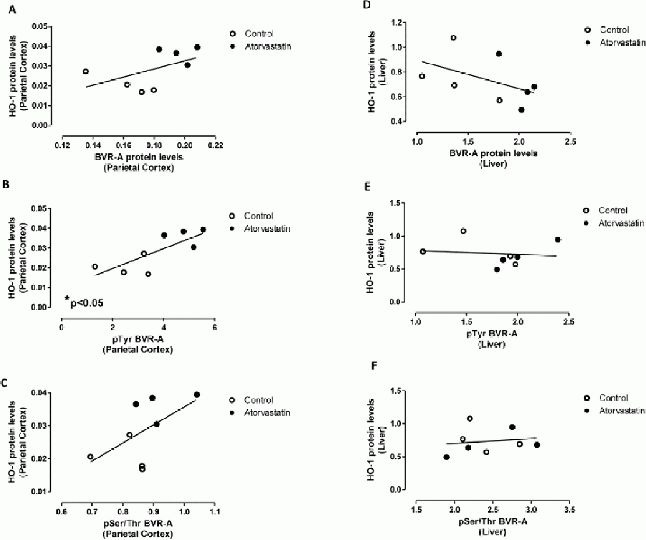
Heme oxygenase-1 (HO-1) protein levels are associated with biliverdin reductase-A (BVR-A) protein levels and phosphorylation in the parietal cortex and liver of aged beagles treated with atorvastatin.
Positive correlations were found between HO-1 protein levels and (A) BVR-A protein levels (r = 0.69; P = 0.06), (B) phospho-tyrosine (pTyr) on BVR-A (r = 0.78, P < 0.05) and (C) phosphor-Serine/Threonine (pSer/Thr) on BVR-A (r = 0.57; P = 0.14) in the parietal cortex.
A negative correlation was found between HO-1 protein levels and (D) BVR-A protein levels (r = −0.66; P = 0.08) in the liver. Any association was found between HO-1 protein levels and (E) pTyr on BVR-A or (F) pSer/Thr on BVR-A in the liver.
Similarly, phospho-tyrosine on biliverdin reductase-A (Pearson r = 0.78; P < 0.05) and phosphor-Seine/Threonine on biliverdin reductase-A (Pearson r = 0.57; P = 0.14) positively correlated with heme oxygenase-1 protein levels in the parietal cortex (Figures 4B, C, respectively). On the contrary, in the liver, biliverdin reductase-A protein levels negatively correlated with heme oxygenase-1 protein levels (Pearson r = −0.66; P = 0.08, Figure 4D) and an association was found between the extent of biliverdin reductase-A phospshorylation and heme oxygenase-1 protein levels (Figure 4E, F). Thus, higher levels and activation of biliverdin reductase-A were associated with increased heme oxygenase-1 protein levels only in the parietal cortex, and these data resemble those obtained for inducible nitric oxide synthase.
However, there is a discrepancy between the results of inducible nitric oxide synthase and heme oxygenase-1 changes in response to atorvastatin treatment in the parietal cortex and liver, despite a significant and consistent increase of biliverdin reductase-A protein[10]. Although it is well known that several and distinct mechanisms could participate in the regulation of inducible nitric oxide synthase and heme oxygenase-1 expression, negative associations between the oxidation of biliverdin reductase-A (4-hydroxy-2-nonenal on biliverdin reductase-A) and (i) inducible nitric oxide synthase (Spearman r = −0.76, P < 0.05; Figure 5A) or (ii) heme oxygenase-1 protein levels (Spearman r = −0.45, P = 0.32; Figure 5B) in the liver, were observed. These data support the hypothesis that an oxidative impairment of biliverdin reductase-A in the liver could be responsible, at least in part, for the loss of the effect on inducible nitric oxide synthase and heme oxygenase-1 following atorvastatin treatment.
Figure 5.
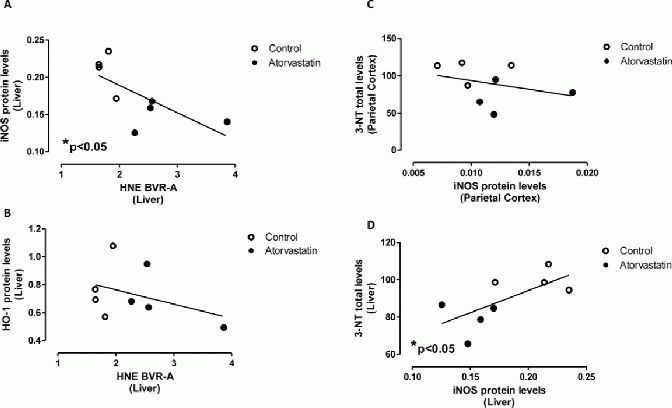
Association between biliverdin reductase-A (BVR-A) oxidation and (A) inducible nitric oxide synthase (iNOS), (B) heme oxygenase-1 (HO-1) protein levels in the liver of aged beagles treated with atorvastatin.
Association between 3-nitrotyrosine (3-NT) levels and iNOS protein levels in the (C) parietal cortex and (D) liver of aged beagles treated with atorvastatin. Negative correlations were found between BVR-A protein levels and (A) iNOS protein levels (r = −0.76, P < 0.05), (B) HO-1 protein levels (r = −0.45, P = 0.32) in the liver. Any correlation was found between iNOS levels and (C) 3-NT levels in the parietal cortex. A negative correlation was found between iNOS protein levels and (D) 3-NT levels (r = 0.69, P < 0.05) in the liver.
Atorvastatin-induced changes of inducible nitric oxide synthase protein levels were associated with 3-nitrotyrosine levels only in the liver
An evaluation of the association between inducible nitric oxide synthase protein levels and a well-known biomarker of nitrosative stress, i.e., 3-nitrotyrosine, was performed. As we previously reported, atorvastatin produced a significant decrease of 3-nitrotyrosine in the parietal cortex[9] (Table 1), whereas no changes were observed in the liver[11] of the animals used for this study (Table 1). Since inducible nitric oxide synthase is one of the main sources of nitric oxide during conditions responsible for an increase of oxidative/nitrosative stress levels (e.g., inflammation), this study aimed to understand if changes produced by atorvastatin also are associated with nitrosative stress levels.
As expected, no significant association between inducible nitric oxide synthase and 3-nitrotyrosine in the parietal cortex (Pearson r = −0.29, P = 0.42; Figure 5C), was found. On the contrary, inducible nitric oxide synthase protein levels were negatively associated with 3-nitrotyrosine levels in the liver (Pearson r = 0.69, P < 0.05, Figure 5D). Thus, lower levels of inducible nitric oxide synthase protein were associated with reduced nitrosative damage in the liver.
DISCUSSION
Aging is one of the greatest risk factors for cognitive decline and Alzheimer's disease. Among the elderly, Alzheimer's disease represents one of the main causes of severe cognitive decline and dementia[1].
Increased oxidative and nitrosative stress levels with age and in the pathogenesis of neurodegenerative disorders could be a significant contributor to cognitive impairment and dementia[31,32,33,34,35,36,37,38,39,40]. Among the defenses that the human brain possesses, a majorrole is played by the heme oxygenase-1/biliverdin reductase-A system, whose up-regulation (i) is one of the earlier events in the adaptive response to stress[41], and (ii) was proposed as a useful pathway to manipulate to counteract Alzheimer's disease-induced oxidative/nitrosative damage[39,42,43,44,45,46].
We previously described phosphorylation and oxidative modifications to the levels and activity of the heme oxygenase-1/biliverdin reductase-A system in the brain and plasma of Alzheimer's disease or mild cognitive impairment subjects[47,48,49,50], opening a debate on its possiblepathophysiological and clinical significance. Indeed, we found that biliverdin reductase-A protein levels were significantly increased in the brain of Alzheimer's disease and or mild cognitive impairment subjects, whereas the activity of the protein was significantly decreased[47]. The explanation we provided to clarify this apparent paradox was that biliverdin reductase-A undergoes oxidative/nitrosative post-translational modifications which affects its structure and thus, its activity. Very interestingly, by using a well characterized method to identify protein oxidatively modified[10,47,50,51], we found that the levels of 3-nitrotyrosine on biliverdin reductase-A were significantly increased in the hippocampi of both Alzheimer's disease or or mild cognitive impairment subjects whereas the levels of phosphorylation of serine/threonine/tyrosine residues were reduced, making biliverdin reductase-A dysfunctional[47,50].
Since it is well known that the oxidative/nitrosa-tivepost-translational modificationsalter proteins structure[52,53] and result in a marked decrease of their function[45,51,54], it is plausible to argue that the formation of biliverdin reductase's 3-nitrotyrosine-adducts are responsible for the the significant reduction of biliverdin reductase activity in Alzheimer's disease and MCI hippocampi.
Furthermore, by using a well characterized pre-clinical model of Alzheimer's disease, such as aged beagles[15], we showed that parietal-resident biliverdin reductase-A may be a target for atorvastatin, which was able to increase biliverdin reductase-A protein levels as well as to improve its activities with positive effects on cognition[10]. In this study, we extend the neurobiological benefits of atorvastatin in the brain of aged beagles by showing that inducible nitric oxide synthase protein levels are associated with changes of biliverdin reductase-A levels and activities.
Novel data are provided in this study of age-dependent changes in biliverdin reductase-A protein levels in the parietal cortex of beagles. These results, are in good agreement with those found previously in rat brain[25], and strengthen the concept that, in the brain, biliverdin reductase-A is tightly regulated across the lifespan. One possible explanation, is that, since biliverdin reductase-A is an oxidative stress-induced protein[16], its levels rise together with those of oxidative stress as also observed in Alzheimer's disease and or mild cognitive impairment hippocampus[47,50]. Based on these findings, our studies with aging dogs[10] suggest that atorvastatin could promote biliverdin reductase-A up-regulation in the parietal cortex even at an old age (11–13 years old) when levels naturally decrease as shown in Figure 1. Thus, despite the observed decrease of biliverdin reductase-A protein levels at 14 years old, which may be due to progressive neurodegeneration, it may still be possible to improve biliverdin reductase-A functions with atorvastatin. The mechanisms through which biliverdin reductase-A could exert its neuroprotective effects are those noted above, and it is conceivable to think that a therapeutic approach able to recover biliverdin reductase-A activities could represent a good strategy aimed to improve cognitive function.
Consistent with this hypothesis, there appears to be a very interesting link between biliverdin reductase-A and inducible nitric oxide synthase[16,26,27]. Nitric oxide, the end-product of Nitric Oxide Synthase(s) activity, has an important role in the regulation of synaptic plasticity, which in turn is critically involved in cognition, including memory[42,55,56]. Inducible nitric oxide synthase levels in the central nervous system are generally fairly low; however, an increased expression of inducible nitric oxide synthase in astrocytes and microglia occurs due to pro-inflammatory conditions[42,57]. Thus, increased production of nitric oxide becomes harmful involving the formation of RNS, such as peroxynitrite, which in turn leads to the formation of 3-nitrotyrosine adducts[53,58,59]. Our results clearly showed that following atorvastatin treatment, the levels of inducible nitric oxide synthase protein are differentially affected in the brain, and between brain and periphery (Figure 2A–C). Despite an increase of inducible nitric oxide synthase in parietal cortex (Figure 2A), the levels of 3-nitrotyrosine in this area were significantly lower than those observed in the control dogs[9] (Table 1). This result is surprising given the patho-physiological role of inducible nitric oxide synthase, but it may be conceivable that the improvement of biliverdin reductase-A activities[10] together with the concomitant increased production of the antioxidant and anti-nitrosative molecule BR[10,24,60] are able to maintain nitric oxide levels under the pathological threshold, providing a direct neuroprotective effect. Furthermore, these finding are in good agreement with those found in Alzheimer's disease brain where an increase of 3-nitrotyrosine levels together with an impairment of biliverdin reductase-A activities were observed[50]. In this scenario, the biliverdin reductase-A-dependent modulation of inducible nitric oxide synthase expression becomes fascinating. In fact, as recently demonstrated in an elegant paper by Maines and colleagues[26], biliverdin reductase, as an intracellular scaffold/bridge/anchor protein, is required for placing ERK1/2 in proximity to its kinases, MEK1/2, in the cytoplasm and bringing Elk1 in contact with the activated ERK1/2 in the nucleus[26]. Through this mechanism it regulates the activation of an estimated 50 nuclear factors and proteins that influence cell differentiation, proliferation, stress response, and promote tumor growth including inducible nitric oxide synthase and heme oxygenase-1[16,26]. That said, the positive correlations found in the parietal cortex with regard to biliverdin reductase-A protein levels or phosphorylation and inducible nitric oxide synthase protein levels seem to support in an in vivo model the mechanism proposed above. To strengthen this hypothesis further, we analyzed the association between biliverdin reductase-A and another target of its activity, heme oxygenase-1. Similarly to the results obtained for inducible nitric oxide synthase, heme oxygenase-1 protein levels were positively associated with biliverdin reductase-A protein levels and phospshorylation in the parietal cortex, althought a significant association was found only for phospho-tyrosine on biliverdin reductase-A. In combination, these observations could suggest that the increase of biliverdin reductase-A (i) protein levels and (ii) phosphor-Serine/Threonine/Tyrosine represents an important feature in the signaling pathway stimulated by atorvastatin and involving biliverdin reductase-A-dependent regulation of stress-responsive gene such as heme oxygenase-1 or inducible nitric oxide synthase[16].
In paradoxical support of the role of biliverdin reductase-A, are the results observed in the liver where it appears that other mechanisms affect inducible nitric oxide synthase regulation. In fact, an increase of biliverdin reductase-A protein levels and significant reduction of inducible nitric oxide synthase was observed. In our previous study, we showed that no differences exist for 3-nitrotyrosine in the liver between controls and atorvstatin-treated beagles[11], and we proposed that this effect could be related to the oxidative-induced impairment of biliverdin reductase-A (increased 4-hydroxy-2-nonenal on biliverdin reductase-A, Table 1). This hypothesis is further corroborated by several reports that many proteins involved in the regulation of important cellular functions became dysfunctional in Alzheimer's disease brain due to oxidative and nitrosative modifications[61,62,63,64,65,66,67,68,69]. The novelty of our results is that an increase of biliverdin reductase-A oxidation (4-hydroxy-2-nonenal on biliverdin reductase-A) is associated with decreased levels of inducible nitric oxide synthase (Figure 5A), supporting once again the role of biliverdin reductase-A in inducible nitric oxide synthase regulation. Thus, if biliverdin reductase-A does not function properly, this could have consequences on its downstream target (Figure 6).
Figure 6.
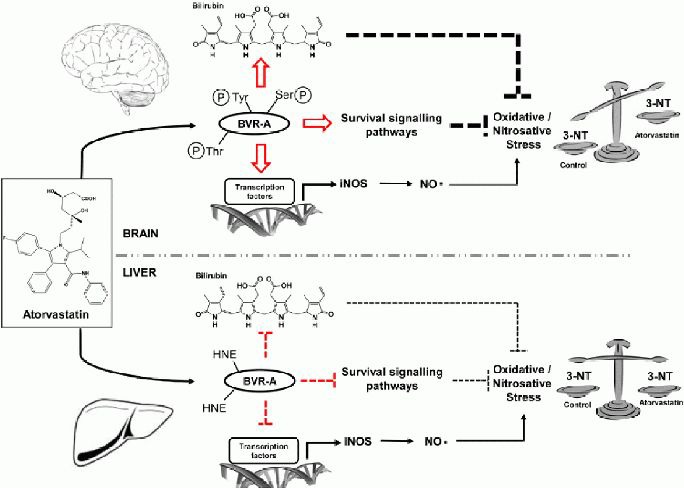
Proposed mechanisms for atorvastatin-induced biliverdin reductase-A-dependent modulation of inducible Nitric Oxide Synthase and oxidative stress levels in the brain and liver of aged beagles.
PC: protein carbonyls; HNE: 4-hydroxy-2-nonenal; 3-NT: 3-nitrotyrosine; BVR-A: biliverdin reductase-A; BVR-A-3NT: 3-nitrotyrosine on BVR-A; BVR-A-HNE: 4-hydroxy-2-nonenal on BVR-A; HO-1: heme oxygenase-1; iNOS: inducible nitric oxide synthase; pTyr-BVR-A: phospho-tyrosine on BVR-A; pSer/Thr-BVR-A: phosphor-Serine/Threonine on BVR-A.
Atorvastatin treatment promoted two different effects in the brain and liver of aged beagles. In the brain, atorvastatin increased biliverdin reductase-A protein levels and phosphorylation (phosphor-Serine/Threonine/Tyrosine), thus promoting its reductase and kinase activities. Through its reductase activity biliverdin reductase-A produces the antioxidant and antinitrosative molecule bilirubin. Through its kinase activity, biliverdin reductase-A is able to activate downstream survival signaling pathways[16], as well as to promote the transcription of oxidative-induced genes such as heme oxygenase-1 and inducible nitric oxide synthase[16]. It is conceivable to hypothesize that the neuroprotective effects mediated by bilirubin and the activated survival signaling pathways, overcome the neurotoxic increase of nitric oxide due to the concomitant up-regulation of inducible nitric oxide synthase. Thus, the final effect is a reduction of oxidative/nitrosative stress levels, e.g. 3-nitrotyrosine as previously demonstrated[9]. In the liver, atorvastatin increased biliverdin reductase-A protein levels, but also its oxidation (4-hydroxy-2-nonenal on biliverdin reductase-A) leading to an impairment of biliverdin reductase-A activities. Furthermore, biliverdin reductase-A oxidation was associated with a reduction of inducible nitric oxide synthase protein levels. In this case, the lack of biliverdin reductase-A antioxidant power which should favor an increase of the oxidative/nitrosative stress levels, is balanced by the decrease of inducible nitric oxide synthase protein levels (less nitric oxide production) with the final effect that no changes were observed for the oxidative/nitrosative stress levels e.g., 3-nitrotyrosine.
Also for heme oxygenase-1 we observed the same profile (Figure 5B). Increased heme oxygenase-1 protein levels in parietal cortex were positively associated with biliverdin reductase-A protein levels and phosphorylation (Figures 4A–C). On the contrary, the oxidative-induced impairment of biliverdin reductase-A seems to be associated with a decrease of heme oxygenase-1 protein levels (Figure 5B). In addition, in light of these results, the observation that any significant change of 3-nitrotyrosine levels in the liver was found between control and atorvastatin-treated dog (Table 1,[11]) requires a novel explanation. This latter, could account not only the lack of effect on biliverdin reductase-A activities as previously postulated[10,11], but also the observation of a reduction of inducible nitric oxide synthase protein levels following atorvastatin treatment. As shown in Figure 5D, a significant correlation was found between the levels of 3-nitrotyrosine and those of inducible nitric oxide synthase in the liver suggesting that reduced levels of inducible nitric oxide synthase are associated with lower levels of 3-nitrotyrosine. That said, why we did not find any significant difference between controls and atorvastatin treated dogs (Table 1,[11])? An intuitive explanation could be that the lack of effect of atorvastatin treatment on 3-nitrotyrosine levels in the liver is due to the small sample size used for this study. However, although the sample size was relatively small, consistent effects with (i) other oxidative stress markers[9]; (ii) biliverdin reductase-A protein levels, post-translational modifications and activity[10]; (iii) heme oxygenase-1 protein levels[11] and inducible nitric oxide synthase protein levels were observed. Therefore, it seems much more consistent with the hypothesis, that in the liver, the impairment of biliverdin reductase-A is an important event which maybe affect oxidative/nitrosative stress levels through different mechanisms. Keeping in mind the role of biliverdin reductase-A in the regulation of inducible nitric oxide synthase transcription into the nucleus[26], the results obtained in the liver could be explained as the follows: despite it should expect an increase of 3-nitrotyrosine due to the decrease of biliverdin reductase-A antioxidant power, this did not happen because the reduction of inducible nitric oxide synthase levels (less nitric oxide production) (Figure 6).
In other words, like in a compensation mechanism, the lack of biliverdin reductase-A antioxidant power which should favor an increase of the oxidative/nitrosative stress levels, is balanced by the decrease of inducible nitric oxide synthase protein levels (Figure 6). Of course, based only on these results, we cannot exclude that other mechanisms participate in atorvastatin-mediated changes of inducible nitric oxide synthase in the brain and liver of aged beagles, and more mechanistic studies are required. Ad hoc designed experiments to address this point are ongoing in our laboratories.
In conclusion, this work supports a role for biliverdin reductase-A in the regulation of inducible nitric oxide synthase following atorvastatin treatment in aged beagles. Atorvastatin-induced neuroprotective effects in the brain appear to be mediated by biliverdin reductase-A which, although was associated with an increase of inducible nitric oxide synthase protein levels (Figure 3A–C), because of its antioxidant and antinitrosative features may serve to overcome nitric oxide-induced neurotoxic effects, thus supporting a potential therapeutic role of biliverdin reductase-A in Alzheimer's disease.
MATERIALS AND METHODS
Design
The study design of the atorvastatin treatment of aged beagles has been described previously[8,9,10,11] and is briefly summarized here.
Time and setting
This work was performed at the Department of Chemistry, Center of Membrane Sciences, and Department of Molecular and Biomedical Pharmacology, Sanders-Brown Center on Aging, University of Kentucky, Lexington, Kentucky, USA between 2010 and 2011.
Materials
Beagles were obtained from the Lovelace Respiratory Research Institute and Harlan (Indianapolis, IN, USA). For the age-dependence analysis of biliverdin reductase-A in the parietal cortex, 5 beagles ranging from 4.49 to 14 years were used. For the atorvastatin study, eight beagles ranging in age from 8.9–13.2 years were used. Based on our previous work, dogs of this age show cognitive decline and significant amounts of brain amyloid-β-peptide[70]. All animals had documented dates of birth, comprehensive medical histories and a veterinary examination ensuring that the animal was in good health prior to the start of the study. At the end of the study, all the animals had received treatment for 14.5 months and they ranged in age from 10.1–14.6 years. All research was conducted in accordance with approved Institutional Animal Care and Use Committee protocols following National Institutes of Health Guidelines. Animals were ranked by cognitive test scores and placed into equivalent groups. These groups were randomly designated as either the placebo-treated control group or the atorvastatin-treated group.
Methods
Drug treatment
Atorvastatin calcium (Lipitor®, 40 mg/tablet) and placebo tablets were kindly provided by Pfizer Inc (New York, NY, USA). Atorvastatin-treated animals received 2 × 40 mg tablets per day for a daily dose of 80 mg/day and control animals received 2 placebo tablets per day. Atorvastatin was chosen for this study because long term studies using an 80 mg/day dose in dogs did not report adverse events such as cataracts[71]. As previously demonstrated, in beagles treated with 6 mg/kg atorvastatin (approximately 90 mg/dog), plasma concentrations of atorvastatin were approximately 500 ng/mL[72]. This plasma concentration is in the same order of magnitude of those achieved in hypercholesterolemic people treated with 80 mg atorvastatin/day (187–252 ng/mL)[73,74]. This differs from rodent studies that have reported reduced brain amyloid-β-peptide in response to statin treatment. In these studies, doses are typically between 200- and 400-time higher than those used in humans[75], which leads to some concern regarding the translation of these outcomes to Alzheimer's disease clinical trials.
Tissue collection
Brain tissues (parietal cortex and cerebellum) and liver samples were collected as previously described[9,76].
Sample preparation
Brain (the parietal cortex and cerebellum) and liver samples from control and atorvastatin-treated dogs were thawed and placed in Media I lysis buffer (pH 7.4) containing 0.32 mol/L sucrose, 0.10 mmol/L Tris-HCl (pH = 8.8), 0.10 mmol/L MgCl2, 0.08 mmol/L EDTA, proteinase inhibitors leupeptin (0.5 mg/mL), pepstatin (0.7 μg/mL), aprotinin (0.5 mg/mL) and PMSF (40 μg/mL) and phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Samples were homogenized by 20 passes of a Wheaton tissue homogenizer and the resulting homogenate was centrifuged at 14 000 × g for 10 minutes to remove cellular debris. Total protein concentration of the supernatant was determined by BCA method (Pierce, Rockford, IL, USA).
Western blot analysis
Western blot analysis was performed as previously described[47]. The following primary antibodies were used: polyclonal anti-rabbit biliverdin reductase-A (Sigma-Aldrich, dilution 1:1000); polyclonal anti-rabbit inducible nitric oxide synthase (Lifespan Biosciences, Seattle, WA, USA, dilution 1:500); and polyclonal anti-rabbit β-actin (Sigma-Aldrich; dilution 1:2 000).
Statistical analysis
Data are expressed as mean ± SD of four individual samples per group. All statistical analyses were performed using a two-tailed Student's t-test. P < 0.05 was considered significantly different from control. Pearson or Spearman correlations were calculated to test the linear associations between the outcome measures in this study. The values of biliverdin reductase-A, heme oxygenase-1 and 3-nitrotyrosine to which we refer for the correlation analysis were obtained in previous works from our laboratory on the same animals used for this study[9,10,11].
Acknowledgments
This work was carried out at the University of Kentucky, Lexington, KY, USA. The authors are very grateful to Prof. D. Allan Butterfield (Department of Chemistry, Center of Membrane Sciences, Sanders-Brown Center on Aging) and Prof. Elizabeth Head (Sanders Brown Center on Aging, Department of Molecular and Biomedical Pharmacology) who provided samples, supplies, and scientific support for the success of this project.
Footnotes
Funding: Funding for the canine atorvastatin study was through the Alzheimer's Association IIRG-03-5673 to Elizabeth Head.
Conflicts of interest: None declared.
Ethical approval: The entire research was conducted in accordance with approved IACUC protocols following NIH Guidelines.
(Reviewed by Ropireddy D, Guo QX)
(Edited by Liu WJ, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362(4):329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- [2].Braak H, Braak E. Neuropathological stageing of Alzheimerrelated changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- [3].Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33(6):403–408. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- [4].Kandiah N, Feldman HH. Therapeutic potential of statins in Alzheimer's disease. J Neurol Sci. 2009;283(1-2):230–234. doi: 10.1016/j.jns.2009.02.352. [DOI] [PubMed] [Google Scholar]
- [5].Jick H, Zornberg GL, Jick SS, et al. Statins and the risk of dementia. Lancet. 2000;356(9242):1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- [6].Butterfield DA, Barone E, Mancuso C. Cholesterol-independent neuroprotective and neurotoxic activities of statins: Perspectives for statin use in Alzheimer disease and other age-related neurodegenerative disorders. Pharmacol Res. 2011;64(3):180–186. doi: 10.1016/j.phrs.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kurata T, Miyazaki K, Kozuki M, et al. Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res. 2011;1371:161–170. doi: 10.1016/j.brainres.2010.11.067. [DOI] [PubMed] [Google Scholar]
- [8].Murphy MP, Morales J, Beckett TL, et al. Changes in cognition and amyloid-beta processing with long term cholesterol reduction using atorvastatin in aged dogs. J Alzheimers Dis. 2010;22(1):135–150. doi: 10.3233/JAD-2010-100639. [DOI] [PubMed] [Google Scholar]
- [9].Barone E, Cenini G, Di Domenico F, et al. Long-term high-dose atorvastatin decreases brain oxidative and nitrosative stress in a preclinical model of Alzheimer disease: a novel mechanism of action. Pharmacol Res. 2011;63(3):172–180. doi: 10.1016/j.phrs.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barone E, Mancuso C, Di Domenico F, et al. Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J Neurochem. 2012;120(1):135–146. doi: 10.1111/j.1471-4159.2011.07538.x. [DOI] [PubMed] [Google Scholar]
- [11].Butterfield DA, Barone E, Di Domenico F, et al. Atorvastatin treatment in a dog preclinical model of Alzheimer's disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int J Neuropsychopharmacol. 2012;15(7):981–987. doi: 10.1017/S1461145711001118. [DOI] [PubMed] [Google Scholar]
- [12].Chen J, Zhang ZG, Li Y, et al. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53(6):743–751. doi: 10.1002/ana.10555. [DOI] [PubMed] [Google Scholar]
- [13].Lu D, Qu C, Goussev A, et al. Statins increase neurogenesis in the dentate gyrus, reduce delayed neuronal death in the hippocampal CA3 region, and improve spatial learning in rat after traumatic brain injury. J Neurotrauma. 2007;24(7):1132–1146. doi: 10.1089/neu.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McGuinness B, O’Hare J, Craig D, et al. Statins for the treatment of dementia. Cochrane Database Syst Rev. 2010;8:CD007514. doi: 10.1002/14651858.CD007514.pub2. [DOI] [PubMed] [Google Scholar]
- [15].Cotman CW, Head E. The canine (dog) model of human aging and disease: dietary, environmental and immunotherapy approaches. J Alzheimers Dis. 2008;15(4):685–707. doi: 10.3233/jad-2008-15413. [DOI] [PubMed] [Google Scholar]
- [16].Kapitulnik J, Maines MD. Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol Sci. 2009;30(3):129–137. doi: 10.1016/j.tips.2008.12.003. [DOI] [PubMed] [Google Scholar]
- [17].Lerner-Marmarosh N, Miralem T, Gibbs PE, et al. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc Natl Acad Sci U S A. 2008;105(19):6870–6875. doi: 10.1073/pnas.0800750105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tudor C, Lerner-Marmarosh N, Engelborghs Y, et al. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem J. 2008;413(3):405–416. doi: 10.1042/BJ20080018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maines MD. Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid Redox Signal. 2007;9(12):2187–2195. doi: 10.1089/ars.2007.1805. [DOI] [PubMed] [Google Scholar]
- [20].Sacktor TC. How does PKMzeta maintain long-term memory? Nat Rev Neurosci. 2011;12(1):9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- [21].Stocker R, Glazer AN, Ames BN. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci U S A. 1987;84(16):5918–5922. doi: 10.1073/pnas.84.16.5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Stocker R, Yamamoto Y, McDonagh AF, et al. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235(4792):1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- [23].Dore S, Takahashi M, Ferris CD, et al. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A. 1999;96(5):2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Barone E, Trombino S, Cassano R, et al. Characterization of the S-denitrosylating activity of bilirubin. J Cell Mol Med. 2009;13(8B):2365–2375. doi: 10.1111/j.1582-4934.2008.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ewing JF, Maines MD. Immunohistochemical localization of biliverdin reductase in rat brain: age related expression of protein and transcript. Brain Res. 1995;672(1-2):29–41. doi: 10.1016/0006-8993(94)01290-x. [DOI] [PubMed] [Google Scholar]
- [26].Gibbs PE, Miralem T, Lerner-Marmarosh N, et al. Formation of ternary complex of human biliverdin reductase-protein kinase Cdelta-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-kappaB, and inducible nitric-oxidase synthase (iNOS) J Biol Chem. 2012;287(2):1066–1079. doi: 10.1074/jbc.M111.279612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gibbs PE, Maines MD. Biliverdin inhibits activation of NF-kappaB: reversal of inhibition by human biliverdin reductase. Int J Cancer. 2007;121(11):2567–2574. doi: 10.1002/ijc.22978. [DOI] [PubMed] [Google Scholar]
- [28].Kravets A, Hu Z, Miralem T, et al. Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J Biol Chem. 2004;279(19):19916–19923. doi: 10.1074/jbc.M314251200. [DOI] [PubMed] [Google Scholar]
- [29].Ahmad Z, Salim M, Maines MD. Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J Biol Chem. 2002;277(11):9226–9232. doi: 10.1074/jbc.M108239200. [DOI] [PubMed] [Google Scholar]
- [30].Maines MD, Ewing JF, Huang TJ, et al. Nuclear localization of biliverdin reductase in the rat kidney: response to nephrotoxins that induce heme oxygenase-1. J Pharmacol Exp Ther. 2001;296(3):1091–1097. [PubMed] [Google Scholar]
- [31].Butterfield DA, Stadtman ER. Paula S.T, Bittar E.E, editors. Protein Oxidation Processes in Aging Brain, in Advances in Cell Aging and Gerontology. 1997:161–191. [Google Scholar]
- [32].Butterfield DA, Galvan V, Lange MB, et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010;48(1):136–144. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Butterfield DA. beta-Amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer's disease. Chem Res Toxicol. 1997;10(5):495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- [34].Butterfield DA, Poon HF, St Clair D, et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22(2):223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- [35].Butterfield DA, Reed T, Newman SF, et al. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007;43(5):658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Butterfield DA, Reed T, Perluigi M, et al. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006;397(3):170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- [37].Hensley K, Hall N, Subramaniam R, et al. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65(5):2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- [38].Lyras L, Cairns NJ, Jenner A, et al. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J Neurochem. 1997;68(5):2061–2069. doi: 10.1046/j.1471-4159.1997.68052061.x. [DOI] [PubMed] [Google Scholar]
- [39].Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23(1):134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- [40].Varadarajan S, Yatin S, Aksenova M, et al. Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130(2-3):184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- [41].Poon HF, Calabrese V, Scapagnini G, et al. Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontol A Biol Sci Med Sci. 2004;59(5):478–493. doi: 10.1093/gerona/59.5.m478. [DOI] [PubMed] [Google Scholar]
- [42].Calabrese V, Mancuso C, Calvani M, et al. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8(10):766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- [43].Mancuso C, Scapagini G, Curro D, et al. Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front Biosci. 2007;12:1107–1123. doi: 10.2741/2130. [DOI] [PubMed] [Google Scholar]
- [44].Calabrese V, Sultana R, Scapagnini G, et al. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer's disease. Antioxid Redox Signal. 2006;8(11-12):1975–1986. doi: 10.1089/ars.2006.8.1975. [DOI] [PubMed] [Google Scholar]
- [45].Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid be-ta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32(11):1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- [46].Smith MA, Perry G, Pryor WA. Causes and consequences of oxidative stress in Alzheimer's disease. Free Radic Biol Med. 2002;32(11):1049. doi: 10.1016/s0891-5849(02)00793-1. [DOI] [PubMed] [Google Scholar]
- [47].Barone E, Di Domenico F, Cenini G, et al. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim Biophys Acta. 2011;1812(4):480–487. doi: 10.1016/j.bbadis.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Di Domenico F, Barone E, Mancuso C, et al. HO-1/BVR-a system analysis in plasma from probable Alzheimer's disease and mild cognitive impairment subjects: a potential biochemical marker for the prediction of the disease. J Alzheimers Dis. 2012;32(2):277–289. doi: 10.3233/JAD-2012-121045. [DOI] [PubMed] [Google Scholar]
- [49].Barone E, Di Domenico F, Sultana R, et al. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med. 2012;52(11-12):2292–2301. doi: 10.1016/j.freeradbiomed.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barone E, Di Domenico F, Cenini G, et al. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer's disease and amnestic mild cognitive impairment. J Alzheimers Dis. 2011;25(4):623–633. doi: 10.3233/JAD-2011-110092. [DOI] [PubMed] [Google Scholar]
- [51].Owen JB, Sultana R, Aluise CD, et al. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Abeta accumulation in AD brain. Free Radic Biol Med. 2010;49(11):1798–1803. doi: 10.1016/j.freeradbiomed.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Subramaniam R, Roediger F, Jordan B, et al. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997;69(3):1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- [53].Butterfield DA, Reed T, Sultana R. Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer's disease. Free Radic Res. 2011;45(1):59–72. doi: 10.3109/10715762.2010.520014. [DOI] [PubMed] [Google Scholar]
- [54].Lauderback CM, Hackett JM, Huang FF, et al. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem. 2001;78(2):413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- [55].Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8(2):211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- [56].Shibuki K, Okada D. Endogenous nitric oxide release required for long-term synaptic depression in the cerebellum. Nature. 1991;349(6307):326–328. doi: 10.1038/349326a0. [DOI] [PubMed] [Google Scholar]
- [57].Siciliano R, Barone E, Calabrese V, et al. Experimental research on nitric oxide and the therapy of Alzheimer disease: a challenging bridge. CNS Neurol Disord Drug Targets. 2011;10(7):766–776. doi: 10.2174/187152711798072356. [DOI] [PubMed] [Google Scholar]
- [58].Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Radi R, Beckman JS, Bush KM, et al. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266(7):4244–4250. [PubMed] [Google Scholar]
- [60].Barone E, Di Domenico F, Cenini G, et al. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim Biophys Acta. 2011;1812(4):480–487. doi: 10.1016/j.bbadis.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sultana R, Perluigi M, Butterfield DA. Oxidatively modified proteins in Alzheimer's disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol. 2009;118(1):131–150. doi: 10.1007/s00401-009-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sultana R, Butterfield DA. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. J Bioenerg Biomembr. 2009;41(5):441–446. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sultana R, Perluigi M, Newman SF, et al. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer's disease. Antioxid Redox Signal. 2010;12(3):327–336. doi: 10.1089/ars.2009.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Reed T, Perluigi M, Sultana R, et al. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol Dis. 2008;30(1):107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- [65].Butterfield DA, Reed TT, Perluigi M, et al. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer's disease. Brain Res. 2007;1148:243–248. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Butterfield DA, Abdul HM, Opii W, et al. Pin1 in Alzheimer's disease. J Neurochem. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- [67].Sultana R, Poon HF, Cai J, et al. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22(1):76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- [68].Castegna A, Thongboonkerd V, Klein JB, et al. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85(6):1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- [69].Cenini G, Sultana R, Memo M, et al. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008;45(1):81–85. doi: 10.1016/j.freeradbiomed.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Head E, McCleary R, Hahn FF, et al. Region-specific age at onset of beta-amyloid in dogs. Neurobiol Aging. 2000;21(1):89–96. doi: 10.1016/s0197-4580(00)00093-2. [DOI] [PubMed] [Google Scholar]
- [71].Walsh KM, Albassam MA, Clarke DE. Subchronic toxicity of atorvastatin, a hydroxymethylglutaryl-coenzyme A reductase inhibitor, in beagle dogs. Toxicol Pathol. 1996;24(4):468–476. doi: 10.1177/019262339602400409. [DOI] [PubMed] [Google Scholar]
- [72].Shen HR, Li ZD, Zhong MK. HPLC assay and pharmacokinetic study of atorvastatin in beagle dogs after oral administration of atorvastatin self-microemulsifying drug delivery system. Pharmazie. 2006;61(1):18–20. [PubMed] [Google Scholar]
- [73].Cilla DD, Jr, Whitfield LR, Gibson DM, et al. Multiple-dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG-CoA reductase, in healthy subjects. doi: 10.1016/S0009-9236(96)90218-0. [DOI] [PubMed] [Google Scholar]
- [74].Stern RH, Yang BB, Hounslow NJ, et al. Pharmacodynamics and pharmacokinetic- pharmacodynamic relationships of atorvastatin, an HMG-CoA reductase inhibitor. J Clin Pharmacol. 2000;40(6):616–623. [PubMed] [Google Scholar]
- [75].Petanceska SS, DeRosa S, Olm V, et al. Statin therapy for Alzheimer's disease: will it work? J Mol Neurosci. 2002;19(1-2):155–161. doi: 10.1007/s12031-002-0026-2. [DOI] [PubMed] [Google Scholar]
- [76].Murphy MP, Morales J, Beckett TL, et al. Changes in cognition and amyloid-β processing with long term cholesterol reduction using atorvastatin in aged dogs. J Alzheimers Dis. 2010;22(1):135–150. doi: 10.3233/JAD-2010-100639. [DOI] [PubMed] [Google Scholar]