

Abstract
X-linked methyl-CpG binding protein 2 mutations can induce symptoms similar to those of Parkinson's disease and dopamine metabolism disorders, but the specific role of X-linked methyl-CpG binding protein 2 in the pathogenesis of Parkinson's disease remains unknown. In the present study, we used 6-hydroxydopamine-induced human neuroblastoma cell (SH-SY5Y cells) injury as a cell model of Parkinson's disease. The 6-hydroxydopamine (50 μmol/L) treatment decreased protein levels for both X-linked methyl-CpG binding protein 2 and tyrosine hydroxylase in these cells, and led to cell death. However, overexpression of X-linked methyl-CpG binding protein 2 was able to ameliorate the effects of 6-hydroxydopamine, it reduced 6-hydroxydopamine-induced apoptosis, and increased the levels of tyrosine hydroxylase in SH-SY5Y cells. These findings suggesting that X-linked methyl-CpG binding protein 2 may be a potential therapeutic target for the treatment of Parkinson's disease.
Keywords: neural regeneration, neurodegenerative diseases, Parkinson's disease, methyl-CpG-binding protein 2, tyrosine hydroxylase, 6-hydroxydopamine, dopaminergic neurons, SH-SY5Y cells, grants-supported paper, neuroregeneration
Research Highlights
-
(1)
X-linked methyl-CpG binding protein 2 and tyrosine hydroxylase expression is reduced in 6-hydroxydopamine-treated cells.
-
(2)
Overexpression of X-linked methyl-CpG binding protein 2 in SH-SY5Y cells reduced cell apoptosis induced by 6-hydroxydopamine and increased tyrosine hydroxylase expression.
-
(3)
X-linked methyl-CpG binding protein 2 inhibited 6-hydroxydopamine-induced apoptosis by increasing tyrosine hydroxylase expression in SH-SY5Y cells.
-
(4)
X-linked methyl-CpG binding protein 2 may be a potential therapeutic target for Parkinson's disease.
INTRODUCTION
Parkinson's disease is the second most common neurodegenerative disorder, affecting nearly 2% of the population over the age of 60[1]. Parkinson's disease is characterized by the pronounced degeneration of dopaminergic neurons in the substantia nigra pars compacta, resulting in a reduction of striatal dopamine levels. Following the loss of approximately 50% of dopamine neurons and 75–80% of striatal dopamine levels, patients begin to exhibit classical symptoms of Parkinson's disease[2]. Although there are several palliative treatments that can be effective over a number of years, their usefulness wanes over time and their use can be accompanied by significant side effects[3]. Thus, the development of novel therapies for this disorder is crucial[4].
Several novel therapeutic agents have been shown to play a role in the prevention of age-related dementias and Parkinson's disease, such as nicotine[5], anti-inflammatory molecules[6], and iron chelators[7]. According to a recent review[8], among the numerous potential genes that have been evaluated as potential therapeutic targets for Parkinson's disease, those encoding tyrosine hydroxylase, guanosine triphosphate cyclohydrolase I, and aromatic L-amino acid decarboxylase all allow for an increase in the production of dopamine[9,10]. However, currently available therapies can neither arrest nor reverse the progression of the disease either in the clinic or in experimental models.
The X-linked methyl-CpG binding protein 2 (MeCP2) gene, located on the long arm of the X chromosome (Xq28)[11], encodes a nuclear protein, MeCP2, that binds specifically to methylated DNA and functions as a general transcriptional repressor by associating with chromatin-remodeling complexes. MeCP2 is expressed at high levels in the mammalian brain[12]. Mutations in MeCP2 result in Rett syndrome (RTT)[13], and girls with RTT develop normally until 6–18 months of age. Parkinsonian symptoms are often observed in older RTT patients[14,15]. Additionally, in both MeCP2 mutant mice and RTT patients, levels of biogenic amines are decreased, indicating a disturbance in dopaminergic transmission[16,17,18]. The conditional loss of MeCP2 from tyrosine hydroxylase-expressing catecholaminergic neurons has been associated with a reduction in locomotion in mice[17]. This is interesting because, in the dopamine synthetic pathway, tyrosine is first hydroxylated and converted to levodopa by tyrosine hydroxylase. Thus, tyrosine hydroxylase is the rate-limiting enzyme in catecholamine synthesis and has been extensively studied in several human pathologies including Parkinson's disease, hypertension and depression[19,20,21].
In immature neurons, the MeCP2 expression level is low but increases during neuronal maturation and reaches its highest level in postmitotic neurons[22,23]. This developmental profile of MeCP2 expression suggests that MeCP2 is involved in neuronal maturation and dendritic arborization[24]. Interestingly, the high level of MeCP2 expression in postmitotic neurons continues throughout adulthood, suggesting that MeCP2 also plays an important role in mature neurons. In olfactory neurons, the MeCP2 expression level also appears to increase in relation to neuronal maturation, during the period between neurogenesis and synaptogenesis[25]. In humans, neuronal maturation and synaptogenesis are key developmental processes that occur as early as embryonic weeks 12 and 20, respectively[26]. The loss of MeCP2 expression within this time window may be responsible for the observed decrease in neuronal and overall brain size in referral to treatment patients[27]. Disruptions in MeCP2 function might therefore interfere with neuronal maturation and synaptogenesis, culminating in abnormal development of the central nervous system.
MeCP2 plays an important role in neuronal development, proliferation, mature synapse regeneration and the regulation of apoptosis[28]. One study found that mice experiencing MeCP2 gene knockout at an early age showed similar symptoms to those in Parkinson's disease, and confirmed that MeCP2 could regulate the metabolism of catecholamines and the expression of tyrosine hydroxylase in dopamine neurons[29]. However, the specific role of MeCP2 in dopaminergic signaling and the pathogenesis of Parkinson's disease remain unclear.
To characterize the relationship between MeCP2 and tyrosine hydroxylase, as well as the role of this interaction in the etiology of Parkinson's disease, we examined the expression of MeCP2 and tyrosine hydroxylase in 6-hydroxydopamine-treated SH-SY5Y neuroblastoma cells, a cellular model of Parkinson's disease. In addition, we analyzed the effect of MeCP2 expression on tyr osine hydroxylase expression in this cellular model of Parkinson's disease[30].
RESULTS
6-Hydroxydopamine reduced the viability of SH-SY5Y cells
To determine the optimal dose for this study, different concentrations of 6-hydroxydopamine (25, 50, 75, and 100 μmol/L) were applied to SH-SY5Y cells and 6-hydroxydopamine-induced cell death was evaluated using the Cell Counting Kit-8 (CCK-8) assay. The CCK-8 assay showed that 25–100 μmol/L 6-hydroxydopamine significantly reduced SH-SY5Y cell viability, and the higher the concentration of 6-hydroxydopamine, the stronger the effect. A concentration of 50 μmol/L 6-hydroxydopamine, which decreased cell viability to 69.4 ± 3.5% and 54.1 ± 3.3% at 12 and 24 hours, respectively (Figure 1), was chosen for subsequent experiments.
Figure 1.
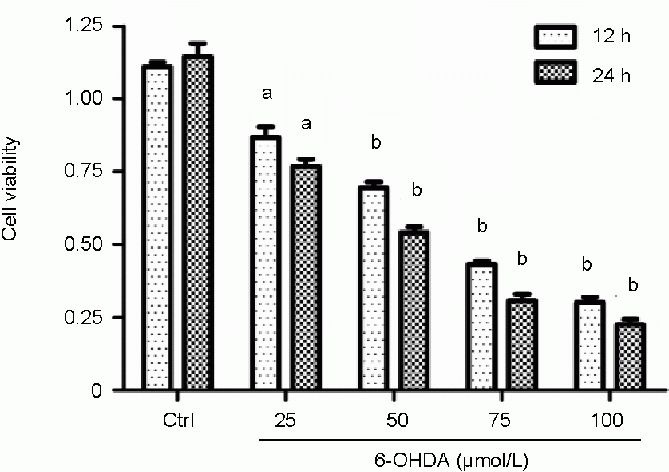
Effect of 6-hydroxydopamine (6-OHDA) on the viability of SH-SY5Y cells.
Different doses of 6-OHDA (25, 50, 75, and 100 μmol/L) were applied to SH-SY5Y cells and cell viability was assessed using the Cell Counting Kit-8 assay at 12 and 24 hours. Absorbance at 540 nm was monitored and the results were normalized to those of control cells incubated with vehicle alone. Data are expressed as mean ± SD of three independent experiments. aP < 0.05, bP < 0.01, vs. control group (Ctrl) at the same time point using one-way analysis of variance followed by the Student Newman-Keuls post hoc test. h: Hours.
6-Hydroxydopamine decreased the expression of MeCP2 and tyrosine hydroxylase in SH-SY5Y cells
To determine whether MeCP2 is involved in the pathogenesis of 6-hydroxydopamine-induced death in SH-SY5Y cells, we first measured the levels of MeCP2 and tyrosine hydroxylase proteins in SH-SY5Y cells treated with 50 μmol/L 6-hydroxydopamine for 3, 6, 12, and 24 hours using immunocytofluorescence staining. We observed a marked down-regulation of MeCP2 and tyrosine hydroxylase proteins from 6 to 24 hours after treatment with 50 μmol/L 6-hydroxydopamine (Figure 2). In addition, we assessed the expression of MeCP2 and tyrosine hydroxylase in parallel cultures using western blot analysis. Consistent with the results of our immunocytofluorescence staining, MeCP2 and tyrosine hydroxylase protein levels began to decrease as early as 3 hours following 6-hydroxydopamine treatment and continued to decrease until the last time point, at 24 hours (P < 0.05 or P < 0.01; Figure 3). These findings show, for the first time, that MeCP2 levels are decreased in the 6-hydroxy dopamine-treated SH-SY5Y cell model of Parkinson's disease.
Figure 2.
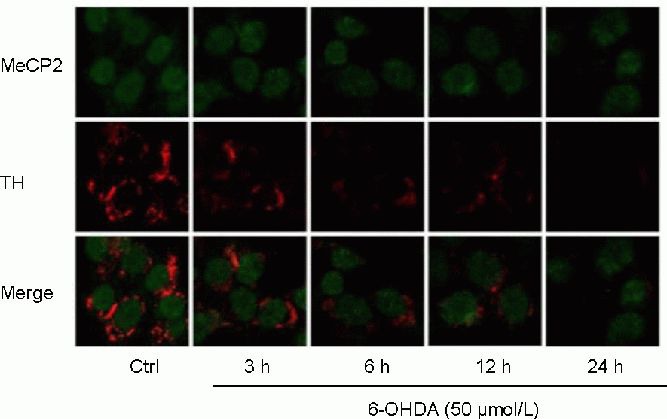
Effect of 6-hydroxydopamine (6-OHDA) on the expression of X-linked methyl-CpG binding protein 2 (MeCP2) and tyrosine hydroxylase (TH) in SH-SY5Y cells (immunocytofluorescence staining, × 1 000).
SH-SY5Y cells treated with 50 μmol/L 6-OHDA for 3, 6, 12, and 24 hours were visualized by confocal microscopy. Green and red fluorescence represent MeCP2 and TH, respectively. The longer SH-SY5Y cells were treated with 50 μmol/L 6-OHDA, the weaker the green and red fluorescence became. Ctrl: Control group.
Figure 3.
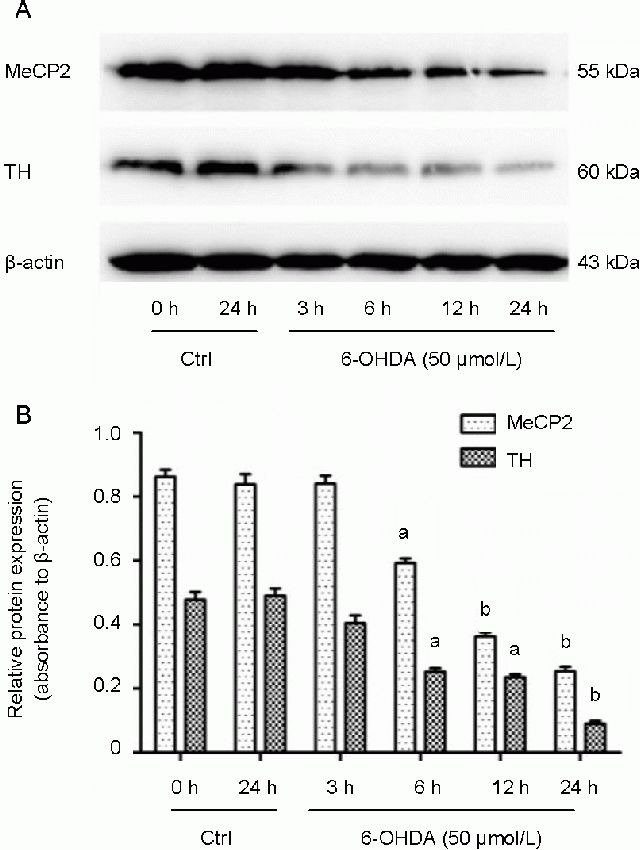
X-linked methyl-CpG binding protein 2 (MeCP2) and tyrosine hydroxylase (TH) protein levels in 6-hydroxydopamine (6-OHDA)-treated SH-SY5Y cells.
SH-SY5Y cells were treated with 50 μmol/L 6-OHDA for 3, 6, 12, and 24 hours and protein levels were assessed by western blot.
(A) Representative western blot of MeCP2 and TH proteins.
(B) Quantitative analysis of western blots. The quantity of target proteins was normalized to β-actin. Data are expressed as mean ± SD of three independent experiments. aP < 0.05, bP < 0.01, vs. 0 hours in the control group (Ctrl) using one-way analysis of variance followed by the Student Newman-Keuls post hoc test. h: Hours.
Identification of recombinant pEGFP-N1-MeCP2 vector and MeCP2 expression
To further elucidate the possible role of MeCP2 in the regulation of tyrosine hydroxylase expression, pEGFP-N1-MeCP2 was constructed. The plasmid pEGFP-N1-MeCP2 was identified by digestion with Pst I and Xho I, and subsequent sequencing. As shown in Figure 4A, the size of the fragment was consistent with the length of the MeCP2 gene (1 531 bp). When pEGFP-N1-MeCP2 and pEGFP-N1 were separately transfected into SH-SY5Y cells, O-MeCP2-SH-SY5Y and EGFP-SH-SY5Y cells were processed for western blot using an anti-EGFP antibody. The EGFP-MeCP2 fusion protein was evident as an immunoreactive band with a relative molecular weight of 82 kDa in O-MeCP2-SH-SY5Y cells, and was not evident in control EGFP-SH-SY5Y cells. However, a band with a molecular weight of 27 kDa was seen in extracts from EGFP-SH-SY5Y cells (Figure 4B).
Figure 4.
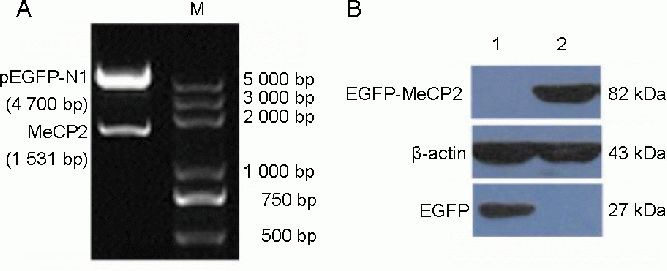
Identification and expression of plasmid pEGFP-N1-MeCP2.
(A) The pEGFP-N1-MeCP2 plasmid was identified by digestion with Pst I and Xho I. M: Marker.
(B) The EGFP-MeCP2 fusion protein was detected by western blot using anti-EGFP antibody. 1: EGFP-SH-SY5Y cells (SH-SY5Y cells transfected with pEGFP-N1); 2: O-MeCP2-SH-SY5Y cells (SH-SY5Y cells transfected with pEGFP-N1-MeCP2). MeCP2: X-linked methyl-CpG binding protein 2.
MeCP2 protected against 6-hydroxydopamine-induced neurotoxicity
We then examined the effects of MeCP2 overexpression on the viability of 6-hydroxydopamine-treated SH-SY5Y cells. Using the CKK-8 assay, we found that the upregulation of MeCP2 in SH-SY5Y cells increased cell viability following 6-hydroxydopamine treatment to levels comparable to those in the untreated control (Figure 5A). It has been reported that 6-hydroxydopamine-induced cell death involves apoptotic features such as DNA fragmentation and phosphatidylserine exposure[31]. To assess the impact of MeCP2 overexpression upon 6-hydroxydopamine-induced apoptosis in SH-SY5Y cells, we observed that 52.6 ± 3.2% of control cells underwent apoptosis following exposure to 50 μmol/L 6-hydroxydopamine for 24 hours. The overexpression of MeCP2 resulted in a marked reduction of 6-hydroxydopamine-induced death in these cells (P < 0.01; Figure 5B, C).
Figure 5.
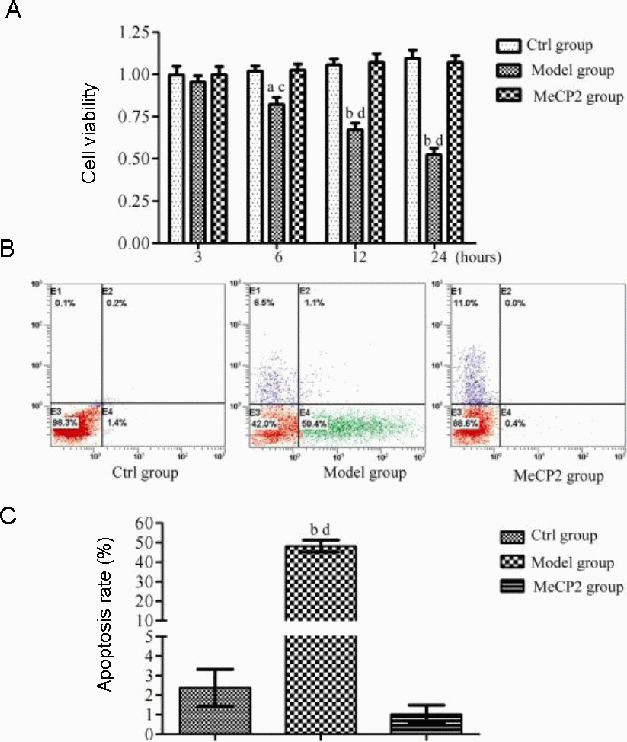
Effect of X-linked methyl-CpG binding protein 2 (MeCP2) on the viability and apoptosis of 6-hydroxydopamine-treated SH-SY5Y cells.
Control group (Ctrl): Untreated cells; model group: cells incubated with 6-hydroxydopamine (50 μmol/L) for 24 hours; MeCP2 group: cells transfected with pEGFP-N1-MeCP2 for 24 hours, followed by treatment with 6-hydroxydopamine (50 μmol/L) for 24 hours.
(A) Cell viability was quantified using the Cell Counting Kit-8 assay. Absorbance at 540 nm was monitored and the results were normalized to those of control cells.
(B) Flow cytometry analysis of apoptosis.
(C) Quantitative analysis of cell apoptosis.
Data are expressed as mean ± SD of three independent experiments. aP < 0.05, bP < 0.01, vs. control group; cP < 0.05, dP < 0.01, vs. MeCP2 group using one-way analysis of variance followed by the Student Newman-Keuls post hoc test.
MeCP2 increased tyrosine hydroxylase expression in 6-hydroxydopamine-treated SH-SY5Y cells
To examine whether MeCP2 overexpression has an impact on tyrosine hydroxylase expression, we used immunocytofluorescence to examine the protein levels of tyrosine hydroxylase in SH-SY5Y cells. As shown in Figure 2, there was a marked decrease in tyrosine hydroxylase immunoreactivity in SH-SY5Y cells treated with 6-hydroxydopamine compared with controls. However, in cells transfected with pEGFP-N1-MeCP2, the expression of tyrosine hydroxylase following 6-hydroxydopamine treatment was at least partially rescued (Figure 6). This finding was confirmed by western blot of extracts from 6-hydroxydopamine-treated cells with and without MeCP2 overexpression (Figure 7).
Figure 6.
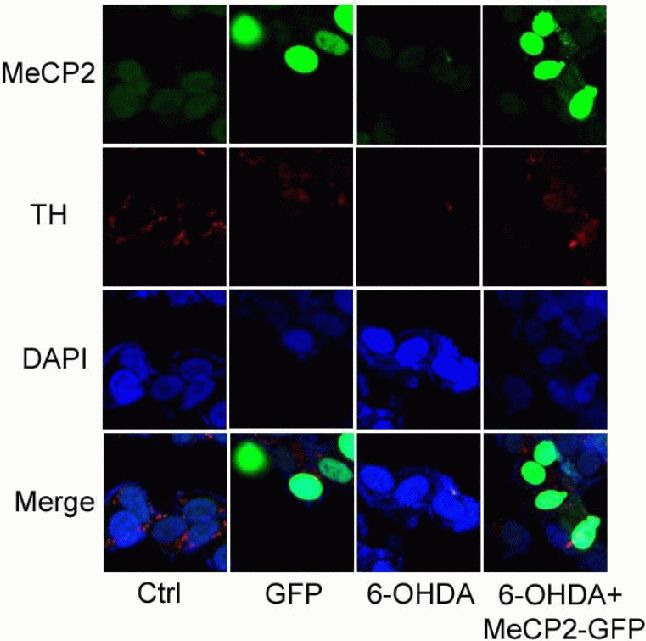
Effects of upregulated X-linked methyl-CpG binding protein 2 (MeCP2) on 6-hydroxydopamine (6-OHDA)-treated SH-SY5Y cells (immunocytofluorescence staining, × 1 000).
Control (Ctrl): Untreated cells; GFP: cells transfected with pEGFP-N1 alone; 6-OHDA: cells incubated with 6-OHDA (50 μmol/L) for 24 hours; 6-OHDA + MeCP2-GFP: cells transfected with pEGFP-N1-MeCP2, followed by 6-OHDA (50 μmol/L) for 24 hours. Blue, green, and red fluorescence represent DAPI, MeCP2, and tyrosine hydroxylase (TH), respectively. After SH-SY5Y cells were incubated with 6-OHDA (50 μmol/L) for 24 hours, the red fluorescence became weaker, but after SH-SY5Y cells were transfected with pEGFP-N1-MeCP2, followed by 6-OHDA treatment for 24 hours, the red fluorescence did not become weaker.
Figure 7.
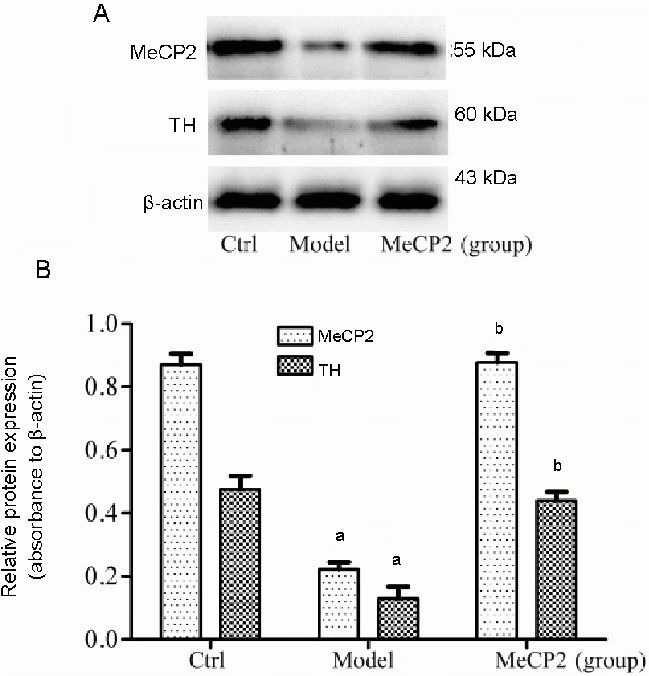
Expression of X-linked methyl-CpG binding protein 2 (MeCP2) and tyrosine hydroxylase (TH) proteins in SH-SY5Y cells.
Control group (Ctrl): Untreated cells; model group: cells incubated with 6-OHDA (50 μmol/L) for 24 hours; MeCP2 group: cells transfected with pEGFP-N1-MeCP2 for 24 hours, followed by 6-OHDA (50 μmol/L) treatment for 24 hours. MeCP2 and TH proteins were detected by western blot. The data are expressed as mean ± SD of three independent experiments. aP < 0.01, vs. Ctrl; bP < 0.01, vs. model group using one-way analysis of variance followed by the Student Newman-Keuls post hoc test.
DISCUSSION
Parkinson's disease is a chronic neurodegenerative disease, primarily occurring in middle-aged individuals, in which the main pathological feature is the death of substantia nigra dopaminergic neurons. The underlying causes of this cell death are not yet understood, and treatment for Parkinson's disease relies mainly on levodopa replacement therapy, a palliative treatment whose long-term use may render it ineffective in patients, or may lead to dopamine-induced dyskinesia.
Therefore, an increased understanding of the etiology of Parkinson's disease, as well as novel therapeutic approaches, is crucial. Despite many years of intense research, the exact mechanisms by which neuronal death occurs in Parkinson's disease remain unknown. Understanding the underlying cause of Parkinson's disease is necessary to direct research that will lead to the development of new and potent therapies. 6-hydroxydopamine causes parkinsonism in animals by decreasing dopamine levels and tyrosine hydroxylase activity, impairing dopamine uptake, and eliciting dopaminergic neuronal loss[32]. Also, 6-hydroxydopamine has been extensively used in vivo in mammalian species and in in vitro paradigms as experimental models of Parkinson's disease[33]. Given the parallels with Parkinson's disease, further understanding of the mechanisms by which 6-hydroxydopamine leads to dopaminergic neuronal death could provide insights into therapeutic targets for Parkinson's disease[33].
MeCP2 is a ubiquitous protein. Mutations in MeCP2 cause Rett syndrome, a severe neurodevelopmental disorder that mainly affects females. MeCP2 binds to methylated DNA and associates with various co-repressor complexes, thereby working as a methylation-dependent transcriptional repressor[15], although recent results indicate that it might also activate gene expression[34]. The MeCP2 protein is thought to function as a transcriptional repressor in methylated regions of DNA via two distinct domains, a methyl-CpG-binding domain[35], which binds to symmetrically methylated cytosines, and a transcriptional repression domain[36], which interacts with co-repressor proteins, including specific histone deacetylases and mSin3a[37]. New evidence suggests that MeCP2 may also bind to active genes[38,39]. MeCP2 has also been shown to interact with RNAs to influence alternative splicing[40]. Recent findings suggest that MeCP2 may mediate increases in the expression of some genes[34,39]. For example, in MeCP2-null mice, the loss of MeCP2 causes decreases in the mRNA and protein levels of tyrosine hydroxylase and tryptophan hydroxylase 2[41].
The present study is the first to examine the role of MeCP2 in a Parkinson's disease cell model. We assessed MeCP2 expression using both immunocytofluorescence and western blot. The results showed that both tyrosine hydroxylase and MeCP2 protein expression were significantly reduced following 6-hydroxydopamine treatment in SH-SY5Y cells. Specifically, the protein levels of both tyrosine hydroxylase and MeCP2 decreased significantly from 6 to 24 hours following 6-hydroxy-dopamine treatment. Subsequent CCK-8 assays and flow cytometry showed that 6-hydroxy dopamine treatment induced apoptosis in this cell line. Previous studies have shown that the promoter for the tyrosine hydroxylase gene contains an MeCP2-binding site[38]. In addition, in MeCP2-deficient mouse models of Rett syndrome, a deficit in tyrosine hydroxylase is observed in various peripheral catecholaminergic tissues[42] and the central nervous system, including the brainstem[43,44] and locus coeruleus[45]. In MeCP2-/y mice, the number of tyrosine hydroxylase-positive neurons was reduced in the nervous system, as was the amount of dopamine[43,44].
Thus, there is a clear relationship between MeCP2 and dopamine catalysis. In this study, we sought to test the hypothesis that the reduced expression of tyrosine hydroxylase following 6-hydroxydopamine treatment in SH-SY5Y cells contributes to the reduced expression of MeCP2 in SH-SY5Y cells[46].
To determine whether the decreased expression of MeCP2 in 6-hydroxydopamine-treated SH-SY5Y cells underlies the reduction in tyrosine hydroxylase expression, we overexpressed MeCP2 in these cells by means of transfection with MeCP2-expressing or control vectors, followed by exposure to 6-hydroxydopamine. Remarkably, both the increased expression of tyrosine hydroxylase and decreased cell viability following 6-hydroxy dopamine treatment were rescued by the overexpression of MeCP2 in SH-SY5Y cells. An MeCP2- binding site in the tyrosine hydroxylase promoter was recently described, opening the possibility that the tyrosine hydroxylase mRNA level could be regulated by MeCP2[38]. Since MeCP2 can function as both an activator and a repressor of transcription, it is only speculative to assign a clear role for MeCP2 in the regulation of the tyrosine hydroxylase promoter[36]. The level of brain-derived neurotrophic factor, which is clearly regulated by MeCP2, is also a key factor in the survival of tyrosine hydroxylase neurons located in the brainstem as well as in the chemoafferent pathway[47,48,49,50]. It was previously reported that the lack of MeCP2 disrupts transsynaptic brain-derived neurotrophic factor regulation in the adrenal medulla and the chemoafferent pathway, and this could participate in the tyrosine hydroxylase deficit[51,52]. In neuronal cultures, MeCP2 binds to brain-derived neurotrophic factor promoter III to repress transcription of brain-derived neurotrophic factor[47,53]. Brain-derived neurotrophic factor protein and mRNA levels are decreased by 70% in the brains of MeCP2 mutant mice compared with the levels in wild-type mice[54], and are significantly reduced in the brains of RTT patients[55]. Our studies show that the upregulation of MeCP2 expression greatly inhibited 6-hydroxydopamine-induced apoptosis in SH-SY5Y cells, and increased the protein expression levels of tyrosine hydroxylase. Thus, the upregulation of MeCP2 in the dopaminergic SH-SY5Y cell line is protective against 6-hydroxydopamine toxicity.
Consistent with previous reports[13], we found that, following 6-hydroxydopamine treatment, the levels of MeCP2 decreased significantly, leading to decreases in the levels of tyrosine hydroxylase protein in SH-SY5Y cells. The mechanism underlying this decrease in tyrosine hydroxylase may involve the occupation of MeCP2 on the tyrosine hydroxylase and Tph2 promoters[38,41]. These previous findings, combined with our expression data, point to a potential role for MeCP2 in the upregulation of genes involved in dopamine synthesis, a concept consistent with recent reports[38,41]. Also, pretreatment with MeCP2 upregulation under conditions in which SH-SY5Y cells showed complete neuroprotection could almost completely block 6-hydroxydopamine-induced apoptosis in SH-SY5Y cells. Our findings suggest a novel yet unknown role for MeCP2 in SH-SY5Y cells in the control of neuronal response to cell death. These results demonstrate that the decrease in cell death occurred in 6-hydroxydopamine-induced SH-SY5Y cells following upregulation of MeCP2 expression may have been due to inhibition of both caspase- and apoptosis inducing factor-dependent apoptotic mechanisms[56]. Recently, MeCP2 phosphorylation at Ser 80, Ser 229 and Ser 421 was shown to occur in the brain and modulate MeCP2 silencing activities[57,58]. Homeodomain-inte- racting protein kinase 2 was found to be a kinase that binds MeCP2 and phosphorylates it at Ser 80 in vitro and in vivo and modulates cell proliferation and apoptosis, and the neurological defects of Hipk2-null mice indicate its role in normal brain functions[59]. The role of MeCP2 in regulating cell growth and apoptosis may occur in 6-hydroxydopamine-induced SH-SY5Y cells, but further study is required to confirm this.
In conclusion, the MeCP2 level is reduced in 6-hydroxydopamine-induced dopaminergic cell death in vitro, and the upregulation of MeCP2 in SH-SY5Y cells restores tyrosine hydroxylase expression and is protective against apoptosis. Targeting MeCP2 activity may thereby be a viable therapeutic approach to rescue dopaminergic neurons in Parkinson's disease.
MATERIALS AND METHODS
Design
In vitro comparative observation of cytology.
Time and setting
The experiments were performed in the Medical Research Center of Zhongnan Hospital, Wuhan University, China from January 1st, 2011 to March 30th, 2012.
Materials
The human dopaminergic neuroblastoma cell line, SH-SY5Y, was purchased from America Type Culture Collection, Manassas, VA, USA.
Methods
Cell culture and drug treatments
SH-SY5Y cells were cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin. 6-hydroxydopamine (Sigma, St. Louis, MO, USA) was dissolved in 0.9% NaCl solution containing 0.01% ascorbic acid to create a stock concentration of 10 mmol/L and was used at a final concentration of 25, 50, 75, and 100 μmol/L. SH-SY5Y cells in the logarithmic growth phase at approximately 75% confluency were treated with 6-hydroxydopamine (25, 50, 75, and 100 μmol/L) for 24 hours.
Construction and transfection of pEGFP-N1-MeCP2 into SH-SY5Y cell
Construction of recombinant vector pEGFP-N1-MeCP2: Total cellular RNA was extracted from SH-SY5Y cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the protocol provided by the manufacturer. Total RNA (1 μg) was reverse-transcribed using M-MLV reverse transcriptase (Promega, Madison, WI, USA). Fragments of human MeCP2 cDNA (NM_001110792) were amplified by PCR using the following primers: Forward 5′-GCG CTC GAG GGT AAA AGC CGT CCG GAA AAT-3′; Reverse 5′-CGG CTG CAG GCT AAC TCT CTC GGT CAC GGG C-3′. Pst I and Xho I (Promega, Madison, WI, USA) restriction sites were included in the primers (underlined) to facilitate the ligation of cDNA products into pEGFP-N1 vector (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China). The initial PCR step was performed at 95°C for 5 minutes. PCR reactions were performed using a total of 35 cycles consisting of a 30-second melting phase at 95°C, followed by a 30-second annealing phase at 56°C, then a 30-second extension phase at 72°C. The product of PCR was confirmed by gel electrophoresis. The fragment was attached to pEGFP-N1 vector by T4 ligase (Fermentas Inc, Ontario, Canada). The resulting plasmid was verified by Sanger sequencing[60] and restriction digestion and was termed “pEGFP-N1-MeCP2”.
Transfection of recombinant vector pEGFP-N1-MeCP2 into SH-SY5Y cells: SH-SY5Y cells were plated into six-well plates at 4 × 105 cells/mL, grown overnight to approximately 60% confluency and transfected with pEGFP-N1 or pEGFP-N1-MeCP2. Briefly, 10 μL of Lipofectamine 2000 (Invitrogen) was mixed with 250 μL of opti-MEM (Gibco) at room temperature for 5 minutes to prepare the Lipofectamine 2000 mixture. The DNA mixture was prepared by adding 4 μg of pEGFP-N1-MeCP2 or pEGFP-N1 per well to 250 μL of opti-MEM for 5 minutes. Then, 250 μL of DNA mixture was added to the 250 μL Lipofectamine 2000 mixture and incubated at room temperature for 20 minutes. The mixture was then added to each well and incubated in 5% CO2 incubator at 37°C for 6 hours. The Lipofectamine/DNA mixture was removed and the fresh DMEM supplemented with 10% fetal bovine serum was added for incubation overnight. Cells transfected with pEGFP-N1 were termed “EGFP-SH-SY5Y” cells and those transfected with pEGFP-N1-MeCP2 were termed “O-MeCP2-SH-SY5Y” cells.
CCK-8 assay for cell viability
For quantitative analysis of cell viability, the cytotoxicity and proliferation of the were assessed using the CCK-8 assay (Dojindo, Kumamoto, Japan), based on the reduction MTT by the mitochondrial dehydrogenase of intact cells[61]. The cells were seeded in a 96-well plate at a density of 5 × 104/mL and routinely incubated at 37°C for 24 hours prior to use. The cells were treated with 6-hydroxydopamine (50 μmol/L) for 3, 6, 12, and 24 hours in 100 μL media without serum and 10 μL of CCK-8 was added to each well. Following incubation at 37°C for 2 hours in a humidified CO2 incubator, absorbance at 540 nm was monitored using a microplate reader (SpectraMax Plus 384; Molecular Devices, Santa Clara, CA, USA). The values obtained were normalized to those of control cells incubated with vehicle only.
Flow cytometry analysis of apoptosis
SH-SY5Y cells were plated into six-well plates at 4 × 105 cells/mL and incubated until they reached the logarithmic phase. Following a 24-hour pretreatment with 6-hydroxydopamine, the cells were subsequently trypsinized and centrifuged at 600 × g at 4°C for 5 minutes. The supernatant was removed and the cells were washed with cold PBS. After washes, the cells were resuspended in PBS at a density of 1 × 106/mL. The suspension was subsequently added to fluorescein isothiocyanate-labeled Annexin V (Sigma) and stained with propidium iodide (Sigma). Flow cytometric analysis was performed using a FACSAria flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Immunocytofluorescence staining for MeCP2 and tyrosine hydroxylase expression
SH-SY5Y cells were seeded onto coverslips in six-well plate, fixed with a 4% paraformaldehyde solution at 37°C for 10 minutes, washed three times with PBS, permeabilized with 0.1% Triton X-100 in PBS for 10 minutes, and blocked with 1% bovine serum albumin (Sigma) in PBS for 1 hour. Rabbit anti-MeCP2 polyclonal antibody (HA000593, 1:300 dilution; Sigma) and goat anti-tyrosine hydroxylase polyclonal antibody (sc-7847, 1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were separately diluted in PBS containing 0.1% Triton X-100 and applied overnight at 4°C. After three washes with PBS, cells were exposed to fluorescein isothiocyanate-labeled goat anti-rabbit IgG (1:800; Jackson, San Diego, CA, USA) or CY3 labeled rabbit anti-goat IgG (1:800; Jackson) at room temperature for 1 hour, then labeled with 4′,6-diamidino-2-phenylindole (Sigma) and dried in a dark room before the coverslips were mounted onto glass microscope slides with Vectashield medium (Vector Laboratories, Burlingame, CA, USA). Immunofluorescence was visualized using a confocal laser scanning microscope (Leica TCS SP2, Solms, Germany).
Western blot analysis for MeCP2 and tyrosine hydroxylase expression
SH-SY5Y cells were seeded in six-well plates at a density of 4 × 105 cells/mL. After treatment, the cells were harvested, washed three times with precooled PBS, and lysed for western blot. After centrifugation, the supernatants were collected and stored at −70°C until testing. The protein concentrations of lysates from SH-SY5Y cells were measured using bicinchoninic acid assay kits (Beyotime, Haimen, Jiangsu, China). Equal amounts of protein from each sample were electrophoresed on 12% denaturing sodium dodecyl sulfate polyacrylamide gels. Protein was then transferred to polyvinylidene fluoride membranes for 90 minutes at 200 mA. Membranes were blocked with 5% fat-free powdered milk at room temperature for 1 hour and incubated overnight at 4°C with rabbit anti-MeCP2 polyclonal antibody (1:2 000), goat anti-tyrosine hydroxylase polyclonal antibody (1:500), rabbit anti-EGFP polyclonal antibody (bs-2194R; 1:500; Bioss, Beijing, China) and mouse anti-human β-actin monoclonal antibody (sc-81178; 1:1 000; Santa Cruz Biotechnology). Following 1 hour of incubation separately at room temperature with horseradish peroxidase-coupled goat anti-rabbit IgG, horseradish peroxidase-coupled rabbit anti-goat IgG, and horseradish peroxidase-coupled goat anti-mouse IgG (Jackson), the membranes were washed with PBS and bound antibody was detected using enhanced chemiluminescence detection reagents (Thermo, Boston, MA, USA). Immunoreactivity was quantified with Sigma Scan Pro 5 and the absorbance from bands representing MeCP2 and tyrosine hydroxylase was normalized to the β-actin loading control to standardize for variations in loading.
Statistical analysis
Experiments were conducted in triplicate and data are expressed as mean ± SD. Data were analyzed with one-way analysis of variance followed by the Student Newman-Keuls post hoc test. A value of P < 0.05 was considered statistically significant. All statistical analyses were performed using SPSS 15.0 for Windows (SPSS, Chicago, IL, USA).
Footnotes
Funding: This study was sponsored by the Ph.D. Independent Research Projects of Wuhan University, No. 201130302020017; a grant from the Science and Technology Bureau of Hubei Province, No. 2011CDB511; and the National Natural Science Foundation of China, No. 81170769.
Conflict of Interest: None declared.
(Reviewed by McGowan D, Pack M, Li X, Zhao Y)
(Edited by Wang LM, Su LL, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Gasser T. Genetics of Parkinson's disease. J Neurol. 2001;248(10):833–840. doi: 10.1007/s004150170066. [DOI] [PubMed] [Google Scholar]
- [2].Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114(Pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- [3].Ossig C, Reichmann H. Treatment of Parkinson's disease in the advanced stage. J Neural Transm. 2013;120(4):523–529. doi: 10.1007/s00702-013-1008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Steece-Collier K, Maries E, Kordower JH. Etiology of Parkinson's disease: Genetics and environment revisited. Proc Natl Acad Sci U S A. 2002;99(22):13972–13974. doi: 10.1073/pnas.242594999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Quik M. Smoking, nicotine and Parkinson's disease. Trends Neurosci. 2004;27(9):561–568. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- [6].Kumar A, Sharma N, Gupta A, et al. Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain Res. 2012;1471:13–22. doi: 10.1016/j.brainres.2012.06.050. [DOI] [PubMed] [Google Scholar]
- [7].Yamamoto T, Nair P, Davis P, et al. Design, synthesis, and biological evaluation of novel bifunctional C-terminal-modified peptides for delta/mu opioid receptor agonists and neurokinin-1 receptor antagonists. J Med Chem. 2007;50(12):2779–2786. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen Q, He Y, Yang K. Gene therapy for Parkinson's disease: progress and challenges. Curr Gene Ther. 2005;5(1):71–80. doi: 10.2174/1566523052997505. [DOI] [PubMed] [Google Scholar]
- [9].Carlsson T, Bjorklund T, Kirik D. Restoration of the striatal dopamine synthesis for Parkinson's disease: viral vector-mediated enzyme replacement strategy. Curr Gene Ther. 2007;7(2):109–120. doi: 10.2174/156652307780363125. [DOI] [PubMed] [Google Scholar]
- [10].Shen Y, Muramatsu SI, Ikeguchi K, et al. Triple transduction with adeno-associated virus vectors expressing tyrosine hydroxylase, aromatic-L-amino-acid decarboxylase, and GTP cyclohydrolase I for gene therapy of Parkinson's disease. Hum Gene Ther. 2000;11(11):1509–1519. doi: 10.1089/10430340050083243. [DOI] [PubMed] [Google Scholar]
- [11].Lewis JD, Meehan RR, Henzel WJ, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69(6):905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- [12].Ballas N, Lioy DT, Grunseich C, et al. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12(3):311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- [14].Schanen NC. Molecular approaches to the Rett syndrome gene. J Child Neurol. 1999;14(12):806–814. doi: 10.1177/088307389901401207. [DOI] [PubMed] [Google Scholar]
- [15].Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- [16].Wenk GL, Naidu S, Casanova MF, et al. Altered neurochemical markers in Rett's syndrome. Neurology. 1991;41(11):1753–1756. doi: 10.1212/wnl.41.11.1753. [DOI] [PubMed] [Google Scholar]
- [17].Dunn HG, MacLeod PM. Rett syndrome: review of biological abnormalities. Can J Neurol Sci. 2001;28(1):16–29. doi: 10.1017/s0317167100052513. [DOI] [PubMed] [Google Scholar]
- [18].Panayotis N, Pratte M, Borges-Correia A, et al. Morphological and functional alterations in the substantia nigra pars compacta of the Mecp2-null mouse. Neurobiol Dis. 2011;41(2):385–397. doi: 10.1016/j.nbd.2010.10.006. [DOI] [PubMed] [Google Scholar]
- [19].Fu AL, Wu SP, Dong ZH, et al. A novel therapeutic approach to depression via supplement with tyrosine hydroxylase. Biochem Biophys Res Commun. 2006;351(1):140–145. doi: 10.1016/j.bbrc.2006.10.013. [DOI] [PubMed] [Google Scholar]
- [20].Haavik J, Toska K. Tyrosine hydroxylase and Parkinson's disease. Mol Neurobiol. 1998;16(3):285–309. doi: 10.1007/BF02741387. [DOI] [PubMed] [Google Scholar]
- [21].Hunt SC. Tyrosine hydroxylase: another piece of the genetics of hypertension puzzle. Circulation. 2007;116(9):970–972. doi: 10.1161/CIRCULATIONAHA.107.721779. [DOI] [PubMed] [Google Scholar]
- [22].Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27(3):306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- [23].Matarazzo V, Cohen D, Palmer AM, et al. The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Mol Cell Neurosci. 2004;27(3):44–58. doi: 10.1016/j.mcn.2004.05.005. [DOI] [PubMed] [Google Scholar]
- [24].Francke U. Mechanisms of disease: neurogenetics of MeCP2 deficiency. Nat Clin Pract Neurol. 2006;2(4):212–221. doi: 10.1038/ncpneuro0148. [DOI] [PubMed] [Google Scholar]
- [25].Cohen DR, Matarazzo V, Palmer AM, et al. Expression of MeCP2 in olfactory receptor neurons is developmentally regulated and occurs before synaptogenesis. Mol Cell Neurosci. 2003;22(4):417–429. doi: 10.1016/s1044-7431(03)00026-5. [DOI] [PubMed] [Google Scholar]
- [26].Marsh R, Gerber AJ, Peterson BS. Neuroimaging studies of normal brain development and their relevance for understanding childhood neuropsychiatric disorders. J Am Acad Child Adolesc Psychiatry. 2008;47(11):1233–1251. doi: 10.1097/CHI.0b013e318185e703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Weng SM, Bailey ME, Cobb SR. Rett syndrome: from bed to bench. Pediatr Neonatol. 2011;52(6):309–316. doi: 10.1016/j.pedneo.2011.08.002. [DOI] [PubMed] [Google Scholar]
- [28].Na ES, Nelson ED, Kavalali ET, et al. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology. 2013;38(1):212–219. doi: 10.1038/npp.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gantz SC, Ford CP, Neve KA, et al. Loss of Mecp2 in substantia nigra dopamine neurons compromises the nigrostriatal pathway. J Neurosci. 2011;31(35):12629–12637. doi: 10.1523/JNEUROSCI.0684-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Giuliani P, Romano S, Ballerini P, et al. Protective activity of guanosine in an in vitro model of Parkinson's disease. Panminerva Med. 2012;54(1 Suppl 4):43–51. [PubMed] [Google Scholar]
- [31].Saito Y, Nishio K, Ogawa Y, et al. Molecular mechanisms of 6-hydroxydopamine-induced cytotoxicity in PC12 cells: involvement of hydrogen peroxide-dependent and – independent action. Free Radic Biol Med. 2007;42(5):675–685. doi: 10.1016/j.freeradbiomed.2006.12.004. [DOI] [PubMed] [Google Scholar]
- [32].Bae J, Lee D, Kim YK, et al. Berberine protects 6-hydroxydopamine-induced human dopaminergic neuronal cell death through the induction of heme oxygenase-1. Mol Cells. 2013;35(2):151–157. doi: 10.1007/s10059-013-2298-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Guo S, Bezard E, Zhao B. Protective effect of green tea polyphenols on the SH-SY5Y cells against 6-OHDA induced apoptosis through ROS-NO pathway. Free Radic Biol Med. 2005;39(5):682–695. doi: 10.1016/j.freeradbiomed.2005.04.022. [DOI] [PubMed] [Google Scholar]
- [34].Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21(21):4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88(4):471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- [37].Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- [38].Yasui DH, Peddada S, Bieda MC, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104(49):19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Skene PJ, Illingworth RS, Webb S, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37(4):457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Young JI, Hong EP, Castle JC, et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A. 2005;102(49):17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Samaco RC, Mandel-Brehm C, Chao HT, et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci U S A. 2009;106(51):21966–21971. doi: 10.1073/pnas.0912257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Roux JC, Dura E, Villard L. Tyrosine hydroxylase deficit in the chemoafferent and the sympathoadrenergic pathways of the Mecp2 deficient mouse. Neurosci Lett. 2008;447(1):82–86. doi: 10.1016/j.neulet.2008.09.045. [DOI] [PubMed] [Google Scholar]
- [43].Ide S, Itoh M, Goto Y. Defect in normal developmental increase of the brain biogenic amine concentrations in the mecp2-null mouse. Neurosci Lett. 2005;386(1):14–17. doi: 10.1016/j.neulet.2005.05.056. [DOI] [PubMed] [Google Scholar]
- [44].Kerr AM, Armstrong DD, Prescott RJ, et al. Rett syndrome: analysis of deaths in the British survey. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):71–74. [PubMed] [Google Scholar]
- [45].Zhang X, Su J, Rojas A, et al. Pontine norepinephrine defects in Mecp2-null mice involve deficient expression of dopamine beta-hydroxylase but not a loss of catecholaminergic neurons. Biochem Biophys Res Commun. 2010;394(2):285–290. doi: 10.1016/j.bbrc.2010.02.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Roux JC, Dura E, Moncla A, et al. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007;25(7):1915–1922. doi: 10.1111/j.1460-9568.2007.05466.x. [DOI] [PubMed] [Google Scholar]
- [47].Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302(5646):885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- [48].Finley JC, Katz DM. The central organization of carotid body afferent projections to the brainstem of the rat. Brain Res. 1992;572(1-2):108–116. doi: 10.1016/0006-8993(92)90458-l. [DOI] [PubMed] [Google Scholar]
- [49].Guo H, Hellard DT, Huang L, et al. Development of pontine noradrenergic A5 neurons requires brain-derived neurotrophic factor. Eur J Neurosci. 2005;21(7):2019–2203. doi: 10.1111/j.1460-9568.2005.04016.x. [DOI] [PubMed] [Google Scholar]
- [50].Hertzberg T, Fan G, Finley JC, et al. BDNF supports mammalian chemoafferent neurons in vitro and following peripheral target removal in vivo. Dev Biol. 1994;166(2):801–811. doi: 10.1006/dbio.1994.1358. [DOI] [PubMed] [Google Scholar]
- [51].Ogier M, Wang H, Hong E, et al. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27(40):10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang H, Chan SA, Ogier M, et al. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26(42):10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- [54].Chang Q, Khare G, Dani V, et al. The disease progression of Mecp2mutantmice is affected by the level of BDNF expression. Neuron. 2006;49(3):341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- [55].Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14(4):483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Russell JC, Blue ME, Johnston MV, et al. Enhanced cell death in MeCP2 null cerebellar granule neurons exposed to excitotoxicity and hypoxia. Neuroscience. 2007;150(3):563–574. doi: 10.1016/j.neuroscience.2007.09.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chao HT, Zoghbi HY. The yin and yang of MeCP2 phosphorylation. Proc Natl Acad Sci U S A. 2009;106(12):4577–4578. doi: 10.1073/pnas.0901518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tao J, Hu K, Chang Q, et al. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci U S A. 2009;106(12):4882–4887. doi: 10.1073/pnas.0811648106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bracaglia G, Conca B, Bergo A, et al. Methyl-CpG-binding protein 2 is phosphorylated by homeodomain-interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 2009;10(12):1327–1333. doi: 10.1038/embor.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94(3):441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- [61].Ishiyama M, Miyazono Y, Sasamoto K, et al. A highly water-soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta. 1997;44(7):1299–1305. doi: 10.1016/s0039-9140(97)00017-9. [DOI] [PubMed] [Google Scholar]