

Abstract
Maternal alcohol consumption during pregnancy has detrimental effects on fetal central nervous system development. Maternal alcohol consumption prior to and during pregnancy significantly affects cognitive functions in offspring, which may be related to changes in cyclin-dependent kinase 5 because it is associated with modulation of synaptic plasticity and impaired learning and memory. In this study, we examined adult offspring in a maternal alcohol consumption model in rats. Y-maze test results showed that in utero exposure to alcohol impairs learning and memory capacities. Cyclin-dependent kinase 5 mRNA and protein expressions in the hippocampus of the offspring were significantly elevated, as assayed by quantitative real-time PCR and reverse transcription-PCR, immunofluorescence, and immuno-precipitation. Our experimental findings strongly suggest that altered cyclin-dependent kinase 5 may mediate impaired learning and memory in adult rats that were exposed to alcohol by maternal consumption while in utero.
Keywords: neural regeneration, neurogenesis, pregnancy, ethanol, hippocampus, development, offspring, learning and memory, cyclin-dependent kinase 5, grants-supported paper, neuroregeneration
Research Highlights
(1) The majority of studies addressing prenatal ethanol exposure have examined neuronal apoptosis, offspring development, and social behaviors. Little research has examined effects on nerve injury signals, which are related to nitric oxide signaling pathways.
(2) In this study, we aimed to explore the correlation between cyclin-dependent kinase 5 expression in the hippocampus and neurological impairments following prenatal ethanol exposure.
(3) We propose new insights into the mechanisms underlying the role of ethanol exposure in central nervous system injuries, and provide a new strategy for treating the consequences of prenatal ethanol exposure.
(4) Cyclin-dependent kinase 5 likely mediates impaired learning and memory capacities in rats following prenatal ethanol exposure.
INTRODUCTION
Fetal alcohol syndrome is the most common nongenetic cause of mental retardation. It is well known that excessive maternal alcohol consumption during pregnancy has teratogenic and other detrimental effects on fetal development, and prenatal alcohol exposure leads to brain injuries that can potentially manifest as life-long learning and cognitive deficits[1,2,3,4]. Fetal alcohol syndrome resides at the most severe end of the continuum of fetal alcohol spectrum disorders, which includes all alcohol-induced abnormalities. The clinical features of fetal alcohol syndrome disorders are variable and include an array of morphologic defects, as well cognitive and behavioral disabilities, which range from severe mental retardations to subtle deficits that become apparent only under stressful conditions. The prevalence of fetal alcohol syndrome disorders has been reported to be 1 in 100 births, but recent studies have estimated that fetal alcohol syndrome disorders may be much more prevalent, at 2–5% of school children in the United States and some western European countries. The prevalence of fetal alcohol syndrome in the United States population was estimated to be at least 2 to 7 per 1 000. One estimate is that 5% of the children of mothers who drank heavily during pregnancy have fetal alcohol syndrome, and animal studies have shown that the dose, timing, and duration of ethanol consumption are critical. The hippocampus is a particularly vulnerable brain region to prenatal ethanol exposure[5,6]. It is generally accepted that prenatal ethanol exposure can alter hippocampal synaptic plasticity and adversely affect hippocampal-dependent learning, and cause other mental defects[7,8]. The neuronal plasticity biomarker cyclin-dependent kinase 5, a proline-directed serine/threonine kinase, plays important roles in various aspects of neural development, including neuronal migration, axonal guidance, synaptic development, and plasticity[9,10,11]. However, the effects of alcohol on cyclin-dependent kinase 5, and particularly the effects of in utero alcohol exposure on cyclin-dependent kinase 5 in adulthood, are largely unknown.
Cyclin-dependent kinase 5 may be specifically activated by p35 and p39 in neurons. The p35 is expressed predominantly in the cerebral cortex, and p39 is largely expressed in the cerebellum. Both calmodulin and elevated concentrations of intracellular Ca2+ are necessary to cleave p35 and p39 into p25 and p29, respectively. The binding of cyclin-dependent kinase 5 with p35 and p39 and their cleavage products forms a heterodimer, resulting in the activation of cyclin-dependent kinase 5. The N-terminal truncation products of p35 and p39 are p25 and p29, and they have longer half-lives and are more stable than p35 and p39. Thus, they can overexcite cyclin-dependent kinase 5 signaling, triggering downstream substrates that exert pathological changes[12]. The expression of p35 peaks at 1 or 2 weeks after birth. In the adult brain, p35 undergoes a process of physiological reduction[13]. Throughout every stage of brain development, cyclin-dependent kinase 5 activity is correlated with the expression of p35 rather than that of cyclin-dependent kinase 5[14].
Maternal alcohol consumption during pregnancy may lead to neurodevelopmental disorders and abnormal psychology and behavior. The mechanism underlying lesions related to alcohol remains to be explored. To elucidate the consequences of maternal alcohol consumption on offspring, in this study, we examined its effects on learning and memory, p35 expression, and cyclin-dependent kinase 5 activity in rats. We then examined whether subsequent changes in synaptic plasticity led to impaired learning and memory. To these ends, an animal model of alcohol consumption during pregnancy was established. Learning and memory were evaluated by ethology in adult offspring. Cyclin-dependent kinase 5 protein expression and activity were detected by immunofluorescence and PCR. Our experimental findings suggest a new strategy and target for drugs to treat alcoholism, and may elucidate the causes of central nervous system impairments.
RESULTS
Quantitative analysis of experimental animals
Eighteen pregnant, female rats were randomly divided into control, isocaloric group and ethanol-treated groups, with six rats in each group. The day that a positive plug was present was defined as E0. Usually, the control and isocaloric groups gave birth after E19, but the ethanol-treated rats typically gave birth 1–2 days later at E20 or E21. Postnatal offspring were produced from timed pregnancies (P = postnatal days; P0 = the first 24 hours after birth). At P14, three pups from each female in each group were randomly assigned and were used for the study. A total of 54 pups were included in the final analysis, without any loss.
Maternal alcohol consumption impaired learning and memory in offspring
The learning and memory capacities of the pups at P14 were determined with Y-maze test. As shown in Table 1 and Figure 1, the training times of the offspring from the ethanol-treated group were significantly increased compared with the control and isocaloric groups (P < 0.05).
Table 1.
Effects of maternal alcohol consumption on learning and memory (Y-maze test) and cyclin-dependent kinase 5/p35 mRNA expression (quantitative real-time PCR) in their offspring
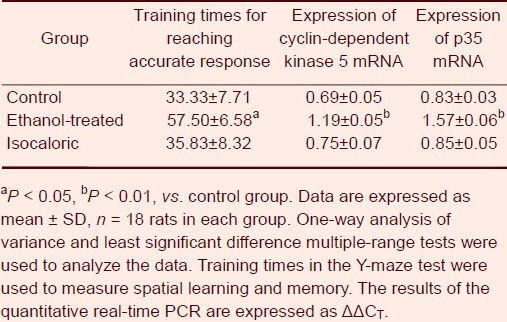
Figure 1.
Effects of maternal alcohol consumption on mRNA expression of cyclin-dependent kinase 5 (Cdk5) and p35 in the hippocampus of offspring (quantitative reverse transcription-PCR).
Total RNA isolated and dissociated from the hippocampus of rats was reverse transcribed to produce PCR products then visualized on agarose gels. (A) Bands of p35 mRNA; (B) bands of Cdk5 mRNA.
CN: Control group; IC: isocaloric group; ET: ethanol-treated group; M: marker.
Maternal alcohol consumption increased the expression of hippocampal cyclin-dependent kinase 5 and p35 mRNA in offspring
Following the Y-maze test, reverse transcription-PCR and quantitative real-time PCR were performed to establish the cyclin-dependent kinase 5 and p35 expression pattern in the hippocampus, and reveal any changes of the cyclin-dependent kinase 5 and p35 expression levels on maternal alcohol consumption during pregnancy. As shown in Table 1 and Figure 1, the expression level of cyclin-dependent kinase 5 and p35 mRNA were significantly increased in the hippocampus of offspring from the ethanol-treated group compared with offspring from the control and isocaloric groups. In contrast, the differences in cyclin-dependent kinase 5 and p35 mRNA in the hippocampus between the offspring from the control and isocaloric groups were not significant.
Maternal alcohol consumption increased the expression of hippocampal cyclin-dependent kinase 5 and p35 in offspring
Following the Y-maze test, immunofluorescence was performed to confirm cyclin-dependent kinase 5 and p35 expression in the hippocampus. As shown in Figure 2, cyclin-dependent kinase 5 and p35 were expressed in the hippocampus. Compared with the control and isocaloric groups, more hippocampal neurons in the offspring from the ethanol-treated group showed cyclin-dependent kinase 5 and p35 fluorescence staining (P < 0.01), exhibiting dense distributions and increased fluorescence intensity.
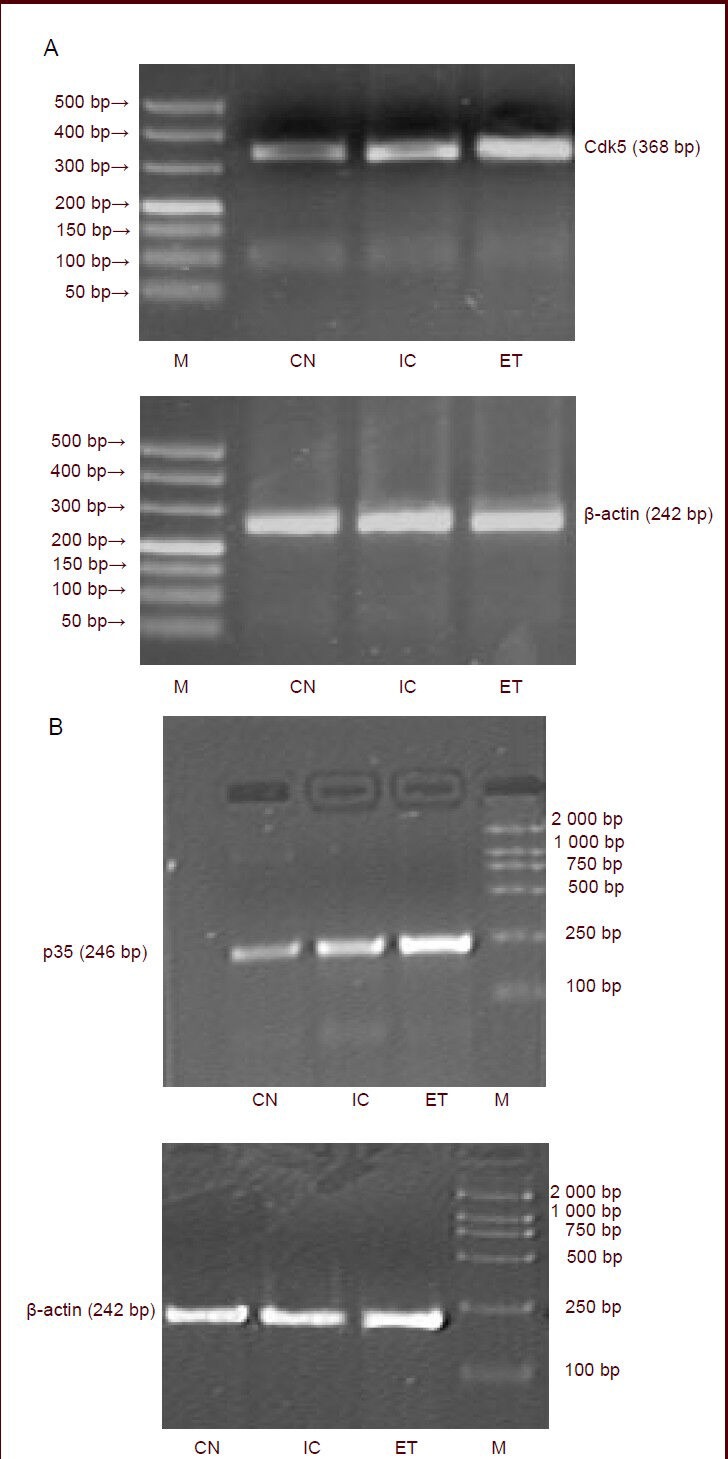
Figure 2.
Effects of maternal alcohol consumption on cyclin-dependent kinase 5 (Cdk5) and p35 expression in the hippocampus of rat offspring (immunofluorescence, × 400).
(A, B) Bar graph of Cdk5- and p35-positive cell numbers in the hippocampus. aP < 0.01, vs. CN and IC. Data are expressed as mean ± SD,n= 18 rats in each group. The experiment was repeated three times, and analyzed using one-way analysis of variance and the least significant difference multiple-range test.
(C) Representative photomicrographs of Cdk5 (Alexa Fluor 488-labeled, green fluorescence) and p35 (Cy3-labeled, red fluorescence) with immunofluorescent staining in the rat hippocampus. Scale bars: 10 μm. Cells with Cdk5 expression are green (white arrows). In contrast, cells with p35 expression are red (yellow arrows). Compared with the control and isocaloric groups, a large number of hippocampal neurons were stained with fluorescence in the offspring from the ethanol-treated group, and the density and fluorescence intensity increased.
CN: Control group; ET: ethanol-treated group; IC: isocaloric group.
Maternal alcohol consumption increased cyclin-dependent kinase 5 activity in the hippocampus of offspring
Following the Y-maze test, to determine cyclin-dependent kinase 5 activity in hippocampal neurons, cyclin-dependent kinase 5 was immunoprecipitated with an anti-cyclin-dependent kinase 5 antibody, and kinase activity was measured with histone H1 as a substrate. As shown in Figure 3, cyclin-dependent kinase 5 kinase activity in the hippocampus of offspring from the ethanol-treated group was significantly increased compared with the control group (P < 0.01).
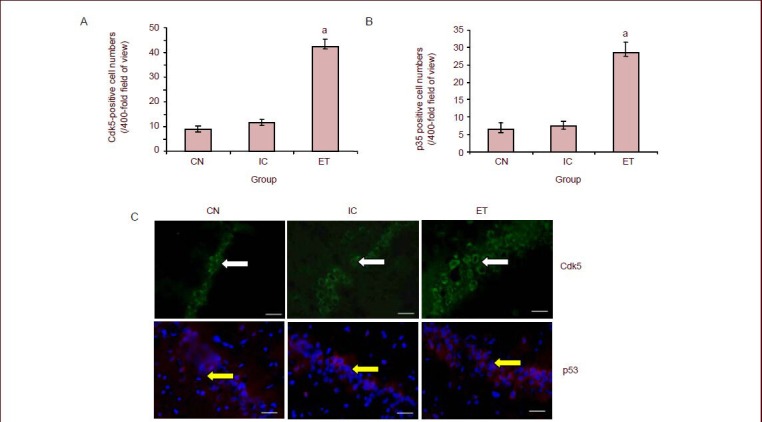
Figure 3.
Effects of maternal alcohol consumption during pregnancy on cyclin-dependent kinase 5 (Cdk5) activity in the hippocampus of rat offspring.
aP < 0.01, vs. control group. Cdk5 activity is expressed as p moles of ATP incorporated/mg protein per minute and is plotted as mean ± SD,n = 18 rats in each group. The experiment was repeated three times over five independent experiments. One-way analysis of variance and the least significant difference multiple-range test were used to determine differences between means.
CN: Control group; ET: ethanol-treated group; IC: isocaloric group.
DISCUSSION
Ethanol, the active ingredient of alcoholic beverages, is the most abused drug in the world, perhaps second only to nicotine. Partially related to its legal status and availability, it causes widespread addiction and dependency problems with devastating health consequences, including numerous premature deaths. Ethanol abuse and addiction are two of the most important public health problems worldwide, and they can lead to tolerance, dependence, and memory impairments. Ethanol is therefore a widely studied molecule in many research institutes[15,16,17]. Fetal alcohol syndrome is a leading cause of birth defects and developmental disabilities. In the United States, an estimated 2 000 to 8 000 new cases occur each year. Fetal alcohol syndrome represents the most severe manifestation of alcohol-related birth defects and developmental disabilities. The number of children with fetal alcohol syndrome and other fetal alcohol spectrum disorders, predominantly alcohol-related neurodevelopmental disorders, is estimated to be three times higher than that of children with fetal alcohol syndrome alone. Although prevention of all fetal alcohol syndrome disorders should be a public health priority, the need for prevention of fetal alcohol syndrome is particularly urgent. To prevent fetal alcohol syndrome, it is necessary to identify and characterize women who are at risk for having children with fetal alcohol syndrome.
Prenatal ethanol exposure in humans can produce a wide range of toxic effects, including teratogenicity and lethality, manifesting as spontaneous abortion or stillbirth[18,19,20]. Such ethanol teratogenicity can manifest as fetal alcohol syndrome, which consists of central nervous system dysfunctions, growth deficiency, and a characteristic cluster of facial abnormalities in offspring[5]. Of these three principal features, central nervous system dysfunction–manifesting as persistent intellectual, behavioral, and neurological deficits–is probably the most debilitating for the individual with fetal alcohol syndrome. Central nervous system dysfunctions can be related to permanent neuronal cell loss in target brain regions, including the hippocampus and cerebellum, as demonstrated in experimental animal models of ethanol central nervous system teratogenicity[21]. Previous studies have identified a number of characteristics and behaviors in mothers that predict the presentation of fetal alcohol syndrome in their children. Some of these risk factors are heavy drinking (including binge drinking), drinking among members of the extended family and social network, more years of drinking, cigarette smoking, previous treatment for alcohol and/or drug abuse, age, high gravidity, lower educational attainments, lower income, a lower intelligence quotient, and a history of sexual or physical abuse or mental illness. In this study, we used rat models to determine the effects of maternal alcohol consumption on their offspring, and found that maternal alcohol consumption prior to and during pregnancy significantly affected the learning capacities of the offspring, and increased p35 expression and cyclin-dependent kinase 5 activity in the hippocampus.
Mammalian brains are flexible, being able to change their functions and structures in response to internal and external stimuli. Such changes are called neuronal plasticity[22]. Synapses enable the transmission of information within neural circuits and allow the brain to change in response to experiences[23]. Neuronal plasticity in animals is essential for their daily lives because they must adapt to changes in their outside environment[24]. Long-term plasticity, either depression or potentiation of synaptic transmission, typically lasts for hours or days, but can possibly last for years in a living animal. The induction mechanisms underlying synaptic plasticity can be presynaptic or post-synaptic[25]. The hippocampus is an essential region for neuronal plasticity, and it is critically involved in learning and memory in mammals. The hippocampus has been a major experimental system for studies of synaptic plasticity in the context of putative information-storage mechanisms in the brain. The simple laminar pattern of its neurons and neural pathways enables the use of extracellular recording techniques to record synaptic events for virtually unlimited periods in vivo[26].
Long-term potentiation is a much-studied model of synaptic plasticity that was first identified in the hippocampus, and has been extensively characterized using electrophysiological, biochemical and molecular techniques. Several recent studies have detected long-term potentiation-like synaptic changes in the hippocampus[27,28] and the amygdala[29] following learning. Other forms of activity-dependent plasticity include long-term depression, potentiation of excitatory postsynaptic potential spikes, spike-timing-dependent plasticity, depotentiation, and reversal of depression[30]. Long-term potentiation in the hippocampus is considered to be important for elucidating the molecular mechanisms of learning and memory, because these processes may be based on long-lasting potentiation of synaptic efficacy. Correlations have been observed between defective hippocampal synaptic plasticity and defective hippocampal-dependent memory tasks following perturbation of a number of proteins involved in synaptic plasticity, either pharmacologically or through gene knockout[31,32]. For example, rodents in which N-methyl-D-aspartate receptor antagonists are infused into the hippocampus[33], and mice lacking expression in the forebrain of the N-methyl-D-aspartate receptor subunit 1[34], do not show long-term potentiation and are impaired on certain types of spatial learning. Furthermore, mice in which the specific N-methyl-D-aspartate receptor subunit NR2B was overexpressed display enhanced long-term potentiation and enhanced spatial learning[35]. In addition to their role in the encoding of declarative memories, hippocampal N-methyl-D-aspartate receptor-dependent long-term potentiation and depression may provide important insights into the pathophysiology and treatment of important mental illnesses. For example, a leading hypothesis for the pathophysiology of schizophrenia posits dysfunctional glutamatergic synapses, in particular hypofunction of N-methyl-D-aspartate receptors[36,37]. Mouse strain differences in learning and memory ability are well documented. A wide variety of paradigms have been studied measuring a range of learning from complex to simple associative learning. However, interstrain comparisons of learning and memory abilities are potentially complicated by both host (genetic) and environmental (experimental parameters) factors that may significantly impact performance, while not pertaining directly to learning and memory constructs per se. Performance levels in a variety of learning and memory procedures may be influenced by motivation to explore novelty, attentional processes, and memories for acquired information. Specifically, the use of appetitive or aversive stimuli can enhance differences in motivation or reactivity between strains that are difficult to dissociate from associative and mnemonic processes. Because of the increasing use of a broad range of mouse genetic backgrounds to investigate normal and disrupted behavior, establishing a paradigm that permits the evaluation of novelty exploration and memory independently and validating it across a variety of genetic backgrounds and procedural variables would be highly valuable. The Y-maze is a simple two-trial recognition test for measuring spatial recognition memory, it does not require learning a rule, and thus is useful for studying memory in rodents, and in particular for the study of genetic influences on responses to novelty and recognition processes[38]. Maternal alcohol consumption during pregnancy may modify hippocampal plasticity and thereby affect learning and memory in the offspring. Chronic prenatal ethanol exposure results in loss of hippocampal CA1 pyramidal cells in young postnatal offspring[39] and decreases synaptic plasticity in the hippocampus of adult offspring[40]. Our results further suggest that learning and memory are impaired by chronic in utero ethanol exposure. These impairments persisted even after the ethanol exposure was discontinued, which may be related to modified synaptic plasticity, leading to the inhibition of long-term potentiation and facilitation of long-term depression[41].
Cyclin-dependent kinase 5, also called tau protein kinase II, is a member of the Ser/Thr family and proline-directed protein kinases that are related to the cyclin-dependent cell cycle regulatory kinases. Despite its high degree of homology to other cyclin-dependent kinases, cyclin-dependent kinase 5 is the only member of this family that is not activated by cyclins[42]. Although cyclin-dependent kinase 5 is expressed in many tissues, its highest kinase activity coincides with the expression pattern of the activators p35 and p39, which are present mainly in post-mitotic neurons of the central nervous system[43]. The emerging evidence that cyclin-dependent kinase 5 is involved in several neuronal functions such as neuronal migration, neurotransmitter release, neuronal plasticity, memory, learning, addiction, and apoptosis as a potential master regulator of many vital neuronal functions[44]. Cyclin-dependent kinase 5 may be a constitutively active kinase. Its activity depends on its direct association with one of two noncyclin cofactors, p35 and p39[45]. Cyclin-dependent kinase 5 is mainly active in postmitotic neurons because of the selective neuronal expression of p35 and p39[46]. These cofactors are subject to cleavage by calpain. Thus, increases in intracellular calcium, which may occur during neuronal injuries or neurotoxic insults, results in conversion of p35 or p39 to p25 or p27, respectively[47]. Transgenic animal studies have demonstrated a critical role of cyclin-dependent kinase 5/p35 in neurite outgrowth and neuronal differentiation during development, likely being regulated by specific protein-protein interactions because cyclin-dependent kinase 5 interacts with a diverse family of regulatory proteins[48]. Cyclin-dependent kinase 5 has been implicated in learning and synaptic plasticity[49] and the pathogenesis of neurodegenerative disorders such as Alzheimer's disease and neuropsychiatric illnesses such as addiction[50]. Learning and memory are vital to animal development and the acquisition of modifiable, plastic neuronal circuits allows for robust adaptation to dynamic surroundings. The ability to learn from experience and employ stored knowledge in the form of memories to modify behavior in response to selective environmental pressures is crucial to animal physiology and cognition[51]. Cyclin-dependent kinase 5 is required in the hippocampus and septum for associative learning to occur closely following training[52]. The first hint that loss of cyclin-dependent kinase 5 directly affects plasticity came from p35-knockout mice in which long-term potentiation was induced by tetanic stimulation (100 Hz), in that it appears to be normal in the hippocampal CA1 region, but low-frequency stimulation (1 Hz, 15 minutes) revealed a marked deficit in long-term depression[53]. Cyclin-dependent kinase 5 is seen to influence pre- and post-synaptic processes integral to synaptic plasticity, including vesicle cycling, ion channel modulation and intra-cellular signaling[54]. Previous reports also implicated both cyclin-dependent kinase 5 and CA1 hippocampal N-methyl-D-aspartate receptors in spatial learning and synaptic plasticity. A cyclin-dependent kinase 5 conditional knockout line was derived under a prion promoter and regulated temporally by the estrogen-receptor transgene[47]. In these studies, cyclin-dependent kinase 5 conditional knockout mice displayed enhanced hippocampal-dependent spatial memory as assayed by Morris water maze task, enhanced contextual fear memories, and enhanced long-term potentiation compared with control mice. Evoked excitatory postsynaptic currents showed that currents mediated by the N-methyl-D-aspartate, but not the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor are enhanced in these mutant mice, which is attributable to greater surface N-methyl-D-aspartate receptor subunit 2B levels and a reduction in N-methyl-D-aspartate receptor degradation. Cyclin-dependent kinase 5 may function structurally to increase interactions between N-methyl-D-aspartate receptor subunit 2B and calpain and to facilitate N-methyl-D-aspartate receptor subunit 2B degradation in response to synaptic activity[55].
Cyclin-dependent kinase 5 activity is linked to the activator p35. Neurotoxic stress induces calcium influx and calpain-directed proteolysis of p35 resulting in the release of p25 from an N-terminal membrane tether[56]. The α-actinin-1 kinase and the Ca2+/calmodulin-dependent protein kinase II interact with cyclin-dependent kinase 5 via their associations with p35 and p39. Ca2+/calmodulin-dependent protein kinase II and α-actinin-1 bind to distinct regions of p35 and p39, and can also interact with each other. The associations between the Ca2+/calmodulin-dependent protein kinase II and α-actinin-1 with the cyclin-dependent kinase 5 activators are stimulated by calcium. Furthermore, the activation of glutamate receptors increases the association of p35 and p39 with Ca2+/calmodulin-dependent protein kinase II, and the inhibition of Ca2+/calmodulin-dependent protein kinase II activation diminishes this effect. The glutamate-mediated increase in association of p35 and Ca2+/calmodulin-dependent protein kinase II is mediated in large part by N-methyl-D-aspartate receptors, suggesting that cross talk between cyclin-dependent kinase 5 and Ca2+/calmodulin-dependent protein kinase II signal transduction pathways may be a component of the complex molecular mechanisms contributing to synaptic plasticity, memory, and learning[57]. When trained in the Morris water maze, p35 null mutants showed increased escape latencies and non-selective searching for the platform at the probe trial, indicating an absence of a spatial learning strategy[53]. To identify the role of cyclin-dependent kinase 5 activators, we examined the expression of p35 regulatory protein of cyclin-dependent kinase 5. Maternal alcohol consumption can stimulate cyclin-dependent kinase 5 activity by enhancing p35 expression. In this report, we found that maternal alcohol consumption prior to and during pregnancy resulted in a significant increase in the cyclin-dependent kinase 5 kinase activity in the hippocampus of offspring (P < 0.05).
Taken together, our results demonstrated that maternal alcohol consumption prior to and during pregnancy leads to modification of plasticity and impaired learning and memory in offspring rats. As cyclin-dependent kinase 5 activity was also significantly increased, these results strongly suggest that the increased expression of cyclin-dependent kinase 5 may be the mechanism responsible for impaired learning and memory in offspring exposed to alcohol in utero.
MATERIALS AND METHODS
Design
A randomized, controlled animal experiment.
Time and setting
This study was performed at the Neurobiology Laboratory, School of Basic Medical Sciences of Xinxiang Medical University in China from March 2011 to November 2012.
Materials
Eighteen healthy female and eighteen healthy male Sprague-Dawley rats of clean grade, aged 2 months, and weighing 200 ± 20 g were provided by the Animal Center, Zhengzhou University, China (license No. SCXK (Yu) 2005-0001). The ambient temperature was maintained at 22°C. Rats had free access to food (standard solid feedstuff produced by the Laboratory Animal Center, Xinxiang Medical University, China) and water. All experimental protocols were performed according to the Guidance Suggestions for the Care and Use of Laboratory Animals issued by the Ministry of Science and Technology of China[58].
Methods
Ethanol treatment
The control group was composed of females that were allowed free access to food and water, except in the morning when food was removed for about 2 hours prior to weighing. The isocaloric group was composed of females administered the same quantity of isocaloric and isovolumetric maltose-dextrin (Tianjiu (Shandong) Co., Ltd., China). The ethanol-treated group was composed of females that received a daily intragastric gavage of 25% (w/v) ethanol (anhydrous, Xingke High Purity Solvents (Shanghai) Co., Ltd., China) at a dose of 4.0 g/kg, beginning on E5 and continuing through the birth of the pups. To allow the stomach to empty and facilitate the absorption of the ethanol, all food was removed 4 hours before ethanol dosing. The ethanol was gavaged at the same time each day, as described previously[59]. Postnatal (P = postnatal days; P0 = the first 24 hours after birth) offspring were produced from timed pregnancies. At P14, three pups from each female in each group were randomly chosen and were used for the study.
Maternal alcohol consumption impaired the learning and memory of offspring by Y maze test
Learning and memory were evaluated by electro-stimulus Y-maze[60] in adult offspring. The Y-maze (MC-3, Zhenghua (Anhui) Biological Instrument Equipment Co., Ltd., China) had three equal arms (30 cm length × 15 cm width × 15 cm height) equipped with a grid floor consisting of stainless steel bars (4.5 mm diameter, 1.5 cm apart). The angle between every two arms was 120° and a sliding detachable door was used to separate each arm from the others. The floor of the Y-maze was connected to a voltage regulator. There was a signal light at the end of each arm. In each trial, when the light was turned on in an arm, it meant that the arm was safe. At the same time, a lack of light in the arms indicated the possibility of shock presentation. Based on the light signal, the animals learned to escape the shocks. The light was turned on randomly during the experiment. At the beginning of experiment, all lights were turned on and the rat was allowed to habituate to the surroundings for 3 minutes, which was followed by turning off all three lights. Then, only one light was randomly turned on. After a delay of 5 seconds, the bars of the other two arms were electrified and the rat could receive a footshock. The initial stimulus voltage was 20 V. If there was no response, the stimulus intensity was increased in steps of 5 V unit until a response was observed in the rat. The rat could then escape to a safe area and the light stayed on for 15 seconds. These procedures describe one complete trial. If the rat escaped to a safe area within 10 seconds, it was considered a correct response. Nine correct responses within ten trials was used as the criteria for learning. The training time required for the rat to exhibit learning was our dependent measure of spatial learning and memory. All the learning and memory tests were performed in the dark, in a quiet and controlled environment.
Measurement of the effects of maternal alcohol consumption on the expression of cyclin-dependent kinase 5 and p35 mRNA in the rat hippocampus by reverse transcription-PCR
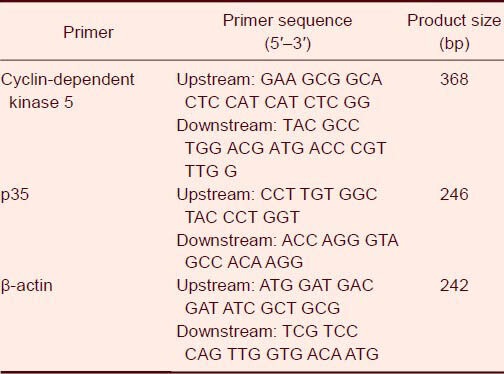
Total mRNA from brain tissues was extracted using Trizol reagent (Takara Biotechnology, Dalian, Liaoning Province, China). RNA 5 μg was reverse transcribed into cDNA using Avian Myeloblastosis Virus reverse transcriptase and oligo dT12 (Takara Biotechnology) as primers. The primer sequences and the size of the PCR products are shown in Table 2.
Table 2.
Cyclin-dependent kinase 5/p35 and β-actin primer sequences
Measurement of the effects of maternal alcohol consumption on the expression of cyclin-dependent kinase 5 and p35 mRNA in rat hippocampus by real-time quantitative PCR
RNA extraction and cDNA conversion: Total RNA was isolated from hippocampal tissues (frozen in liquid nitrogen) by a single-step method using TRIzol Reagent. The RNA samples were treated with RNase free DNase at 37°C for 20 minutes and stored at –80°C for further use. The purity (A260nm/A280nm) and the concentration of RNA were obtained using spectrophotometry. RNA quality was confirmed by gel electrophoresis. The starting material was consistently found to be of good quality. The total RNA (0.5–2.0 μg) was used for cDNA conversion using high capacity cDNA reverse transcription kit. The converted cDNA was stored at –20°C.
Primer design and synthesis: All primers were designed with Primer Premier 5.0 (Premier Inc., Toronto, Canada), based on target sequences obtained from the GenBank database (www.ncbi.nlm.nih.gov/gene) for each gene of interest. For the selection of an ideal primer pair, the factors considered included the melting temperature (60–65°C), GC content (40–60%), and having an amplicon length of about 90–200 bp. All new primers were tested by real-time PCR using a dilution (1 × 106 copies/μL) of plasmid and water as non-template control, followed by gel electrophoresis to determine the amplification efficiency, specificity, and the presence/absence of primer dimmers. Inefficient and/or non-specific primers were excluded. Reaction conditions for each primer pair were optimized to minimize non-specific products or primer dimmers and to maximize the yield of the desired amplicon. The sequence of cyclin-dependent kinase 5 primer is provided (Table 2).
Real-time quantitative PCR optimization: The reaction volume and recipe were performed according to the instructions of iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). Two-step amplification was adopted in the quantitative PCR reactions in which annealing and extension steps were combined as one step. A suitable annealing temperature is very important to generate a good amplifying curve. A serial of gradient annealing temperatures ranging from 65°C to 55°C was tested by real-time PCR using a template plasmid DNA (1 × 106 copies/μL). After analyzing the CT value, amplification efficiency and specificity of the reactions, the optimal annealing temperature was determined. PCR amplification efficiency (E) was calculated according to the following equation: E = 10(-1/slope)−1. With SYBR Green I dye method, amplification and data analysis were investigated in the fluorescence quantitative PCR instrument. With a minimum sample threshold cycle number (CT value) and the highest fluorescence value as the standard, the primer concentration, annealing temperature, and concentration of fluorescent dye premixed enzyme SYBR PreMix were optimized. Real-time quantitative PCR amplification and analysis were performed with software version 3.1 (FTC-2000, Funglyn Biotech Inc, Toronto, Canada). The annealing temperature of the quantitative PCR assay using the primers was optimized. All samples, including the standards and non-template control, were determined in duplicate. The cDNA samples were thawed and mixed with a magnesium chloride solution (vial 2) and primers by vortexing, followed by centrifuging at 4°C. All substances were kept on ice. A serial dilution (1:10, 1:100, and 1: 1 000) of the cDNA samples was prepared with nuclease-free or PCR-grade water. First, 1 μL cDNA was pipetted into a 0.5-mL reaction tube (autoclaved) and mixed with 9 μL PCR grade water. Then 1 μL diluted cDNA was added into a new reaction tube and mixed with 9 μL H2O. Then 10 μL vial 1a (LightCycler Fast-Start Enzyme) was added into vial 1b (LightCycler Fast-Start Reaction Mix SYBR Green I). A ready-to-use PCR mixture was obtained (10 × magnification), which contained FastStart Taq DNA polymerase, reaction buffer, dNTP mix, SYBR Green I dye, and magnesium chloride (10 mmol/L). The vial with SYBR Green I was protected from light because it is light sensitive. The ready-to-use PCR mixture was stored in the dark at 4°C for 1 month. Mixtures of all cDNA samples were prepared, including a negative control (H2O instead of cDNA) to reduce pipette inaccuracy. Each cDNA sample was measured at least in duplicate. All components were mixed by pipette several times. LightCycler capillaries were precooled for each cDNA sample, including a negative control (H2O instead of cDNA) in the LightCycler centrifuge adapters. A 18 μL mixture was pipetted into each precooled LightCycler capillary. The same pipette was used for the same primer pair. 2 μL of each cDNA was added into a separate capillary, which was sealed with a stopper. Samples were centrifuged at 700 × g for 5 seconds, or briefly at 5 000 r/min. The capillaries were transferred carefully into the sample carousel of the LightCycler instrument and PCR was performed.
Standard curve drawing: The prepared plasmids were treated with a 10-fold serial dilution, and samples at a gradient concentration of 10–2 to 10–1 were used as the standard. The standard curve was obtained in a real-time fluorescence quantitative PCR reaction, while negative temperature coefficient was also set up simultaneously.
Evaluation of relative gene expression: The 2−ΔΔCt method was used to analyze the results. The CT values provided from real-time PCR instrumentation were easily imported into the spreadsheet program Microsoft Excel. The change in expression of the cyclin-dependent kinase 5 and p35 target gene normalized to β-actin was monitored over 8 hours. Real-time PCR was performed on the corresponding cDNA synthesized from each sample. The data were analyzed using the formula ΔΔCT= (CT,Target−CT,Actin)Time x−(CT,Target−CT,Actin)Time 0. In this formula, Timex is any time point and Time 0 represents the 1 × expression of the target gene normalized to β-actin. The mean CT values for both the target and internal control genes were determined at time zero and were used in the formula. The fold change in the target gene, normalized to β-actin and relative to the expression at time 0, was calculated for each sample using the formula.
Measurement of the effects of maternal alcohol consumption on the expression of cyclin-dependent kinase 5 and p35 protein in rat hippocampus by immunofluorescence
Sections were stained with hematoxylin-eosin or with reticulin stain, and imaged on a Leica DMRXA microscope. For immunofluorescence analysis, animal brains were then frozen in isopentane, which was cooled by dry ice. Coronal sections (8-μm thickness) were then obtained on a cryostat. Sections were fixed with 2% paraformaldehyde for 10 minutes and blocked with 0.3% Triton-X 100 (Takara Biotechnology) in PBS and 2% bovine serum albumin for 30 minutes. Rabbit anti-cyclin-dependent kinase 5 polyclonal antibody (Boster Biotechnology, Wuhan, China) or rabbit anti-p35 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was incubated overnight. After incubation, sections were washed with PBS. The Immunol Fluorence Staining Kit with Alexa Fluor 488-Labeled goat anti-rabbit IgG (Beyotime Institute of Biotechnology, Beijing, China) or Cy3-labeled goat anti-rabbit IgG (Beyotime Institute of Biotechnology) incubation was performed. A BX41 microscope (Olympus, Tokyo, Japan) was used to acquire the images.
Measurement of the effects of maternal alcohol consumption on cyclin-dependent kinase 5 activity in rat hippocampus by immunoprecipitation
Cyclin-dependent kinase 5 activity was assayed as previously described[61]. Briefly, hippocampal tissues were prepared in RIPA buffer (Takara Biotechnology; including 150 mmol/L NaCl, 20 mmol/L Tris-HCl, 1 mmol/L EDTA, 0.5% Nonidet P-40, 5 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride, 2.5 mmol/L benzamidine, 20 μg/mL antipain, 20 μg/mL leupeptin, 5 μg/mL chymostatin, 5 μg/mL pepstatin, 50 mmol/L NaF, and 5 mmol/L Na3VO4 at pH 7.4) containing a protease inhibitor tablet and phosphatase inhibitor cocktails I and II (Sigma). Protein lysates (Takara Biotechnology; 500 μg) were dissolved in lysis buffer to achieve a 1 μg/μL concentration and then precleared with normal rabbit IgG followed with 50–100 μL of 50% protein A–agarose slurry prepared in lysis buffer. These lysates were then incubated overnight at 4°C with 0.01 μg/μL of anti-cyclin-dependent kinase 5 IgG (Sigma). The next day, lysates were incubated with 50 μL of a 50% protein A–agarose slurry for 3 hours at 4°C. Immunoprecipitates were washed three times with lysis buffer followed with kinase buffer (20 mmol/L Tris-HCl, pH 7.4, 10 mmol/L MgCl2, 1 mmol/L ethylenediaminetetraactic acid (EDTA), 10 μmol/L NaF, and 1 μmol/L Na2VO3) and resuspended in 10 μL of 5 × kinase assay mixture (100 mmol/L Tris-HCl, pH 7.4, 50 mmol/L MgCl2, 5 mmol/L EDTA, 50 μmol/L NaF, 5 μmol/L Na2VO3, and 5 mmol/L DTT), 30 μL of water, and 20 μmol/L of the immunoprecipitated coronin 1a. Samples were maintained at 30°C for 60 minutes after adding 5 μCi γ-[32P] adenosine triphosphate (Sigma; 0.5 mmol/L), and the reaction was stopped by adding 10% trichloroacetic acid in peptide kinase assay buffer and by adding sample buffer (2% SDS, 10% glycerol, 80 mmol/L Tris, pH 6.8, and 1 mmol/L dithiothreitol) and boiling for 10 minutes, for the coronin 1a kinase reaction. This step was repeated one more time, and two supernatant fractions were combined. The final mixture was incubated with 0.08 μg/μL histone H1 protein and 0.2 μCi/μL [γ-32P] adenosine triphosphate at 30°C for 30 minutes. Radioactivity was measured with a scintillation counter (PerkinElmer, Waltham, MA, USA) and normalized to basal activity.
Statistical analysis
All data are expressed as mean ± SD of three replicated determinations, and analyzed with SPSS 11.5 software (SPSS, Chicago, IL, USA). One-way analysis of variance and least significant difference multiple-range tests were used to determine statistically significant differences between means. Differences at a probability of P < 0.05 level were considered to be significant.
Footnotes
Shuang Li, Master, Lecturer.
Funding: This project was supported by the National Nature Science Foundation of China, No. 81171261; a grant from Henan Science Technology Committee of China, No. 112300410162; Fund for Talents with Innovation in Medical Science and Technology of Henan Province of China, No. 3052.
Conflicts of interest: None declared.
Ethical approval: Experimental protocols were approved by the Animal Ethics Committee of Xinxiang Medical University, China.
(Reviewed by Maxwell R, Norman C, Li WL, Li DL)
(Edited by Wang J, Yang Y, Li CH, Song LP)
REFERENCES
- [1].Sobrian SK, Holson RR. Social behavior of offspring following prenatal cocaine exposure in rodents: a comparison with prenatal alcohol. Front Psychiatry. 2011;2:1–17. doi: 10.3389/fpsyt.2011.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Popova NK, Morozova MV, Amstislavskaya TG. Prenatal stress and ethanol exposure produces inversion of sexual partner preference in mice. Neurosci Lett. 2011;489(1):48–52. doi: 10.1016/j.neulet.2010.11.064. [DOI] [PubMed] [Google Scholar]
- [3].Piper BJ, Corbett SM. Executive function profile in the offspring of women that smoked during pregnancy. Nicotine Tob Res. 2012;14(2):191–199. doi: 10.1093/ntr/ntr181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fagerlund A, Autti-Rämö I, Hoyme HE, et al. Risk factors for behavioural problems in foetal alcohol spectrum disorders. Acta Paediatr. 2011;100(11):1481–1488. doi: 10.1111/j.1651-2227.2011.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alfonso-Loeches S, Guerri C. Molecular and behavioral aspects of the actions of alcohol on the adult and developing brain. Crit Rev Clin Lab Sci. 2011;48(1):19–47. doi: 10.3109/10408363.2011.580567. [DOI] [PubMed] [Google Scholar]
- [6].Naseer MI, Ullah N, Ullah I, et al. Vitamin C protects against ethanol and PTZ-induced apoptotic neurodegeneration in prenatal rat hippocampal neurons. Synapse. 2011;65(7):562–571. doi: 10.1002/syn.20875. [DOI] [PubMed] [Google Scholar]
- [7].Gil-Mohapel J, Boehme F, Patten A, et al. Altered adult hippocampal neuronal maturation in a rat model of fetal alcohol syndrome. Brain Res. 2011;1384:29–41. doi: 10.1016/j.brainres.2011.01.116. [DOI] [PubMed] [Google Scholar]
- [8].Medina AE. Fetal alcohol spectrum disorders and abnormal neuronal plasticity. Neuroscientist. 2011;17(3):274–287. doi: 10.1177/1073858410383336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012;22(3):169–175. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- [10].Contreras-Vallejos E, Utreras E, Gonzalez-Billault C. Going out of the brain: non-nervous system physiological and pathological functions of Cdk5. Cell Signal. 2012;24(1):44–52. doi: 10.1016/j.cellsig.2011.08.022. [DOI] [PubMed] [Google Scholar]
- [11].Lopes JP, Agostinho P. Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol. 2011;94(1):49–63. doi: 10.1016/j.pneurobio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- [12].Drerup JM, Hayashi K, Cui H, et al. Attention-deficit/hyperactivity phenotype in mice lacking the cyclin-dependent kinase 5 cofactor p35. Biol Psychiatry. 2010;68(12):1163–1171. doi: 10.1016/j.biopsych.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang P, Yu PC, Tsang AH, et al. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J Neurosci. 2010;30(43):14366–14370. doi: 10.1523/JNEUROSCI.3899-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ou CY, Poon VY, Maeder CI, et al. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell. 2010;41(5):846–858. doi: 10.1016/j.cell.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Robinson BG, Khurana S, Kuperman A, et al. Neural Adaptation Leads to Cognitive Ethanol Dependence. Curr Biol. 2012;22(24):2338–2341. doi: 10.1016/j.cub.2012.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Robinson BG, Khurana S, Pohl JB, et al. A low concentration of ethanol impairs learning but not motor and sensory behavior in Drosophila larvae. PLoS One. 2012;7(5):e37394. doi: 10.1371/journal.pone.0037394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Parry CD, Patra J, Rehm J. Alcohol consumption and non-communicable diseases: epidemiology and policy implications. Addiction. 2011;106(10):1718–1724. doi: 10.1111/j.1360-0443.2011.03605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ramadoss J, Magness RR. Vascular effects of maternal alcohol consumption. Am J Physiol Heart Circ Physiol. 2012;303(4):H414–421. doi: 10.1152/ajpheart.00127.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bosco C, Diaz E. Placental hypoxia and fetal development versus alcohol exposure in pregnancy. Alcohol Alcohol. 2012;47(2):109–117. doi: 10.1093/alcalc/agr166. [DOI] [PubMed] [Google Scholar]
- [20].Foltran F, Gregori D, Franchin L, et al. Effect of alcohol consumption in prenatal life, childhood, and adolescence on child development. Nutr Rev. 2011;69(11):642–659. doi: 10.1111/j.1753-4887.2011.00417.x. [DOI] [PubMed] [Google Scholar]
- [21].Jégou S, El Ghazi F, de Lendeu PK, et al. Prenatal alcohol exposure affects vasculature development in the neonatal brain. Ann Neurol. 2012;72(6):952–960. doi: 10.1002/ana.23699. [DOI] [PubMed] [Google Scholar]
- [22].Caroni P, Donato F, Muller D. Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci. 2012;13(7):478–490. doi: 10.1038/nrn3258. [DOI] [PubMed] [Google Scholar]
- [23].Bavelier D, Green CS, Pouget A, et al. Brain plasticity through the life span: learning to learn and action video games. Annu Rev Neurosci. 2012;35:391–416. doi: 10.1146/annurev-neuro-060909-152832. [DOI] [PubMed] [Google Scholar]
- [24].Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100(5):433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- [25].Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33(1):18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- [26].Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9(1):65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- [27].Gruart A, Muñoz MD, Delgado-García JM. Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J Neurosci. 2006;26(4):1077–1087. doi: 10.1523/JNEUROSCI.2834-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Whitlock JR, Heynen AJ, Shuler MG, et al. Learning induces long-term potentiation in the hippocampus. Science. 2006;313(5790):1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- [29].Rumpel S, LeDoux J, Zador A, et al. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308(5718):83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- [30].Hsu JC, Cheng SJ, Yang HW, et al. Bidirectional synaptic plasticity induced by conditioned stimulations with different number of pulse at hippocampal CA1 synapses: roles of N-methyl-D-aspartate and metabotropic glutamate receptors. Synapse. 2011;65(8):795–803. doi: 10.1002/syn.20906. [DOI] [PubMed] [Google Scholar]
- [31].Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84(1):87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- [32].Wang H, Megill A, He K, et al. Consequences of inhibiting amyloid precursor protein processing enzymes on synaptic function and plasticity. Neural Plast 2012. 2012 doi: 10.1155/2012/272374. 272374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Morris RG. Elements of a neurobiological theory of hippocampal function: the role of synaptic plasticity, synaptic tagging and schemas. Eur J Neurosci. 2006;23(11):2829–2846. doi: 10.1111/j.1460-9568.2006.04888.x. [DOI] [PubMed] [Google Scholar]
- [34].Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87(7):1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- [35].Pan B, Wang W, Zhong P, et al. Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci. 2011;31(38):13420–13430. doi: 10.1523/JNEUROSCI.2075-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Coyle JT, Tsai G. NMDA receptor function, neuroplasticity, and the pathophysiology of schizophrenia. Int Rev Neurobiol. 2004;59:491–515. doi: 10.1016/S0074-7742(04)59019-0. [DOI] [PubMed] [Google Scholar]
- [37].Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010;83(3-4):108–121. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Martin S, Jones M, Simpson E, et al. Impaired spatial reference memory in aromatase-deficient (ArKO) mice. Neuroreport. 2003;14(15):1979–1982. doi: 10.1097/00001756-200310270-00020. [DOI] [PubMed] [Google Scholar]
- [39].Monk BR, Leslie FM, Thomas JD. The effects of perinatal choline supplementation on hippocampal cholinergic development in rats exposed to alcohol during the brain growth spurt. Hippocampus. 2012;22(8):1750–1757. doi: 10.1002/hipo.22009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Richardson DP, Byrnes ML, Brien JF, et al. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur J Neurosci. 2002;16(8):1593–1598. doi: 10.1046/j.1460-9568.2002.02214.x. [DOI] [PubMed] [Google Scholar]
- [41].Yang J, Hou C, Ma N, et al. Enriched environment treatment restores impaired hippocampal synaptic plasticity and cognitive deficits induced by prenatal chronic stress. Neurobiol Learn Mem. 2007;87(2):257–263. doi: 10.1016/j.nlm.2006.09.001. [DOI] [PubMed] [Google Scholar]
- [42].Liu J, Kipreos ET. Evolution of cyclin-dependent kinases (CDKs) and CDK-activating kinases (CAKs): differential conservation of CAKs in yeast and metazoa. Mol Biol Evol. 2000;17(7):1061–1074. doi: 10.1093/oxfordjournals.molbev.a026387. [DOI] [PubMed] [Google Scholar]
- [43].Kino T, Jaffe H, Amin ND, et al. Cyclin-dependent kinase 5 modulates the transcriptional activity of the mineralocorticoid receptor and regulates expression of brain-derived neurotrophic factor. Mol Endocrinol. 2010;24(5):941–952. doi: 10.1210/me.2009-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhu J, Li W, Mao Z. Cdk5: mediator of neuronal development, death and the response to DNA damage. Mech Ageing Dev. 2011;132(8-9):389–394. doi: 10.1016/j.mad.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cruz JC, Tseng HC, Goldman JA, et al. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40(3):471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- [46].Tsai LH, Delalle I, Caviness VS, Jr, et al. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371(6496):419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- [47].Hawasli AH, Benavides DR, Nguyen C, et al. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10(7):880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Paglini G, Cáceres A. The role of the Cdk5-p35 kinase in neuronal development. Eur J Biochem. 2001;268(6):1528–1533. [PubMed] [Google Scholar]
- [49].Kesavapany S, Li BS, Amin N, et al. Neuronal cyclin-dependent kinase 5: role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochim Biophys Acta. 2004;1697(1-2):143–153. doi: 10.1016/j.bbapap.2003.11.020. [DOI] [PubMed] [Google Scholar]
- [50].Bibb JA. Role of Cdk5 in neuronal signaling, plasticity, and drug abuse. Neurosignals. 2003;12(4-5):191–199. doi: 10.1159/000074620. [DOI] [PubMed] [Google Scholar]
- [51].Barnett DG, Bibb JA. The role of Cdk5 in cognition and neuropsychiatric and neurological pathology. Brain Res Bull. 2011;85(1-2):9–13. doi: 10.1016/j.brainresbull.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hawasli AH, Bibb JA. Alternative roles for Cdk5 in learning and synaptic plasticity. Biotechnol J. 2007;2:941–948. doi: 10.1002/biot.200700093. [DOI] [PubMed] [Google Scholar]
- [53].Ohshima T, Ogura H, Tomizawa K, et al. Impairment of hippocampal long-term depression and defective spatial learning and memory in p35 mice. J Neurochem. 2005;94(4):917–925. doi: 10.1111/j.1471-4159.2005.03233.x. [DOI] [PubMed] [Google Scholar]
- [54].Angelo M, Plattner F, Giese KP. Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J Neurochem. 2006;99(2):353–370. doi: 10.1111/j.1471-4159.2006.04040.x. [DOI] [PubMed] [Google Scholar]
- [55].Odajima J, Wills ZP, Ndassa YM, et al. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell. 2011;21(4):655–668. doi: 10.1016/j.devcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Vahidnia A, van der Straaten RJ, Romijn F, et al. Mechanism of arsenic-induced neurotoxicity may be explained through cleavage of p35 to p25 by calpain. Toxicol In Vitro. 2008;22:682–687. doi: 10.1016/j.tiv.2007.12.010. [DOI] [PubMed] [Google Scholar]
- [57].Miao Y, Dong LD, Chen J, et al. Involvement of calpain/p35-p25/Cdk5/NMDAR signaling pathway in glutamate-induced neurotoxicity in cultured rat retinal neurons. PLoS One. 2012;7(8):e42318. doi: 10.1371/journal.pone.0042318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006 Sep 30; [Google Scholar]
- [59].Deng JX, Liu X, Zang JF, et al. The effects of prenatal alcohol exposure on the developmental retina of mice. Alcohol Alcohol. 2012;47(4):380–385. doi: 10.1093/alcalc/ags025. [DOI] [PubMed] [Google Scholar]
- [60].Ma MX, Chen YM, He J, et al. Effects of morphine and its withdrawal on Y-maze spatial recognition memory in mice. Neuroscience. 2007;147(4):1059–1065. doi: 10.1016/j.neuroscience.2007.05.020. [DOI] [PubMed] [Google Scholar]
- [61].Chen PC, Chen JC. Enhanced Cdk5 activity and p35 translocation in the ventral striatum of acute and chronic methamphetamine-treated rats. Neuropsychopharmacology. 2005;30(3):538–549. doi: 10.1038/sj.npp.1300604. [DOI] [PubMed] [Google Scholar]