

Abstract
Penehyclidine hydrochloride can promote microcirculation and reduce vascular permeability. However, the role of penehyclidine hydrochloride in cerebral ischemia-reperfusion injury remains unclear. In this study, in vivo middle cerebral artery occlusion models were established in experimental rats, and penehyclidine hydrochloride pretreatment was given via intravenous injection prior to model establishment. Tetrazolium chloride, terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling and immunohistochemical staining showed that, penehyclidine hydrochloride pretreatment markedly attenuated neuronal histopathological changes in the cortex, hippocampus and striatum, reduced infarction size, increased the expression level of Bcl-2, decreased the expression level of caspase-3, and inhibited neuronal apoptosis in rats with cerebral ischemia-reperfusion injury. Xanthine oxidase and thiobarbituric acid chromogenic results showed that penehyclidine hydrochloride upregulated the activity of superoxide dismutase and downregulated the concentration of malondialdehyde in the ischemic cerebral cortex and hippocampus, as well as reduced the concentration of extracellular excitatory amino acids in rats with cerebral ischemia-reperfusion injury. In addition, penehyclidine hydrochloride inhibited the expression level of the NR1 subunit in hippocampal nerve cells in vitro following oxygen-glucose deprivation, as detected by PCR. Experimental findings indicate that penehyclidine hydrochloride attenuates neuronal apoptosis and oxidative stress injury after focal cerebral ischemia-reperfusion, thus exerting a neuroprotective effect.
Keywords: neural regeneration, brain injury, penehyclidine hydrochloride, cerebral ischemia-reperfusion injury, ischemic cerebrovascular disease, apoptosis, excitatory amino acid, oxygen free radicals, superoxide dismutase, N-methyl-D-aspartate receptor, middle cerebral artery occlusion, oxygen-glucose deprivation, photographs-containing paper, neuroregeneration
Research Highlights
(1) Penehyclidine hydrochloride has been used as a novel selective anticholine drug for the clinical treatment of organophosphate poisoning.
(2) In this study, in vitro experiments were designed to explore the effect of penehyclidine hydrochloride on N-methyl-D-aspartate receptors in hippocampal neurons of rats after oxygen-glucose deprivation, and to observe the mechanism of action underlying this drug on excitatory amino acid receptors.
(3) Our results found that penehyclidine hydrochloride could attenuate neuronal apoptosis and oxidative stress damage following focal cerebral ischemia-reperfusion.
INTRODUCTION
Stroke is one of the most common fatal diseases all over the world, and ischemic stroke accounts for more than 80% of cases. The treatment for ischemic injury is to restore blood flow as soon as possible. However, recent studies show that reperfusion may aggravate ischemic injury and induce death of brain cells, a process termed cerebral ischemia-reperfusion injury. Cerebral ischemia-reperfusion injury may further deteriorate cerebral function and reduce the therapeutic effect of drugs. Therefore, treatments for cerebral ischemia- reperfusion injury have attracted increasing attention and become a major focus of cerebral ischemia research. Excitatory amino acid toxicity, oxidative stress, intracellular calcium overload, as well as inflammation and apoptosis, are involved in the pathological process after cerebral ischemia-reperfusion injury[1].
Although a large number of pre-clinical and clinical studies have been performed, the majority of drugs have failed in clinical trials at different stages[2]. Because previous stroke trials have demonstrated that drugs that target one or several signal transduction pathways cannot improve clinical outcomes after stroke, multi-pathway drugs have been suggested to overcome this challenge[3]. In recent years, many narcotics have been used clinically to reduce damage caused by ischemia-reperfusion[4,5,6], such as scopolamine, which is developed from traditional Chinese medicinal plants, and plays a major role in blocking the muscarinic acetylcholine receptor. Moreover, scopolamine has a variety of biological activities, including antioxidant and cytoprotective actions.
Penehyclidine hydrochloride or 3-(2′-phenyl-2′-cyclopentyl-2′-hydroxyl-ethoxy), is a new anti-cholinergic drug derived from scopolamine, having both anti-muscarinic and anti-nicotinic activities, and retains effective central and peripheral anticholinergic activities. Clinical data showed that penehyclidine hydrochloride is effective in treatment for soman poisoning and pulmonary dysfunction of chronic obstructive pulmonary disease[7,8]. More importantly, penehyclidine hydrochloride can improve microcirculation, depress microvascular permeability, attenuate the release of lysome and inhibit lipid peroxidation, and it is also reported to have protective effects against organ injury[9,10,11], and is used widely in the clinic as an antagonist of organic phosphorus poisoning at present in China[12,13]. However, the role and mechanism associated with penehyclidine hydrochloride in cerebral ischemia-reperfusion injury is not clear.
This study was performed to explore the protective effects and the possible mechanisms of penehyclidine hydrochloride on neuronal injury in rats following focal cerebral ischemia-reperfusion, in a broader attempt to guide further studies on ischemic prevention and treatment.
RESULTS
Quantitative analysis of experimental animals
A total of 24 Sprague-Dawley male rats were divided randomly into four groups with six rats in each: sham-surgery group (only the right middle cerebral artery was isolated but not occluded), model group (middle cerebral artery occlusion + intravenous injection of normal saline 20 minutes before surgery), penehyclidine hydrochloride group (middle cerebral artery occlusion + intravenous injection of penehyclidine hydrochloride 20 minutes before surgery), and scopolamine group (middle cerebral artery occlusion + intravenous injection of scopolamine 20 minutes before surgery), with 24-hour reperfusion after middle cerebral artery occlusion for 2 hours. All 24 rats were included in the final analysis without any loss.
Effect of penehyclidine hydrochloride on infarction volume and morphological changes in neuronal cells induced by cerebral ischemia-reperfusion injury
Brain tissues were sliced at a section thickness of 2 mm and were incubated in 2%(v/v) of 2,3,5-triphenyl-tetrazolium chloride solution. Intact mitochondria were stained with 2,3,5-triphenyl-tetrazolium chloride, the dark red area represented the living tissue, whereas infarction areas that in the parietotemporal cortex, hippocampus and striatum remained unstained (gray)[14].
As shown in Figure 1A, the volume of the gray region (infarcted tissue) was significantly reduced in the penehyclidine hydrochloride group compared with the other groups. As shown in Figure 1B, the model group showed obvious vacuoles, ampliative gaps between neurons, and more dead neurons, while the penehyclidine hydrochloride group showed a greater number of surviving neurons and less cell plasma condensation, as well as less dead neurons, as revealed by hematoxylin-eosin staining (Figure 1).
Figure 1.
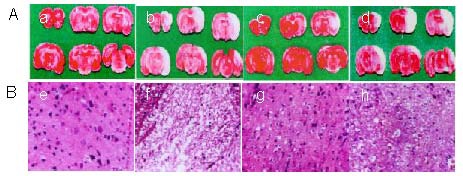
Effects of penehyclidine hydrochloride (PHC) on infarction volume and morphological characterization in neuronal cells (light microscope, × 200).
After 2-hour ischemia and 24-hour reperfusion, Sprague-Dawley rats were randomly divided into four groups according to the interventions: sham-surgery group (a, e), model group (b, f), PHC group (c, g) and scopolamine group (d, h).
(A) 2,3,5-triphenyl- tetrazolium chloride (MTT) stained brain slices. Normal brain tissue is red and the infarct area is gray following MTT staining. The infarct mainly appears in the top temporal cortex, hippocampus and striatum. The gray volume is smaller in the PHC group than that in other injury groups.
(B) Using hematoxylin-eosin staining, PHC could reduce the number of dead neuronal cells. Model group showed obvious vacuoles, ampliative gaps between neurons, and more dead neurons, while the PHC group showed a greater number of surviving neurons, less cell plasma condensation, and less neuron death.
Penehyclidine hydrochloride inhibited neuronal apoptosis in rats with cerebral ischemia-reperfusion injury
The number of positive cells was evaluated by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) and immunohistochemistry. Compared with the model group, the number of TUNEL-positive cells was reduced in the penehyclidine hydrochloride group (P < 0.001), which indicated that penehyclidine hydrochloride could protect nerve cells against focal cerebral ischemia-reperfusion injury, and the effect was associated with its anti-apoptotic action (Figure 2A). Immunohistochemical staining results showed that Bcl-2 expression in the cytoplasm was higher in the penehyclidine hydrochloride group than that in the model group (P < 0.001), while that of caspase-3 expression decreased (P < 0.001; Figure 2B). This result is evidence that penehyclidine hydrochloride can prevent apoptosis through the regulation of apoptosis-related genes, which is one of the possible neuroprotective mechanisms underlying penehyclidine hydrochloride following focal cerebral ischemia-reperfusion.
Figure 2.
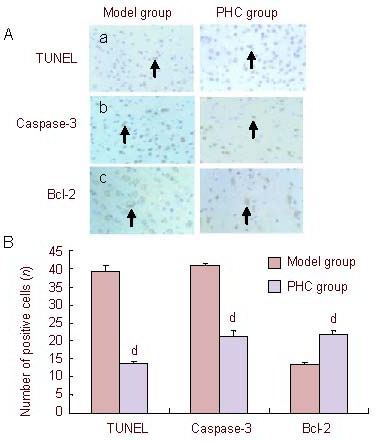
Effects of penehyclidine hydrochloride (PHC) on cell apoptosis in cerebral ischemia-reperfusion injury rats (TUNEL staining).
(A) Cell apoptosis in the ischemic area (× 400). Compared with the model group, the number of TUNEL-positive cells (arrows) was reduced in the PHC group. Immunohistochemical staining showed that Bcl-2 expression (arrows) in the cytoplasm was higher in the PHC group than that in the model group, while the expression of caspase-3 (arrows) was decreased. (a) TUNEL staining; (b) immunohistochemical staining of caspase-3; (c) immunohistochemical staining of Bcl-2.
(B) Quantification of apoptotic cells in the ischemic area. Data are expressed as mean ± SD (n = 6; one-way analysis of variance followed by least significant difference test). dP < 0.001, vs. model group.
TUNEL: Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling.
Effect of penehyclidine hydrochloride on the expression of superoxide dismutase, malondialdehyde and excitatory amino acids in rats with cerebral ischemia-reperfusion injury
The change of oxygen radicals and excitatory amino acids in brain tissue was determined to investigate whether penehyclidine hydrochloride could play a protective role in neurons by influencing the content of oxygen free radicals and excitatory amino acids. Compared with the model group, penehyclidine hydrochloride was shown to upregulate the sensitivity of superoxide dismutase and downregulate the concentration of malondialdehyde in ischemic cerebral tissue. In addition, glutamic acid, aspartate, glycine and gamma-aminobutyric acid in brain tissue decreased in the penehyclidine hydrochloride group compared with the model group (P < 0.05; Figure 3).
Figure 3.
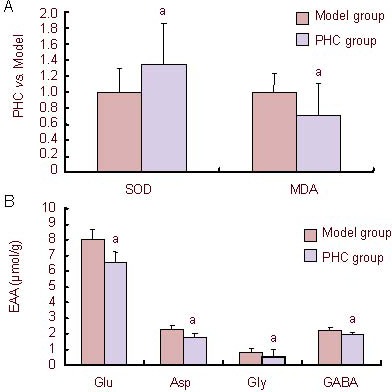
Effect of penehyclidine hydrochloride (PHC) on the concentration of superoxide dismutase (SOD), malondialdehyde (MDA) and excitatory amino acids (EAA) in ischemic cerebral tissue.
PHC could upregulate the sensitivity of SOD and downregulate the content of MDA and EAA in ischemic cerebral tissue. In this study, EAA was detected as glutamic acid (Glu), aspartate (Asp), glycine (Gly) and gamma-aminobutyric acid (GABA).
(A) Compared with the model group, PHC upregulated the sensitivity of SOD and downregulated the concentration of MDA in ischemic cerebral tissue.
(B) Glu, Asp, Gly and GABA in brain tissue in the PHC groups were decreased compared with the model group.
Data are expressed as mean ± SD (n = 6; one-way analysis of variance followed by least significant difference test). aP < 0.05, vs. model group.
Effects of penehyclidine hydrochloride on the expression of NR1 mRNA in cultured hippocampal cells
By measuring the expression of N-methyl-D-aspartate receptor NR1 subunit mRNA in hippocampal cells after oxygen-glucose deprivation and reperfusion, we explored the role of penehyclidine hydrochloride at different time points.
After incubation without oxygen and glucose for 2.5 hours, the cells cultured under normal conditions were randomly divided into six groups: normal control group (normal Hank's buffer), oxygen-glucose deprivation/reperfusion model group, high-dose penehyclidine hydrochloride group (modeling + 1.6 μM penehyclidine hydrochloride), middle-dose penehyclidine hydrochloride group (modeling + 0.4 μM penehyclidine hydrochloride), low-dose penehyclidine hydrochloride group (modeling + 0.1 μM penehyclidine hydrochloride) and MK-801- positive group (modeling + 0.1 μM MK-801), followed by reperfusion for 1, 6, 12, 24 hours. Reverse transcription- PCR was applied to detect NR1 mRNA expression and results showed that except at the 1-hour reperfusion injury period, penehyclidine hydrochloride at each concentration in this study could decrease the expression of the NR1 subunit at the other reperfusion injury times, and with the longer times, the effects were more pronounced (Figure 4). In the MK-801-positive control group, mRNA expression was stably maintained.
Figure 4.
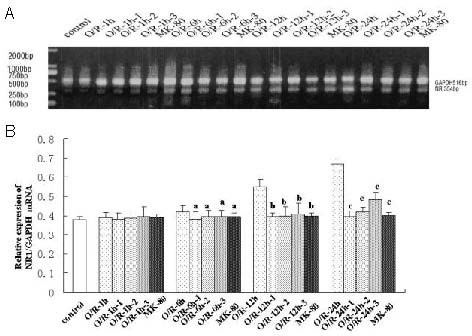
Effect of penehyclidine hydrochloride (PHC) on the expression of NR1 mRNA in hippocampal cells.
1: High-dose PHC group (1.6 μM); 2: middle-dose PHC group (0.4 μM); 3: low-dose PHC group (0.1 μM).
Compared with the O/R model group, except at the 1-hour reperfusion injury period, PHC at each concentration in the study could reduce NR1 subunit expression to varying degrees at different reperfusion injury time. Data are expressed as mean ± SD (n = 3; one-way analysis of variance followed by least significant difference test). aP < 0.05, vs. O/R-6 h group; bP < 0.05, vs. O/R-12 h group; cP < 0.05, vs. O/R-24 h group.
O/R: Oxygen-glucose deprivation and reperfusion; h: hour; GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
DISCUSSION
At present, clinical measures to protect neurons from ischemic damage mainly focus on the following aspects, such as addressing the energy depletion, acidosis, excessive oxygen free radical production, increase in intracellular calcium, enhanced excitatory amino acids and inhibitory amino acids, changes in cytokines, rising levels of inflammatory cytokines and upregulation of apoptosis-related genes. However, the mechanism of damage is unclear and requires further study.
Penehyclidine hydrochloride is a new anti-cholinergic drug derived from hyoscyamine, which has no or poor M2 receptor-associated cardiovascular side effects owing to selectively blocking M1, M3 receptors and N receptors. Moreover, penehyclidine hydrochloride has been reported to have a good protective effect against both soman-induced seizures, especially over the non-cholinergic phase, and against pentylenetetrazo- induced convulsions and N-methyl-D-aspartate-induced mortality in mice[8,15]. It has been well-established that increased acetylcholinesterase expression or activity is detected in apoptotic cells after apoptotic stimuli in vitro and in vivo[16]. In addition, elevated acetylcholinesterase activity was reported after middle cerebral arterial occlusion[17]. In this study, penehyclidine hydrochloride was able to protect against apoptosis, and we propose that penehyclidine hydrochloride may have anti-apoptotic properties similar to acetylcholinesterase inhibitors in the treatment of middle cerebral arterial occlusion injury.
Cell apoptosis, also known as programmed cell death, has obvious morphological and biochemical characteristics. Programmed cell death is carried out in a regulated process and leads to characteristic cell changes and death. These changes include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation. Programmed cell death has fundamental functions during both plant and multicellular animal tissue development. Inhibiting apoptosis has been confirmed to be the most effective way to delayed neuronal death after cerebral ischemia reperfusion[18,19]. Neuronal apoptosis has been divided into caspase-dependent and caspase-independent processed and mitochondria play an important role in both pathways[20]. After cerebral ischemia-reperfusion, a series of transformations may increase the permeability of the inner mitochondrial membrane, resulting in failure of energy metabolism, calcium imbalance, mitochondrial transmembrane potential change and the generation of oxygen free radicals. Apoptosis initiation factors are released from the mitochondrion, which triggers the cell death cascade[21,22]. Despite the increased apoptosis following middle cerebral arterial occlusion, the importance and regulatory mechanisms of apoptosis have not been fully elucidated. Of the apoptosis-related gene products, the caspase family plays a significant role in the incidence of apoptosis, which mediate apoptosis involved in two important pathways: the extrinsic pathway and the intrinsic pathway. Caspase-3, a terminal effector protein, plays a crucial role in the occurrence of apoptosis mediated by the above two pathways, and causes a subsequent cascade that ultimately leads to apoptosis[20,23]. The role of Bcl-2 in the progress of apoptosis has been evaluated in several in vivo and in vitro systems[24,25]. The Bcl-2 oncogene inhibits apoptosis by affecting several cell pathways. One possibility is that Bcl-2 plays a role in desensitizing thymocytes to apoptosis. In contrast, activation of caspase-3 results in apoptosis via chromosome condensation, cell membrane blebbing and DNA fragmentation[26]. Caspase-3 can be activated by cell death signals via three ways: mitochondria (related to bax and Bcl-2), death receptors (FAS, FAS-L), or the endoplasmic reticulum[27,28]. Furthermore, caspase-3 can cleave Bcl-2 protein in vivo, which promotes the release of cytochrome C and activation of caspase-3 to strengthen the apoptotic effect as a positive feedback loop[29,30,31].
The excitatory amino acid theory has received much attention in recent years. Excitatory amino acids, such as glutamate, are important and ubiquitous excitatory neurotransmitters in the mammalian central nervous system and are involved in the mediation of physiological functions such as learning, memory, synaptic plasticity and cardiovascular regulation[32,33,34]. Moreover, excitatory amino acid receptors are known to mediate synaptic excitation throughout the central nervous system. The role of glutamatergic neurotransmission in the central regulation of cardiovascular function is emerging. Excessive accumulation of excitatory amino acids can trigger cerebral ischemic injury and play an important role in the pathophysiology of cerebral ischemia[35,36,37,38]. In addition, excitatory amino acids may activate intracellular signal transduction pathways and trigger inflammatory gene expression second to the ischemia. Therefore, regulating excitatory amino acid levels is the initial goal for the treatment of cerebral ischemia. Recent studies indicate that cerebral ischemia-reperfusion injury of brain cells can trigger damage involving at least four different mechanisms: energy failure, excitotoxicity, peri-infarct depolarization, and inflammation and programmed cell death. This ischemic injury cascade may be initiated by energy barriers and excitatory amino acids[38,39,40,41,42]. In this study, penehyclidine hydrochloride reduced the concentration of extracellular excitatory amino acids during cerebral ischemia-reperfusion injury in brain cells.
The endogenous excitatory amino acids mediate their excitatory actions via multiple synaptic transmitter receptors. According to their selectivity for different agonists, amino acid receptors can generally be divided into N-methyl-D-aspartate type, α-aminohydroxymethyl-oxazole proprionic acid, and other metabolic receptors[43]. N-methyl-D-aspartate receptors are the most important and the most studied receptor. The elevated concentration of glutamate stimulates the over-activation of N-methyl-D-aspartate receptors, and a transient increase in Ca2+ is produced due to influx of calcium from the extracellular environment. Excessive intracellular Ca2+ can activate phospholipase A, and biofilm phospholipid degradation can generate arachidonic acid, prostaglandins, thromboxanes and leukotrienes, resulting in an inflammatory reaction and the generation of oxygen free radicals causing cell damage[40]. In short, abnormally high levels of excitatory amino acids can play a key role in the pathological process of cerebral ischemic neuronal damage. Therefore, inhibition of glutamic acid excitotoxicity in ischemia-reperfusion injury has a defensive role in the prevention and treatment of cerebrovascular disease.
Receptor regulation is one of the most important mechanisms to stabilize the internal environment. Excitatory amino acids must activate excitatory amino acid receptors on postsynaptic membranes. N-methyl-D-aspartate receptors are ligand-gated Ca2+ channels and are highly permeable to Ca2+, which plays an essential role in excitotoxicity. Thus, the N-methyl-D-aspartate receptor family is attracting much attention in cerebral ischemia-reperfusion injury. N-methyl-D-aspartate receptors, the major ionotropic glutamate receptors, are highly expressed in the central nervous system and play a key role in excitatory synaptic transmission. In addition, they are involved in many physiological and pathological processes. N-methyl-D-aspartate receptors are composed of NR1, NR2A, NR2B, NR2C, or NR2D subunits, and the different subunits combine with different N-methyl-D-aspartate receptor subtypes with distinct functional features[44,45]. Overactivation of N-methyl-D-aspartate receptors can lead to excitotoxicity, which is associated with the pathogenesis of neurodegenerative diseases of the central nervous system[46]. The NR1 subunit of the N-methyl-D-aspartate receptor is the basic functional unit and is closely related to cerebrovascular disease[47,48]. In this study, hippocampal cells expressing N-methyl-D-aspartate receptors were selected to study the variation of N-methyl-D-aspartate receptors after oxygen-glucose deprivation and reperfusion. Reverse transcription-PCR was used to detect the mRNA expression levels of the NR1 subunit. Compared with the control group, NR1 subunit mRNA expression was significantly increased in the experiment group. Penehyclidine hydrochloride was able to reduce the expression of NR1 after reperfusion for 6–24 hours, which confirmed that penehyclidine hydrochloride plays a role in the activity of the N-methyl-D-aspartate receptor.
Oxygen is an essential for the maintenance of metabolism and survival. The various causes of hypoxic injury clinically require further investigation. The high demand for oxygen and low tolerance to hypoxia of brain tissue is the root cause of cerebral hypoxic-ischemic injury. A large number of experiments have shown that oxygen free radicals are involved in the pathological process of neuron death after ischemia-reperfusion injury[49,50,51,52]. In hypoxic-ischemic encephalopathy, lipid peroxidation induced by oxygen free radicals is an extremely important factor in pathogenesis and damage. Ischemia-generated free radicals are considered to be an important basis of ischemic damage resulting in cell membrane structure damage and lipid peroxidation. Excessive reactive oxygen species can cause damage to macromolecules in cells, including lipids, proteins, and nucleic acids, culminating in neuronal dysfunction and depression[53,54]. There is an intrinsic antioxidant defense system in cells for scavenging reactive oxygen species to prevent cellular damage. Superoxide dismutase, one of the most important antioxidant enzymes, is the endogenous oxygen free radical scavenger that protects cells from oxidative damage, and its activity may reflect the ability to eliminate free radicals in cells[55]. The use of exogenous superoxide dismutase has been evaluated against various reactive oxygen species-mediated brain injuries, especially those associated with ischemia/reperfusion. In animals with superoxide dismutase overexpression, the infarct volume of cerebral ischemia-reperfusion injury was significantly reduced[51,52,56]. Malondialdehyde, reporting the degree of cell injury, is one of the stable metabolites generated from the lipid peroxidation reaction of oxygen free radicals and biofilm poly-unsaturated fatty acids. Malondialdehyde levels are proportional to lipid peroxidation and oxidative stress[57]. Tissue malondialdehyde levels may indicate the content of oxygen free radicals. Twenty-four hours reperfusion reduced the activity of superoxide dismutase and increased the content of malondialdehyde, which confirmed the presence of free radicals following cerebral ischemia-reperfusion injury. Results in this study showed that penehyclidine hydrochloride could upregulate the sensiticity of superoxide dismutase and downregulate the content of malondialdehyde in ischemic cerebral tissue. These data indicated that penehyclidine hydrochloride reduced the toxicity of free radicals and played an antioxidative role, resulting in the protection of nerve cells during the process of ischemia-reperfusion injury.
In summary, penehyclidine hydrochloride not only altered the apoptotic gene expression of Bcl-2 and caspase-3 to suppress nerve cell apoptosis, it also reduced free radical toxicity by regulating superoxide dismutase and the concentration of malondialdehyde. In addition, penehyclidine hydrochloride protected hippocampal neurons from injury by inhibiting excitotoxicity and N-methyl-D-aspartate receptor overexpression following focal cerebral ischemia-reperfusion injury. The present study demonstrated that penehyclidine hydrochloride provides a protective effect against cerebral ischemia-reperfusion injury and may be a promising therapeutic drug.
MATERIALS AND METHODS
Design
A randomized, controlled animal experiment.
Time and setting
The experiments were performed at the First Affiliated Hospital of China Medical University in China and Yantai Yuhuangding Hospital Central Laboratory in China from October 2009 to January 2012.
Materials
A total of 24 normal male Sprague-Dawley rats, weighing 250–300 g, aged 10 weeks, and of specific pathogen-free grade, were provided by the Experimental Animal Center of China Medical University, China (license No. SCXK (Liao) 2008-0005).
Penehyclidine hydrochloride (C20H29NO2•HCl) was provided by Lisite Pharmaceutical Co., Ltd., Chengdu City, Sichuan Province, China (batch No. 060502-1). The molecular formula is as follows:
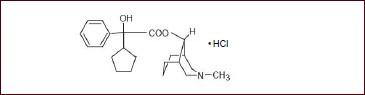
Penehyclidine hydrochloride is a new anticholinergic drug, which can cross the blood-brain barrier and has both anti-muscarinic (M receptor) and anti-nicotinic (N receptor) activities, and retains potent central and peripheral anticholinergic activities. Penehyclidine hydrochloride has been used widely clinically as an antagonist of organic phosphorus and soman poisoning in China. Penehyclidine hydrochloride has little or no effect on M2 receptor subtypes, which has no significant effect on heart rate.
Methods
Establishment of middle cerebral arterial occlusion models in vivo
Middle cerebral arterial occlusion was induced by extracranial vascular occlusion in Sprague-Dawley rats (body weight 250–300 g), according to previously reported procedures with slight modification[58]. Sprague-Dawley rats were anesthetized using 10% (v/v) chloral hydrate via intraperitoneal injection, and body temperature was maintained at 36.5–37.5°C and physiological parameters were monitored continuously during the anesthesia and surgery. Briefly, a 1.5-cm long incision was made at the midline of the neck, the right carotid bifurcation and common carotid artery were separated from the adjacent tissue, avoiding harm to the vagus nerve. After careful isolation of the external carotid artery branches of the occipital and the superior thyroid arteries, as well as the internal carotid artery branch, all of these arteries were exposed. The right common carotid artery and internal carotid artery were carefully separated from the adjacent vagus nerve and connective tissue, and a 0.265-cm diameter nylon intraluminal suture was ligated into the cervical internal carotid artery, advancing intracranially to block blood flow into the middle cerebral artery, and collateral blood flow was also reduced by interrupting all branches of the external carotid artery and all extracranial branches of the internal carotid artery. The suture was withdrawn after occlusion for 120 minutes, and then reperfusion was achieved[59].
Determination of infarction volume in the rat brain
Brain tissues were stored at –70°C for 8 minutes and sliced at a thickness of 2 mm. Slices were incubated in 2% (v/v) 2,[3]5-triphenyl-tetrazolium chloride solution for 30 minutes at 37°C. Samples were then washed in distilled water for 30 seconds and photographed. OSIRIS soft 4.19 (http://www.seekbio.com/soft/176.html) was used to measure the infarct area in each sample, and the percentage of the hemispheric infarction volume was calculated. To minimize errors in the estimation of infarction volume, three measurements were needed on each slice for calculation of the infarction volume. The infarction volume (V) was calculated using the following equation: V= t(A1+A2+…+An), where t is the thickness of the brain tissues (mm), A is the area of the infarcted hemisphere slice (mm2), and n is the number of the brain tissues[60].
Analysis of cell apoptosis in the brain by TUNEL staining
To detect cell apoptosis, TUNEL staining was performed according to the protocol of the TUNEL Detection System (In Site Cell Apoptosis Detection Kit I, POD, MK1020; Boster, Wuhan, Hubei Province, China). Three rats in each group were deeply anesthetized using 10% (v/v) chloral hydrate (300 mg/kg), transcardially perfused with sodium chloride and 4% (w/v) formaldehyde (200 mL), and then decapitated at a given time. Brain samples, including the cortex, hippocampus and striatum, were post-fixed in 4% (w/v) formaldehyde for 2 hours, dehydrated in alcohol, hyalinized by dimethylbenzene, embedded in paraffin, sectioned at a thickness of 4 μm, adhered to superfrost plus slides, and finally stored at room temperature. A standard TUNEL method was employed to detect the fragmented nuclear DNA associated with apoptosis on paraffin sections. After standard deparaffinization, hydration, incubation with proteinase K, and blocking of endogenous peroxidase, tissue sections were incubated with terminal deoxynucleotidyl transferase and digoxigenin-deoxyuridine triphosphate at 37°C for 60 minutes and then with peroxidase converter antibody at 37°C for 30 minutes. For negative controls, some slides were incubated with label solution that did not contain terminal deoxynucleotidyl transferase. Under the optical microscope (Olympus, Tokyo, Japan), the nucleus of apoptotic cells appeared yellow. Positive cells were counted and the mean number in five random views was recorded.
Immunohistochemical staining for Bcl-2 and caspase-3 expression in brain tissues
Formalin-fixed and paraffin-embedded brain sections (5 mm thickness) were first dewaxed in xylene and rehydrated with a graded ethanol series. Endogenous peroxidase was inactivated by incubation in 3% (v/v) H2O2 for 10 minutes at room temperature. Sections were placed in citrate buffer (0.01 M, pH 6.0) for antigen retrieval by microwave heating. Nonspecific binding was blocked by incubation in goat serum (1:10 for 30 minutes at room temperature, and then sections were incubated at 4°C overnight with rabbit anti-Bcl-2 and -caspse-3 polyclonal antibodies (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA). After three washes in PBS for 15 minutes, the slides were incubated with the appropriate secondary antibody at room temperature for 30 minutes. Slides were incubated in horseradish peroxidase-labeled anti-rabbit IgG (1:200; DAKO, Copenhagen, Denmark) at room temperature for 20 minutes and developed with 3,3′-diaminobenzidine (Sigma, St Louis, MO, USA). The sections were finally counterstained with hematoxylin. For negative-control purposes, the same procedure was used on tissue sections in which 1% (v/v) bovine serum albumin in PBS was substituted for the primary antibody[61,62]. Under the optical microscope (Olympus, Tokyo, Japan), brown particles or patches in the cytoplasm indicated positive staining. Ten fields per section were selected in the cortex and striatum. The numbers of positive cells in the grid were counted and a mean value was recorded.
Detection of excitatory amino acid concentrations in the rat brain
Rats were sacrificed by an overdose of chloral hydrate after 24 hours of reperfusion. Brains were removed from the cranium quickly. The right hemisphere (extracted by professional anatomical staff) of each rat was separated, weighed and immediately mixed with 8% (v/v) sulfosalicylic acid, and then freeze centrifuged at 16 000 r/min for 30 minutes. The supernatant was isolated for analysis. Excitatory amino acid concentrations were measured with a high performance liquid chromatography system (Hitachi, Tokyo, Japan) equipped with a 2.6 × 150.0 mm column using O-phthalaldehyde precolumn derivatization. A mixture of potassium dihydrogen phosphate and 35% (v/v) methanol was run through as mobile phase A, and 90% (v/v) methanol as mobile phase B, at a rate of 1.0 mL/min. Excitatory amino acid concentrations were determined by a calibration curve with known amino acid standards[63].
Measurement of malondialdehyde content and superoxide dismutase activity in the rat brain
Rats were killed immediately after 24 hours of reperfusion. Both sides of the cerebral cortex and hippocampus were separately weighed and stored at –70°C. The samples were homogenized in 0.9% (w/v) saline solution using a homogenizer. The homogenate was then centrifuged at 3 000 r/min for 10 minutes at 4°C. The supernatant obtained was used for assays of malondialdehyde content and superoxide dismutase activity. Malondialdehyde content was determined by the thiobarbituric acid method[64], whereas superoxide dismutase activity was evaluated according to the kit instructions (Jiancheng Institute of Biological Products, Nanjing, Jiangsu Province, China).
Primary culture and establishment of oxygen-glucose deprivation and reperfusion models of hippocampal cells in vitro
Primary cultures of the hippocampus were prepared from 1-day-old Wistar rats based on previously reported articles[65,66]. Briefly, the skin and skull were removed and brain tissue was exposed completely. The bilateral hippocampi were bluntly separated and repeatedly washed with D-Hank's buffer with 10 μg/mL gentamycin to remove blood vessels and meninges. The dissected pieces were digested in trypsin (0.125% (w/v)) for 20–25 minutes at 37°C, neutralized with trypsin inhibitor, and washed three times with D-Hank's buffer. Dissociated cell suspensions of 1 × 106/L density were transferred into Dulbecco's Modified Eagle Medium buffer with 15% (v/v) serum at 37°C in a humidified atmosphere of 5% (v/v) CO2 in air. Fresh medium was replaced after overnight incubation to remove dead cells and cytarabine was added into the medium on day 3. Half amount of medium was refreshed after overnight incubation, and medium was completely replaced twice per week thereafter. After 8–12 days, oxygen-glucose deprivation and reperfusion was performed. Neuronal cells was incubated with 100 μL Hank's buffer without glucose at 37°C in 24-well culture plates for 2.5 hours and maintained in a 95% (v/v) N2 and 5% (v/v) CO2 incubator. Oxygen levels in the medium of cultured cells was recovered[67] and the cells were randomly divided into six groups according to treatment with different concentrations of penehyclidine hydrochloride for 20 minutes before oxygen-glucose deprivation/reperfusion: control group with normal Hank's buffer, model group, high-dose penehyclidine hydrochloride group (1.6 μM), middle-dose penehyclidine hydrochloride group (0.4 μM), low-dose penehyclidine hydrochloride group (0.1 μM) and MK-801-positive group (0.1 μM), and incubated for 1, 6, 12, 24 hours after reoxygenation.
N-methyl-D-aspartate-NR1 mRNA expression in primary cultured hippocampal cells by reverse transcription-PCR
Total RNA was directly extracted from primary cultured cells using Trizol reagent according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). The absorbance at 260 and 280 nm was measured regularly. The quality of total RNA was examined by running it on a 1% (w/v) agarose/denaturing formaldehyde gel. Samples with a ratio of absorbance at 260 and 280 nm of 1.8–2.0 and a 28s/18s of approximately 2 were qualified for the next round. Three micrograms of total RNA was reverse-transcribed by M-MuLV reverse transcriptase (MBI, Vilmus, Lithuania) with oligo(dT)16 as a primer. Using equal amounts of cDNA, PCR amplification was performed with 20 nM of each primer, 25 mM Mg2+ and 1 U Taq polymerase with the following N-methyl-D-aspartate-NR1 primers (20 mM each) and glyceraldehyde-3-phosphate dehydrogenase primers (20 mM each; Table 1).
Table 1.
Sequence of NMDA-NR1 and GAPDH primers

All experiments were performed in triplicate. The mRNA level of N-methyl-D-aspartate-NR1 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was assessed by measuring the density of the PCR generated amplicon on the gel by densitometric analysis. The final expression level was the average value of the three amplicons. Relative N-methyl-D-aspartate-NR1 expression is given as N-methyl-D-aspartate-NR1/GAPDH absorbance ratio.
Statistical analysis
Statistical analysis was performed using SPSS 13.0 software (SPSS, Chicago, IL, USA). Data were expressed as mean ± SD. Statistical comparison was performed by one-way analysis of variance followed by least significant difference test. P values of less than 0.05 were considered statistically significant.
Acknowledgments
We are grateful for the excellent pathology assistance from Professor Weidong Yao, Department of Pathology in the Affiliated Yuhuangding Hospital of Medical College of Qingdao University, China.
Footnotes
Conflicts of interest: None declared.
Ethical approval: The experimental procedures were approved by the Animal Use and Care Advisory Committee of Experimental Center of China Medical University in China, and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996).
(Edited by Guan HB, Chen X/Yang Y/Song LP)
REFERENCES
- [1].Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28(9):2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87(1):179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- [3].Minnerup J, Schabitz WR. Multifunctional actions of approved and candidate stroke drugs. Neurotherapeutics. 2009;6(1):43–52. doi: 10.1016/j.nurt.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Annecke T, Kubitz JC, Kahr S, et al. Effects of sevoflurane and propofol on ischaemia-reperfusion injury after thoracic- aortic occlusion in pigs. Br J Anaesth. 2007;98(5):581–590. doi: 10.1093/bja/aem049. [DOI] [PubMed] [Google Scholar]
- [5].Chen Q, Zeng Y. Anisodamine protects against neuronal death following cerebral ischemia in gerbils. Chin Med J (Engl) 2000;113(7):636–639. [PubMed] [Google Scholar]
- [6].Guzmán-De La Garza FJ, Cámara-Lemarroy CR, Ballesteros-Elizondo RG, et al. Ketamine reduces intestinal injury and inflammatory cell infiltration after ischemia/reperfusion in rats. Surg Today. 2010;40(11):1055–1062. doi: 10.1007/s00595-009-4177-4. [DOI] [PubMed] [Google Scholar]
- [7].Shen W, Gan J, Xu S, et al. Penehyclidine hydrochloride attenuates LPS-induced acute lung injury involvement of NF-kappaB pathway. Pharmacol Res. 2009;60(4):296–302. doi: 10.1016/j.phrs.2009.04.007. [DOI] [PubMed] [Google Scholar]
- [8].Wang LL, Zhan LY, Wu XJ, et al. Effects of penehyclidine hydrochloride on apoptosis of lung tissues in rats with traumatic acute lung injury. Chin J Traumatol. 2010;13(1):15–19. [PubMed] [Google Scholar]
- [9].Han XY, Liu H, Liu CH, et al. Synthesis of the optical isomers of a new anticholinergic drug, penehyclidine hydrochloride (8018) Bioorg Med Chem Lett. 2005;15(8):1979–1982. doi: 10.1016/j.bmcl.2005.02.071. [DOI] [PubMed] [Google Scholar]
- [10].He SS, Lin CS, Gu MN, et al. Protective effects of penehyclidine hydrochloride against acute renal injury induced by hemorrhagic shock and lipopolysaccharides in rats. Nan Fang Yi Ke Da Xue Xue Bao. 2011;31(5):899–902. [PubMed] [Google Scholar]
- [11].Xiao HT, Liao Z, Meng XM, et al. Characterization of the effect of penehyclidine hydrochloride on muscarinic receptor subtypes mediating the contraction of guinea-pig isolated gastrointestinal smooth muscle. J Pharm Pharmacol. 2009;61(7):949–952. doi: 10.1211/jpp/61.07.0015. [DOI] [PubMed] [Google Scholar]
- [12].Wang YA, Zhou WX, Li JX, et al. Anticonvulsant effects of phencynonate hydrochloride and other anticholinergic drugs in soman poisoning: neurochemical mechanisms. Life Sci. 2005;78(2):210–223. doi: 10.1016/j.lfs.2005.04.071. [DOI] [PubMed] [Google Scholar]
- [13].Li JT, Ruan JX, Zhang ZQ, et al. Effects of pretreatment with 8018 on the toxicokinetics of soman in rabbits and distribution in mice. Life Sci. 2003;73(8):1053–1062. doi: 10.1016/s0024-3205(03)00371-0. [DOI] [PubMed] [Google Scholar]
- [14].Gorgulu A, Kins T, Cobanoglu S, et al. Reduction of edema and infarction by Memantine and MK-801 after focal cerebral ischaemia and reperfusion in rat. Acta Neurochir (Wien) 2000;142(11):1287–1292. doi: 10.1007/s007010070027. [DOI] [PubMed] [Google Scholar]
- [15].Xiao HT, Liao Z, Meng XM, et al. Underlying mechanism of penehyclidine hydrochloride on isolated rat uterus. Fundam Clin Pharmacol. 2009;23(4):419–421. doi: 10.1111/j.1472-8206.2009.00712.x. [DOI] [PubMed] [Google Scholar]
- [16].Zhang YM, Yan YS, Wang LN, et al. A novel rice gene, NRR responds to macronutrient deficiency and regulates root growth. Mol Plant. 2012;5(1):63–72. doi: 10.1093/mp/ssr066. [DOI] [PubMed] [Google Scholar]
- [17].Hu T, Fu Q, Liu X, et al. Increased acetylcholinesterase and capase-3 expression in the brain and peripheral immune system of focal cerebral ischemic rats. J Neuroimmunol. 2009;211(1-2):84–91. doi: 10.1016/j.jneuroim.2009.04.002. [DOI] [PubMed] [Google Scholar]
- [18].Liu B, Li J, Li L, et al. Electrical stimulation of cerebellar fastigial nucleus promotes the expression of growth arrest and DNA damage inducible gene β and motor function recovery in cerebral ischemia/reperfusion rats. Neurosci Lett. 2012;520(1):110–114. doi: 10.1016/j.neulet.2012.05.044. [DOI] [PubMed] [Google Scholar]
- [19].Formichi P, Radi E, Battisti C, et al. Cerebrolysin administration reduces oxidative stress-induced apoptosis in limphocytes from healthy individuals. J Cell Mol Med. 2012;16(11):2840–2843. doi: 10.1111/j.1582-4934.2012.01615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].D’Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 2010;17(7):1104–1114. doi: 10.1038/cdd.2009.180. [DOI] [PubMed] [Google Scholar]
- [21].Di Menna L, Molinaro G, Di Nuzzo L, et al. Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol Res. 2012;67(1):1–9. doi: 10.1016/j.phrs.2012.10.004. [DOI] [PubMed] [Google Scholar]
- [22].Gabryel B, Kost A, Kasprowska D. Neuronal autophagy in cerebral ischemia--a potential target for neuroprotective strategies? Pharmacol Rep. 2012;64(1):1–15. doi: 10.1016/s1734-1140(12)70725-9. [DOI] [PubMed] [Google Scholar]
- [23].Wlodkowic D, Skommer J, Darzynkiewicz Z. Cytometry of apoptosis. Historical perspective and new advances. Exp Oncol. 2012;34(3):255–262. [PMC free article] [PubMed] [Google Scholar]
- [24].Cheng EH, Kirsch DG, Clem RJ, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278(5345):1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- [25].Tilly JL, Tilly KI, Kenton ML, et al. Expression of members of the Bcl-2 gene family in the immature rat ovary: equine chorionic gonadotropin-mediated inhibition of granulosa cell apoptosis is associated with decreased bax and constitutive Bcl-2 and bcl-xlong messenger ribonucleic acid levels. Endocrinology. 1995;136(1):232–241. doi: 10.1210/endo.136.1.7828536. [DOI] [PubMed] [Google Scholar]
- [26].Kim HS, Park MS, Lee JK, et al. Time point expression of apoptosis regulatory proteins in a photochemically- induced focal cerebral ischemic rat brain. Chonnam Med J. 2011;47(3):144–149. doi: 10.4068/cmj.2011.47.3.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shtilbans V, Wu M, Burstein DE. Evaluation of apoptosis in cytologic specimens. Diagn Cytopathol. 2010;38(9):685–697. doi: 10.1002/dc.21313. [DOI] [PubMed] [Google Scholar]
- [28].Yu SS, Zhao J, Zheng WP, et al. Neuroprotective effect of 4-hydroxybenzyl alcohol against transient focal cerebral ischemia via anti-apoptosis in rats. Brain Res. 2010;1308:167–175. doi: 10.1016/j.brainres.2009.10.037. [DOI] [PubMed] [Google Scholar]
- [29].Yang B, El Nahas AM, Thomas GL, et al. Caspase-3 and apoptosis in experimental chronic renal scarring. Kidney Int. 2001;60(5):1765–1776. doi: 10.1046/j.1523-1755.2001.00013.x. [DOI] [PubMed] [Google Scholar]
- [30].Kirsch DG, Doseff A, Chau BN, et al. Caspase-3- dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274(30):21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- [31].Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21(1):92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hovelsø N, Sotty F, Montezinho LP, et al. Therapeutic potential of metabotropic glutamate receptor modulators. Curr Neuropharmacol. 2012;10(1):12–48. doi: 10.2174/157015912799362805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Young AB, Fagg GE. Excitatory amino acid receptors in the brain: membrane binding and receptor autoradiographic approaches. Trends Pharmacol Sci. 1990;11(3):126–133. doi: 10.1016/0165-6147(90)90199-i. [DOI] [PubMed] [Google Scholar]
- [34].Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- [35].Wei J, Quast MJ. Effect of nitric oxide synthase inhibitor on a hyperglycemic rat model of reversible focal ischemia: detection of excitatory amino acids release and hydroxyl radical formation. Brain Res. 1998;791(1-2):146–156. doi: 10.1016/s0006-8993(98)00089-4. [DOI] [PubMed] [Google Scholar]
- [36].Dawson LA, Djali S, Gonzales C, et al. Characterization of transient focal ischemia-induced increases in extracellular glutamate and aspartate in spontaneously hypertensive rats. Brain Res Bull. 2000;53(6):767–776. doi: 10.1016/s0361-9230(00)00363-4. [DOI] [PubMed] [Google Scholar]
- [37].Mahesh VB, Brann DW. Regulatory role of excitatory amino acids in reproduction. Endocrine. 2005;28(3):271–280. doi: 10.1385/ENDO:28:3:271. [DOI] [PubMed] [Google Scholar]
- [38].Javitt DC, Schoepp D, Kalivas PW, et al. Translating glutamate: from pathophysiology to treatment. Sci Transl Med. 2011;3(102):102mr2. doi: 10.1126/scitranslmed.3002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- [40].Nishizawa Y. Glutamate release and neuronal damage in ischemia. Life Sci. 2001;69(4):369–381. doi: 10.1016/s0024-3205(01)01142-0. [DOI] [PubMed] [Google Scholar]
- [41].Degracia DJ. Towards a dynamical network view of brain ischemia and reperfusion. Part II: a post-ischemic neuronal state space. J Exp Stroke Transl Med. 2010;3(1):72–89. doi: 10.6030/1939-067x-3.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- [43].Farber NB. The NMDA receptor hypofunction model of psychosis. Ann N Y Acad Sci. 2003;1003:119–130. doi: 10.1196/annals.1300.008. [DOI] [PubMed] [Google Scholar]
- [44].Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258(5082):597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- [45].Monyer H, Burnashev N, Laurie DJ, et al. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12(3):529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- [46].Luo T, Wu WH, Chen BS. NMDA receptor signaling: death or survival? Front Biol. 2011;6(6):468–476. doi: 10.1007/s11515-011-1187-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Santangelo RM, Acker TM, Zimmerman SS, et al. Novel NMDA receptor modulators: an update. Expert Opin Ther Pat. 2012;22(11):1337–1352. doi: 10.1517/13543776.2012.728587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Luo X, Baba A, Matsuda T, et al. Susceptibilities to and mechanisms of excitotoxic cell death of adult mouse inner retinal neurons in dissociated culture. Invest Ophthalmol Vis Sci. 2004;45(12):4576–4582. doi: 10.1167/iovs.04-0166. [DOI] [PubMed] [Google Scholar]
- [49].Reiter RJ, Manchester LC, Tan DX. Neurotoxins: free radical mechanisms and melatonin protection. Curr Neuropharmacol. 2010;8(3):194–210. doi: 10.2174/157015910792246236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Faustino JV, Wang X, Johnson CE, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. 2011;31(36):12992–30001. doi: 10.1523/JNEUROSCI.2102-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Olmez I, Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem Int. 2012;60(2):208–212. doi: 10.1016/j.neuint.2011.11.009. [DOI] [PubMed] [Google Scholar]
- [52].Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4(6):461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- [53].Zhao Z, Wang W, Guo H, et al. Antidepressant-like effect of liquiritin from Glycyrrhiza uralensis in chronic variable stress induced depression model rats. Behav Brain Res. 2008;194(1):108–113. doi: 10.1016/j.bbr.2008.06.030. [DOI] [PubMed] [Google Scholar]
- [54].Bito T, Nishigori C. Impact of reactive oxygen species on keratinocyte signaling pathways. J Dermatol Sci. 2012;68(1):3–8. doi: 10.1016/j.jdermsci.2012.06.006. [DOI] [PubMed] [Google Scholar]
- [55].Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91(1-3):21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- [56].Shi LL, Chen BN, Gao M, et al. The characteristics of therapeutic effect of pinocembrin in transient global brain ischemia/reperfusion rats. Life Sci. 2011;88(11-12):521–528. doi: 10.1016/j.lfs.2011.01.011. [DOI] [PubMed] [Google Scholar]
- [57].Xiao X, Liu J, Hu J, et al. Protective effects of protopine on hydrogen peroxide-induced oxidative injury of PC12 cells via Ca(2+) antagonism and antioxidant mechanisms. Eur J Pharmacol. 2008;591(1-3):21–27. doi: 10.1016/j.ejphar.2008.06.045. [DOI] [PubMed] [Google Scholar]
- [58].Yang L, Shah K, Wang H, et al. Characterization of neuroprotective effects of biphalin, an opioid receptor agonist, in a model of focal brain ischemia. J Pharmacol Exp Ther. 2011;339(2):499–508. doi: 10.1124/jpet.111.184127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- [60].Majid A, He YY, Gidday JM, et al. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke. 2000;31(11):2707–2714. doi: 10.1161/01.str.31.11.2707. [DOI] [PubMed] [Google Scholar]
- [61].Landberg G, Ostlund H, Nielsen NH, et al. Downregulation of the potential suppressor gene IGFBP-rP1 in human breast cancer is associated with inactivation of the retinoblastoma protein, cyclin E overexpression and increased proliferation in estrogen receptor negative tumors. Oncogene. 2001;20(27):3497–505. doi: 10.1038/sj.onc.1204471. [DOI] [PubMed] [Google Scholar]
- [62].Ruan W, Xu E, Xu F, et al. IGFBP7 plays a potential tumor suppressor role in colorectal carcinogenesis. Cancer Biol Ther. 2007;6(3):354–359. doi: 10.4161/cbt.6.3.3702. [DOI] [PubMed] [Google Scholar]
- [63].Zhou W, Yoshioka M, Yokogoshi H. Sub-chronic effects of s-limonene on brain neurotransmitter levels and behavior of rats. J Nutr Sci Vitaminol (Tokyo) 2009;55(4):367–373. doi: 10.3177/jnsv.55.367. [DOI] [PubMed] [Google Scholar]
- [64].Cao Y, Mao X, Sun C, et al. Baicalin attenuates global cerebral ischemia/reperfusion injury in gerbils via anti-oxidative and anti-apoptotic pathways. Brain Res Bull. 2011;85(6):396–402. doi: 10.1016/j.brainresbull.2011.05.002. [DOI] [PubMed] [Google Scholar]
- [65].Katsube N, Sunaga K, Aishita H, et al. ONO-1603, a potential antidementia drug, delays age-induced apoptosis and suppresses overexpression of glyceraldehyde-3-phosphate dehydrogenase in cultured central nervous system neurons. J Pharmacol Exp Ther. 1999;288(1):6–13. [PubMed] [Google Scholar]
- [66].Shkryl VM, Nikolaenko LM, Kostyuk PG, et al. High-threshold calcium channel activity in rat hippocampal neurones during hypoxia. Brain Res. 1999;833(2):319–328. doi: 10.1016/s0006-8993(99)01575-9. [DOI] [PubMed] [Google Scholar]
- [67].Almeida A, Delgado-Esteban M, Bolanos JP, et al. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem. 2002;81(2):207–217. doi: 10.1046/j.1471-4159.2002.00827.x. [DOI] [PubMed] [Google Scholar]