

Abstract
The experimental model of traumatic brain injury was established in Sprague-Dawley rats according to Feeney's free falling method. The brains were harvested at 2, 6 and 24 hours, and at 3 and 5 days after injury. Changes in brain water content were determined using the wet and dry weights. Our results showed that water content of tissue significantly increased after traumatic brain injury, and reached minimum at 24 hours. Hematoxylin-eosin staining revealed pathological impairment of brain tissue at each time point after injury, particularly at 3 days, with nerve cell edema, degenera-tion, and necrosis observed, and the apoptotic rate significantly increased. Immunohistochemistry and western blot analysis revealed that the expression of occludin at the injured site gradually de-creased as injury time advanced and reached a minimum at 3 days after injury; the expression of connexin 43 gradually increased as injury time advanced and reached a peak at 24 hours after in-jury. The experimental findings indicate that changes in occludin and connexin 43 expression were consistent with the development of brain edema, and may reflect the pathogenesis of brain injury.
Keywords: neural regeneration, traumatic brain injury, brain edema, connexin 43, occludin, nerve cells, thology, tight junction, grants-supported paper, neuroregeneration
Research Highlights
(1) Brain water content increased after traumatic brain injury, and nerve cells exhibited edema, degeneration, necrosis and other pathological changes.
(2) Occludin expression in brain tissue decreased after traumatic brain injury while connexin 43 expression increased.
(3) The above changes were most apparent at 2–3 days after injury.
INTRODUCTION
Brain edema is a kind of secondary pathological process, and a major cause of death and disability. Understanding the pathogenesis of brain edema may allow for the prevention and treatment of edema in the field of neurosurgery, thus resulting in better treatments for intracranial diseases[1].
Traumatic brain edema is the most important secondary pathophysiological response following severe traumatic brain injury. The pathogenesis of traumatic brain edema is usually thought to be associated with energy metabolism disturbance, microcirculatory disorders, inflammatory cytokines, oxygen free radicals, calcium overload, and the expression of aquaporin 4. In addition, when brain edema occurs, the inflammatory cytokines, oxygen free radicals, calcium and other toxic metabolites can cause or aggravate edema through intercellular connections
between nerve cells. Recently, studies have focused on the intercellular junctions after brain injury, mainly tight junctions[2,3], while few studies have focused on the correlation between gap junctions and traumatic brain edema. However, some reports have investigated the main component proteins of gap junctions, namely connexin 43[4]. To date, few have investigated the role of occludin, an important protein of tight junctions, and connexin 43 in traumatic brain edema, especially their dynamic changes.
In this study, the dynamic changes of occludin and connexin 43 were observed in rats following traumatic brain injury, and their correlation with the development of brain edema.
RESULTS
Quantitative analysis of experimental animals
A total of 80 Sprague-Dawley rats were randomly divided into a sham-surgery group (surgery only, no injury) and a trauma group (brain injury models were established according to the free falling method of Feeney[5]). Brains were harvested at 2, 6 and 24 hours, and at 3 and 5 days after injury. Eight rats in each group at each time point were selected. In total, 80 rats were involved in the final analysis.
Pathological changes in rat brain tissue after injury
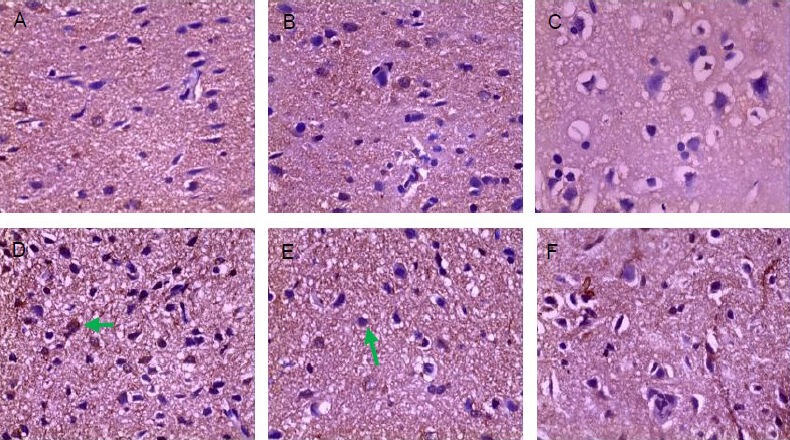
Hematoxylin-eosin staining showed clear brain tissue structure, regular cell arrangement, normal cell contours, centered nuclei and distinct nucleoli present in the sham-surgery group. At 2 hours post-injury, the trauma caused hemorrhaging, the disorderly arrangement of nerve cells with unclear polarity, some nerve cell swelling, and focal nuclei pyknosis. At 6 hours post-injury, hemorrhagic focus was apparent, nerve cells swelled, degenerated, and became necrotic and apoptotic. In addition, nuclear condensation and interstitial edema were observed in some cells. At 24 hours post-injury, the above changes were visible including edema, degeneration, and necrosis of nerve cells, and vacuolar degeneration and unclear nuclear structure. At 3 days post-injury, the above phenomena were aggravated, large amounts of pyknotic nuclei and nuclear fragments were visible in the center of the lesions with surrounding apoptotic cells. At 5 days post-injury, interstitial edema was visible, some swollen nerve cells were observed and focal nuclei pyknosis was visible (Figure 1).
Figure 1.
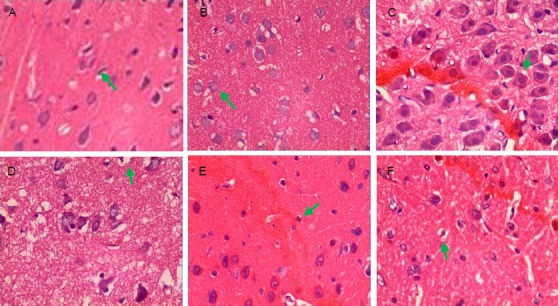
Pathological changes of rat brain tissue after traumatic brain injury (hematoxylin-eosin staining, × 400).
(A) Sham-surgery group: Brain tissue structure was clear, cells arranged regularly, cell contour was normal (arrow), with nuclei in the center and distinct nucleoli. (B) 2 hour trauma group: Hemorrhage foci were visible, neural cells arranged disorderly, with unclear polarity, some cells mildly swelled (arrow), focal nuclei pyknosis was observed. (C) 6 hour trauma group: Hemorrhage foci were apparent, nerve cells began to swell (arrow), degenerate, necrosis and apoptosis, some cells exhibited nuclear condensation and interstitial edema.
(D) 24 hour trauma group: In addition to the above changes, nerve cell edema, degeneration and necrosis were significantly seen (arrow), with vacuolar degeneration and unclear nuclear structure. (E) 3 day trauma group: The above phenomena were aggravated, a large amount of pyknotic nuclei and nuclear fragments were visible in the center of the lesion (arrow), surrounded by apoptotic cells. (F) 5 day trauma group: Interstitial edema was still visible but significantly attenuated, some nerve cells swelled and focal nuclei pyknosis was observed (arrow).
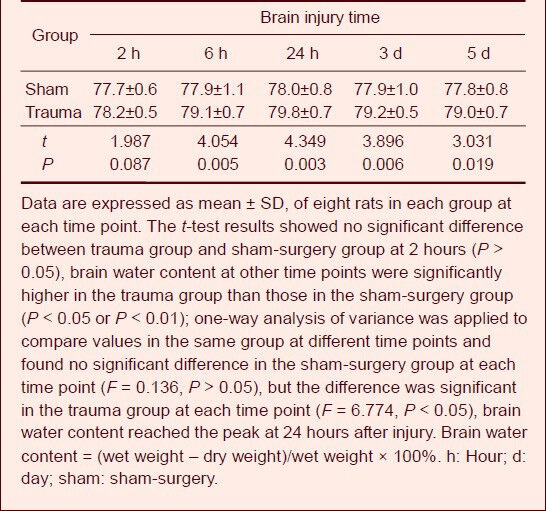
Changes to brain water content in rat brain tissue after injury
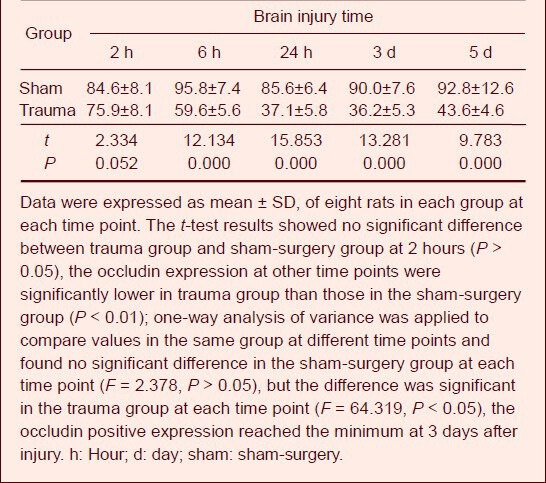
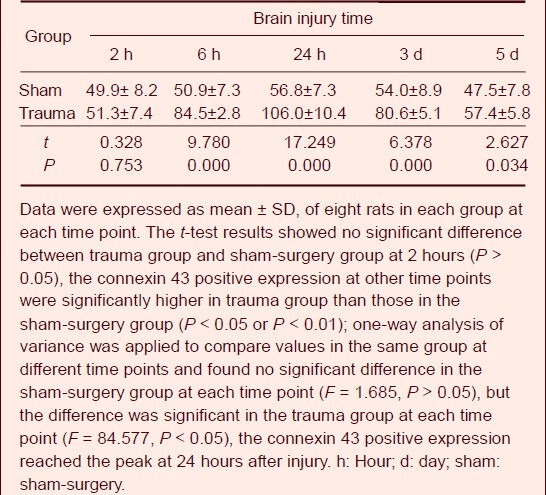
The dry-wet weight method was used to determine the brain water content. Results showed that brain tissue water content in the trauma group was significantly increased when compared with the sham-surgery group at 6 hours post-injury (P < 0.01), reached a peak at 24 hours after injury, and then gradually decreased at 3 days (Table 1).
Table 1.
Change of brain water content (%) after traumatic brain injury in rats
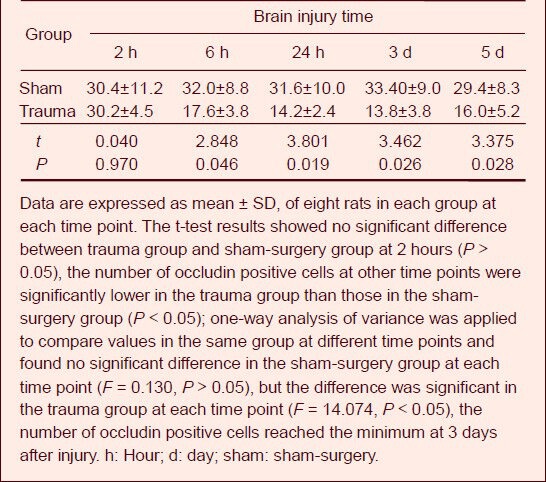
Occludin expression in rat brain tissue after injury
Immunohistochemical staining showed that occludin was expressed in rat brain tissue from both groups, mainly in the cerebral cortex, which exhibited brown granules. In comparison to the sham-surgery group, occludin expression began to decrease at 6 hours after injury and reached a minimum at 3 days (P < 0.05), which was maintained at 5 days after injury (Figure 2, Table 2).
Figure 2.
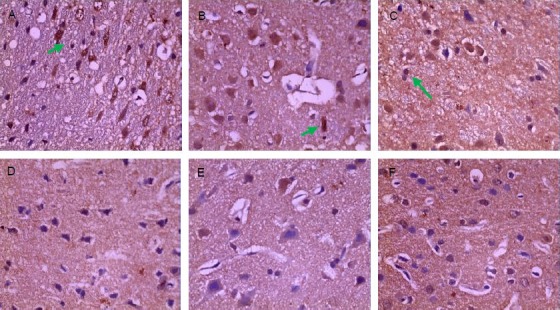
Expression of occludin positive cells in brain tissue after traumatic brain injury (immunohistochemical staining, × 400).
The brown granules (arrows) indicate occludin positive expression.
(A) Sham-surgery group: Occludin was abundantly expressed. (B) 2 hour trauma group: Occludin was abundantly expressed and the expression level was not decreased. (C) 6 hour trauma group: Occludin expression was significantly decreased.
(D) 24 hour trauma group: Occludin was still lowly expressed. (E) 3 day trauma group: Occludin expression was the minimal. (F) 5 day trauma group: Occludin was lowly expressed.
Table 2.
Counting results of occludin positive cells (n/high-power (400 ×) field of view) in brain tissue after traumatic brain injury
Western blot analysis results showed that occludin expression was highly expressed in rat brain tissue from the sham-surgery group, and began to decrease after brain injury, reaching its lowest levels at 3 days, then gradually increasing. However, expression levels remained low at 5 days. Compared with the sham-surgery group, occludin expression was significantly decreased in the trauma group at each time point, except 2 hours after injury (P < 0.01; Figure 3, Table 3).
Figure 3.
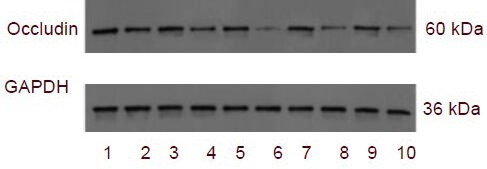
Occludin expression in brain tissue after traumatic brain injury in rats (western blot analysis).
1–10: 2 hour sham-surgery group, 2 hour trauma group, 6 hour sham-surgery group, 6 hour trauma group, 24 hour sham-surgery group, 24 hour trauma group, 3 day sham-surgery group, 3 day trauma group, 5 day sham-surgery group, 5 day trauma group.
Table 3.
Occludin expression in brain tissue (absorbance ratio to GAPDH) after traumatic brain injury by western blot analysis
Connexin 43 expression in rat brain tissue after injury
Immunohistochemical staining showed that connexin 43, which appeared as brown-yellow granules, was expressed in brain tissue of all rats.
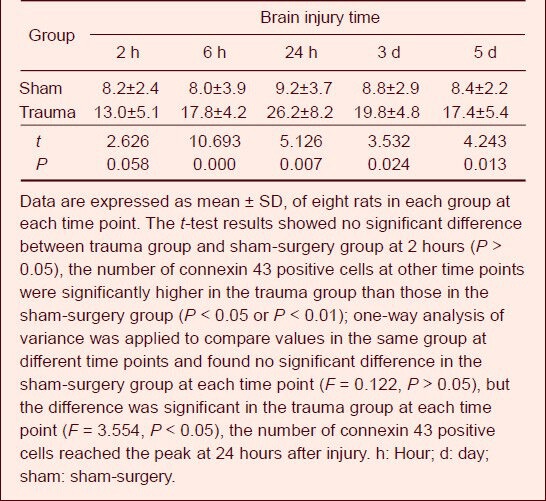
Compared with the sham-surgery group, connexin 43 expression in astrocytes peripheral to the brain edema began to increase at 6 hours post-injury, reached a peak at 24 hours and gradually decreased at 3 days (Figure 4, Table 4).
Figure 4.
Connexin 43 expression in brain tissues of rats after traumatic brain injury (immunohistochemical staining, ×400).
The brown granules (arrows) indicate connexin 43 positive expression.
(A) Sham-surgery group: Connexin 43 was scarcely expressed. (B) 2 hour trauma group: Connexin 43 was lowly expressed and the expression level was not increased. (C) 6 hour trauma group: Connexin 43 expression was significantly increased.
(D) 24 hour trauma group: Connexin 43 expression reached the peak. (E) 3 day trauma group: Connexin 43 was highly expressed. (F) 5 day trauma group: Connexin 43 expression was decreased, which was still higher than that in sham-surgery group.
Table 4.
Counting results of connexin 43 positive cells (n/high-power (400 ×) field of view) in brain tissue after traumatic brain injury
Western blot analysis results showed that connexin 43 in the trauma group began to increase at 6 hours post-injury and continued to increase in a time-dependent manner, reaching a peak at 24 hours and then decreasing gradually. Expression levels were still low at 5 days, which was higher than that of the sham-surgery group (P < 0.05; Figure 5, Table 5).
Figure 5.
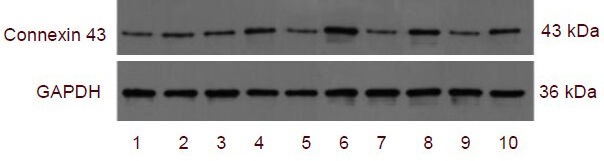
Connexin 43 expression in brain tissue after traumatic brain injury in rats (western blot analysis).
1–10: 2 hour sham-surgery group, 2 hour trauma group, 6 hour sham-surgery group, 6 hour trauma group, 24 hour sham-surgery group, 24 hour trauma group, 3 day sham-surgery group, 3 day trauma group, 5 day sham-surgery group, 5 day trauma group.
Table 5.
Connexin 43 expression in brain tissue (absorbance ratio to GAPDH) after traumatic brain injury by western blot analysis
DISCUSSION
Brain edema may increase intracranial pressure, which aggravates brain edema, resulting in irreversible secondary injury if treatment is not given in time. Many mechanisms contribute to the pathogenesis of traumatic brain injury, but how these lead to death and disability remains unclear. Therefore, understanding the pathogenesis of traumatic brain injury is necessary for an appropriate treatment scheme.
Intercellular junctions are specific structures formed by the local plasma membrane at cell junctions, and can be divided into three types according to their structure and function: tight junctions, desmosomes and gap junctions[6].
Tight junctions and gap junctions act as barriers and passages for intercellular substance transmission and transfer, and they are believed to be associated with the development of secondary brain injury and brain edema[7,8,9]. Intervention with tight and gap junction function may reduce secondary injury and brain edema. Tight junctions are the selective barrier between epithelial cells or between endothelial cells, and are involved in the regulation of solutes and drug transfer[10].
Tight junctions can be found in brain capillary endothelial cells in the central nervous system, where they prevent the mutual diffusion of blood and brain extracellular fluid. Therefore, they are considered to be important factors for maintaining blood-brain barrier structural integrity. Tight junctions are composed of the transmembrane proteins occludin, the atresia protein family (at least 24 members), cell adhesion molecule, tricellulin (a new transmembrane protein family[11]), cytoplasmic adhesion protein (ZO-1, ZO-2, ZO-3), actin, cingulate protein, 7H6, signal transduction components (AF6, agouti signal protein ASIP) and other components[10,12]. A variety of endogenous or exogenous signaling pathways contribute to regulate these proteins, thus modulating the permeability of the blood-brain barrier, which affects brain edema[2].
Occludin (molecular weight 60 kDa), first identified and cloned by Furuse et al[13] in 1993, is a transmembrane protein. Its discovery was considered a milestone in tight junction research and is still considered as the most important transmembrane protein. Occludin expression in the brain vascular endothelium is significantly higher than that in the non-nervous tissue vascular endothelium[14], which causes the permeability of the blood-brain barrier to be significantly lower when compared with other blood tissue barriers. The permeability of the blood-brain barrier is influenced by the amount of occludin and its distribution[9,10,11,12]. Ding et al[15] found that severe acute pancreatitis induced the enhancement of blood-brain barrier permeability, and that occludin expression levels were decreased. In addition, cerebral ischemia-caused tyrosine phosphorylation, which can induce blood-brain barrier dysfunction[16]. Moreover, Wu et al[17] showed that hypoxia and ischemia may induce blood-brain barrier tight junction opening and increase blood-brain barrier permeability, but that occludin expression remained unchanged, which is possibly due to the altered distribution of occludin in cells. However, a recent study showed that when occludin was downregulated or degraded, blood-brain barrier function was damaged[18].
Gap junctions are intercellular channels formed between two hemichannels on the surface of adjacent cell membranes, and are the most prevalent kind of cell junction in animals. Gap junctions allow various molecules and ions to pass between cells[6,19,20]. Ion and water-soluble molecules of low molecular weight (1 000–1 500 kDa) can directly cross, while large molecules such as proteins, nucleic acids and polysaccharides cannot[6]. This is the basis of direct intercellular information exchange, which does not require the help of second messenger pathways. Therefore, it is called direct communication, and is also known as gap junction intercellular communication. In the central nervous system, gap junctions are widely present in astrocytes and between astrocytes and neurons or other glial cells[21]. These junctions are involved in intercellular substance exchange and signal transmission[22,23,24], and also contribute to the regulation of cellular metabolism and maintenance of the inner environment. Using electron microscopy, we observed clusters of tightly arranged particles on the surface of the plasma membrane (outer layer of inner lobe), and these particles were considered to be basic structural units, or connexons. Each connexon is composed of 6 protein molecular subunits, and can be purified into connexin[6]. Six connexins with the same structure can form a gap junction half-channel on the cell membrane in a hexameric form[25]. These gap junction half-channels on the plasma membrane of two adjacent cells connect to form gap junctions and intercellular channels[20].
Connexins are a group of structurally related proteins, with at least 20 identified at present in mammals[26]. Connexin 43 (molecular weight 43 kDa) is the predominant gap junction protein found in the adult brain and is primarily found in astrocytes. Connexin 43 connects astrocytes, allowing them to become functional cell aggregates, while glial cells and neurons also connect to form broader cell aggregates through homologous or heterologous connexins[27,28], allowing ion and small signal molecule transmission among perivascular astrocytic networks between cells, as well as maintaining the integrity of the blood-brain barrier[29]. The function of gap junctions and abnormal expression of connexin 43 are closely related to the pathophysiology of many nervous system diseases[30,31,32].
During early stages of traumatic brain injury, a series of metabolic stress signals such as calcium ions, amino acids, oxygen free radicals, inflammatory factors and metabolites enter surrounding cells through connexin 43, whose expression increases post-injury, and thus contributes to the formation and aggravation of brain edema[4]. Astrocyte swelling is an important indicator of brain edema at early stages[33] (cytotoxic brain edema). Connexin 43 has been shown to regulate the permeability of the blood-brain barrier[34] and connexin 43 blockers can attenuate brain edema[4], which suggests that blocking connexin 43 expression may reduce brain edema. Another study[29] showed that connexin 43 is necessary for maintaining the integrity of the blood-brain barrier, with decreased expression inducing edema in astrocyte foot processes, attenuating blood-brain barrier function, and aggravating cerebral edema. An increasing number of studies regarding the correlation between occludin and brain edema[9,14,35] have shown that occludin is closely related to the occurrence of brain edema. In this study, the expression of occludin and connexin 43 in rats with traumatic brain edema was observed at different time points post-injury. The experimental results showed that brain water content was significantly increased at 6 hours post-injury, reached a peak at 24 hours, and then decreased at 5 days, which was still higher than that of the control group. Immunohistochemistry and western blot analysis results revealed that occludin expression reduced after injury and reached a minimum at 3 days, which was significantly different when compared with the control group at each time point after 6 hours. Immunohistochemistry and western blot analysis results also found that the expression of connexin 43 increased and reached a peak at 24 hours post-injury, which was significantly different when compared with the control group at each time point after 6 hours. This evidence indicates that both occludin and connexin 43 are responsible for the formation of traumatic brain edema.
The changes in brain water content were consistent with previous findings[36,37], but were in contrast to a recent study[38], which showed that the blood-brain barrier was obviously destroyed at 6 hours post-trauma. Moreover, there were two peak periods in expression: 6 hours and 72 hours after injury. Our experimental findings on occludin expression also differed to previous studies[39] that found a reduction in occludin expression at 12 hours to 6 days or 2–4 days post-injury, and the results of Sun et al[14] who showed that occludin expression decreased at 3 hours and was minimal at 24 hours. In addition, Avila et al[40] observed a decrease in connexin 43 expression at 2 hours post-injury, and Ohsumi et al[19] found phosphorylation of connexin 43 expression was induced at 1 hour after brain injury, and reached a peak at 6 hours. The expression of connexin 43 was apparent at 6 hours and peaked at 3 days[41,42]. These differences may be related to the experimental animal used, or the experimental and trauma conditions.
In summary, the changes in occludin expression, connexin 43 and brain water content demonstrate that occludin and connexin 43 participate in post-traumatic blood-brain barrier dysfunction, and astrocyte inner ion and water movement. In addition, connexin 43 contributes to early traumatic brain edema and occludin is associated with post-traumatic brain edema at later periods. To our knowledge, this is the first study to investigate the relationship between connexin 43 and occludin during post-injury in a brain edema model. Our study provides evidence for using connexin 43 and occludin as therapeutic targets for brain edema. However, further studies are required to fully understand the role of connexin 43 and occludin during traumatic brain injury, particularly those associated with identifying the relationship between occludin, connexin 43 and cerebral edema, and monitoring blood-brain barrier function and its relationship to occludin and connexin 43 expression.
MATERIALS AND METHODS
Design
A randomized, controlled animal experiment.
Time and setting
The experiment was performed at Guangdong Medical College from September 2011 to February 2012.
Materials
Eighty clean, healthy, adult Sprague-Dawley rats, male or female, weighing 300–340 g, were provided by the Experimental Animal Center of Guangdong Medical College, China (license No. SCXK (Yue) 2008-0008). The protocols were performed in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, formulated by the Ministry of Science and Technology of China[43].
Methods
Establishment of brain injury models
Brain injury models were produced using Feeney's free falling method[5]. Rats were intraperitoneally anesthetized with 10% (w/v) chloral hydrate (350 mg/kg), and fixed on the operating table, in a prone position. After routine disinfection, a sagittal incision was made on the skull, exposing the parietal bone, and the skull was drilled at 1.5 mm behind the coronal suture and 2 mm left to the sagittal suture[44]. A bone window at 4–5 mm diameter was opened, to expose the dura mater, and a gasket (4.5 mm diameter; homemade) was placed on the bone window. A 40-g weight was allowed to fall from a 30 cm height to hit the gasket, resulting in the traumatic brain injury model (Figure 6). In the sham-surgery group, a bone window was also opened, but no weight was dropped.
Figure 6.
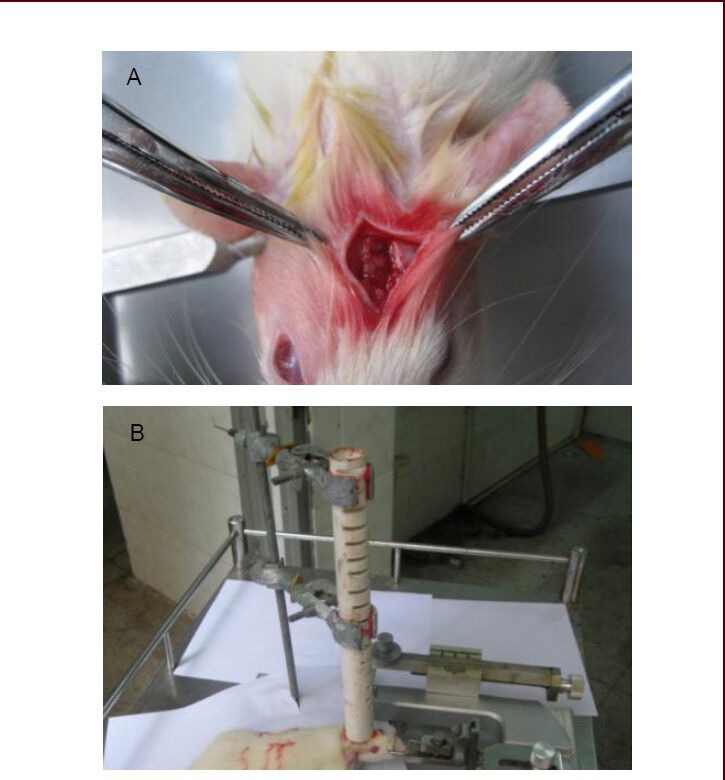
Establishment process of traumatic brain injury models.
(A) The skull was drilled at 1.5 mm behind the coronal suture and 2 mm left to the sagittal suture. A bone window at 4–5 mm diameter was opened, to expose dura mater.
(B) Brain injury model was produced using Feeney's free falling method.
Preparation of brain tissue specimens
After modeling, rats were intraperitoneally anesthetized with 10% (w/v) chloral hydrate (350 mg/kg), and the chest was opened to expose the heart. Following left ventricle cannulation, the right atrial appendage was cut and rinsed with normal saline until no liquid flowed out. Rats were killed and brain tissue was harvested on ice. Three specimens were collected from brain tissues: one at the anterior border of the injured area, which was used to determine brain water content using the dry-wet weight method; one at the injured area, which was fixed with 4% (w/v) paraformaldehyde and used for hematoxylin-eosin staining and immunohistochemical staining; and another specimen was stored at −80°C for western blot analysis for occludin and connexin 43.
Determination of brain water content
Brain tissue at the anterior border of injury was harvested, and wet weight was measured with an analytical balance (Shanghai Sunny Hengping Scientific Instruments Co., Ltd., Shanghai, China). Brain tissue specimens were baked at 105°C using an electrothermostat (Changsha Medical Instrument Factory, Hunan Province, China) for 24 hours until a constant dry weight was achieved. Brain water content was calculated using the dry-wet weight method (Elliot formula): Brain water content = (wet weight – dry weight)/wet weight × 100%.
Hematoxylin-eosin staining for pathological brain injury
Brain tissue was fixed with 4% (w/v) paraformaldehyde, followed by routine dehydration and embedding in paraffin. Tissue was cut into coronal slices at 4 μm thickness, and preserved at 4°C. Slices were subjected to xylene dewaxing, gradient ethanol dehydration, routine hematoxylin-eosin staining, and again dehydrated with gradient ethanol, followed by xylene transparency and mounting. Pathological changes in brain tissue were observed under a light microscope (Olympus, Tokyo, Japan).
Immunohistochemical staining for occludin and connexin 43 expression
Changes in occludin and connexin 43 expression were determined by immunohistochemical staining using the SP method (universal immunohistochemical staining kit, Beijing Zhong-shan Golden Bridge BioTechnology Co., Ltd., Beijing, China). Slices were hydrated, and rinsed with PBS three times, for 3 minutes each. Sections were subjected to antigen retrieval at high temperature and high pressure, incubated with 3% (v/v) methanol-hydrogen peroxide to block endogenous peroxidases at room temperature for 10–15 minutes, and rinsed with cold PBS three times, for 5 minutes each. Slices were then blocked and incubated at room temperature for 20 minutes and incubated with rabbit anti-occludin and connexin 43 polyclonal antibodies (1:200; Beijing Boisynthesis Biotechnology, Co., Ltd., Beijing, China). PBS instead of primary antibody served as a control. Sections were left at 4°C overnight, rinsed with cold PBS three times, for 5 minutes each, incubated with goat anti-rabbit IgG (1:200; Beijing Zhongshan Golden Bridge Biotechnology) at room temperature for 10–15 minutes, rinsed with cold PBS three times, for 5 minutes each, and incubated with streptavidin-biotin protein and peroxidase (1:200; Beijing Zhongshan Golden Bridge Biotechnology) at room temperature for 10–15 minutes. Staining was visualized using diaminobenzidine (Beijing Zhongshan Golden Bridge Biotechnology). Sections were counterstained with hematoxylin and observed under a light microscope.
Counts of positive cells
Positive staining was identified as brown or brown-yellow granule-like cells. The experimental results were independently assessed by two physicians using a double-blind method. The number of positive cells was counted under 400 × magnification. Cells were counted in four slices from each rat at each time point, in five visual fields form each slice, and the average values were measured.
Western blot assay for occludin and connexin 43 expression
After brain tissue was homogenated, total protein was extracted and protein concentration was quantified using the BCA Protein Quantitation Kit (Guangzhou Geneseed Biotech Co., Ltd., Guangzhou, Guangdong Province, China). Total protein was mixed with 5 × SDS protein sample buffer solution (5:1) and heated at 100°C for 5 minutes and stored at −20°C. Protein samples were subjected to SDS-PAGE. The gel was removed and samples were transferred to nitrocellulose membrane. The membrane was immersed in blocking liquid at room temperature for 4 hours, and stored at 4°C overnight. The membrane was incubated with rabbit anti-occludin, connexin 43 (1:300) polyclonal antibodies for 3 hours at room temperature, rinsed with tris buffered saline containing 0.1% (v/v) Tween-20 (TBST) and incubated with goat anti-rabbit IgG (1:1 000) for 1 hour. The membrane was washed with TBST and protein expression was visualized using the chemiluminescence method. Protein expression was quantified using Quantity One software (Bio-Rad, Hercules, CA, USA) and the expression levels of GAPDH in the same specimens were used as an internal reference. Target protein relative value was calculated according to the formula: Relative value = target protein (occludin and connexin 43) absorbance value / GAPDH absorbance value.
Statistical analysis
Statistical analysis was performed with SPSS 17.0 software (SPSS, Chicago, IL, USA) and results were expressed as mean ± SD. Differences between the two groups were compared using the paired t-test, and differences in the same group at different time points were compared by one-way analysis of variance. A P < 0.05 value was considered a significant difference.
Acknowledgments
We would like to thank Professor Chen B, Pan WD, Wang B and Liu YX from the Affiliated Hospital of Guangdong Medical College for their kind instructions and help in the study design, experimental completion and manuscript writing.
Footnotes
Funding: This study was supported by the Natural Science Foundation of Guangdong Province, No. 10151600101000002.
Conflicts of interest: None declared.
Ethical approval: This study was approved by the Animal Ethics Committee of Guangdong Medical College in China.
(Reviewed by Diwakarla S, Norman C, Chi ZF, Zhang XL)
(Edited by Wang LM, Yang Y, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Wang ZC. Hubei: Hubei Science and Technology Press; 2005. Wang Zhongcheng Neurosurgery. [Google Scholar]
- [2].Higashida T, Kreipke CW, Rafols JA, et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114(1):92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- [3].Béziaud T, Ru Chen X, El Shafey N, et al. Simvastatin in traumatic brain injury: effect on brain edema mechanisms. Crit Care Med. 2011;39(10):2300–2307. doi: 10.1097/CCM.0b013e3182227e4a. [DOI] [PubMed] [Google Scholar]
- [4].Wu ZL, Liao CH, Ren N. Study on correlation between connexin 43 and brain edema in experimental brain injury. Zhonghua Shenjing Waike Jibing Yanjiu Zazhi. 2008;7(3):201–204. [Google Scholar]
- [5].Feeney DM, Boyeson MG, Linn RT, et al. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211(1):67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- [6].Ling YP. Beijing: People's Medical Publishing House; 2002. Cell Biology. [Google Scholar]
- [7].ElAli A, Doeppner TR, Zechariah A, et al. Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke. 2011;42(11):3238–3244. doi: 10.1161/STROKEAHA.111.615559. [DOI] [PubMed] [Google Scholar]
- [8].Nakase T, Maeda T, Yoshida Y, et al. Ischemia alters the expression of connexins in the aged human brain. J Biomed Biotechnol 2009. 2009 doi: 10.1155/2009/147946. 147946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lichter-Konecki U, Mangin JM, Gordish-Dressman H, et al. Gene expression profiling of astrocytes from hyperammonemic mice reveals altered pathways for water and potassium homeostasis in vivo. Glia. 2008;56(4):365–377. doi: 10.1002/glia.20624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schulzke JD, Fromm M. Tight junction: molecular structure meets function. Ann N Y Acad Sci. 2009;1165:1–6. doi: 10.1111/j.1749-6632.2009.04925.x. [DOI] [PubMed] [Google Scholar]
- [11].Ikenouchi J, Furuse M, Furuse K, et al. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. 2005;171(6):939–945. doi: 10.1083/jcb.200510043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic storke. Neurobiol Dis. 2008;32(2):200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- [13].Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123(6 Pt 2):1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun YX, Chu GL. Expression changes of tight junction proteins ZO-1 and occludin after hypoxic-ischemic brain damage in neonatal rats. Zhongguo Xiandai Yixue Zazhi. 2010;20(21):3210–3213. [Google Scholar]
- [15].Ding Z, Liu J, Lin R, et al. Experimental pancreatitis results in increased blood-brain barrier permeability in rats: a potential role of MCP-1. J Dig Dis. 2012;13(3):179–185. doi: 10.1111/j.1751-2980.2011.00568.x. [DOI] [PubMed] [Google Scholar]
- [16].Kago T, Takagi N, Date I, et al. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2006;339(4):1197–1203. doi: 10.1016/j.bbrc.2005.11.133. [DOI] [PubMed] [Google Scholar]
- [17].Wu LW, Yin F, Peng J, et al. The tight junction proteins ZO-1, occludin and actin participate in the permeability increasing of blood-brain barrier induced by hypoxia- ischemia. Zhongguo Dang Dai Er Ke Za Zhi. 2008;10(4):513–516. [PubMed] [Google Scholar]
- [18].Liu J, Jin X, Liu KJ, et al. Matrix metalloproteinase-2- mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32(9):3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ohsumi A, Nawashiro H, Otani N, et al. Temporal and spatial profile of phosphorylated connexin43 after traumatic brain injury in rats. J Neurotrauma. 2010;27(7):1255–1263. doi: 10.1089/neu.2009.1234. [DOI] [PubMed] [Google Scholar]
- [20].Brokamp C, Todd J, Montemagno C, et al. Electrophysiology of single and aggregate cx43 hemichannels. PLoS One. 2012;7(10):e47775. doi: 10.1371/journal.pone.0047775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bezzi P, Volterra A. A neuron-glia signalling network in the active brain. Curr Opin Neurobiol. 2001;11(3):387–394. doi: 10.1016/s0959-4388(00)00223-3. [DOI] [PubMed] [Google Scholar]
- [22].Evans WH, Boitano S. Connexin mimetic peptides: specific inhibitors of gap-junctional intercellular communication. Biochem Soc Trans. 2001;29(Pt 4):606–612. doi: 10.1042/bst0290606. [DOI] [PubMed] [Google Scholar]
- [23].Ebong EE, Kim S, DePaola N. Flow regulates intercellular communication in HAEC by assembling functional Cx40 and Cx37 gap junctional channels. Am J Physiol Heart Circ Physiol. 2006;290(5):H2015–2023. doi: 10.1152/ajpheart.00204.2005. [DOI] [PubMed] [Google Scholar]
- [24].Evans WH, Leybaert L. Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell Commun Adhes. 2007;14(6):265–273. doi: 10.1080/15419060801891034. [DOI] [PubMed] [Google Scholar]
- [25].Duval N, Gomès D, Calaora V, et al. Cell coupling and Cx43 expression in embryonic mouse neural progenitor cells. J Cell Sci. 2002;115(Pt 16):3241–3251. doi: 10.1242/jcs.115.16.3241. [DOI] [PubMed] [Google Scholar]
- [26].Liao Y, Day KH, Damon DN, et al. Endothelial cell-specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc Natl Acad Sci U S A. 2001;98(17):9989–9994. doi: 10.1073/pnas.171305298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Anderson MF, Blomstrand F, Blomstrand C, et al. Astrocytes and stroke: networking for survival? Neurochem Res. 2003;28(2):293–305. doi: 10.1023/a:1022385402197. [DOI] [PubMed] [Google Scholar]
- [28].Old EA, Malcangio M. Chemokine mediated neuron-glia communication and aberrant signalling in neuropathic pain states. Curr Opin Pharmacol. 2012;12(1):67–73. doi: 10.1016/j.coph.2011.10.015. [DOI] [PubMed] [Google Scholar]
- [29].Ezan P, André P, Cisternino S, et al. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(8):1457–1467. doi: 10.1038/jcbfm.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Benarroch EE. Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin Proc. 2005;80(10):1326–1338. doi: 10.4065/80.10.1326. [DOI] [PubMed] [Google Scholar]
- [31].Xie M, Yi C, Luo X, et al. Glial gap junctional communication involvement in hippocampal damage after middle cerebral artery occlusion. Ann Neurol. 2011;70(1):121–132. doi: 10.1002/ana.22386. [DOI] [PubMed] [Google Scholar]
- [32].Kozoriz MG, Bechberger JF, Bechberger GR, et al. The connexin43 C-terminal region mediates neuroprotection during stroke. J Neuropathol Exp Neurol. 2010;69(2):196–206. doi: 10.1097/NEN.0b013e3181cd44df. [DOI] [PubMed] [Google Scholar]
- [33].Jayakumar AR, Panickar KS, Curtis KM, et al. Na-K-Cl cotransporter-1 in the mechanism of cell swelling in cultured astrocytes after fluid percussion injury. J Neurochem. 2011;117(3):437–448. doi: 10.1111/j.1471-4159.2011.07211.x. [DOI] [PubMed] [Google Scholar]
- [34].Wilson AC, Clemente L, Liu T, et al. Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochim Biophys Acta. 2008;1782(6):401–407. doi: 10.1016/j.bbadis.2008.02.011. [DOI] [PubMed] [Google Scholar]
- [35].Yang D, Li SY, Yeung CM, et al. Lycium barbarum extracts protect the brain from blood-brain barrier disruption and cerebral edema in experimental stroke. PLoS One. 2012;7(3):e33596. doi: 10.1371/journal.pone.0033596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cui XN, Yin L, Wang YL. Effect of aquaporin 4 on traumatic brain edema. Zhongguo Kangfu Lilun yu Shijian. 2005;11(9):719–721. [Google Scholar]
- [37].Zhang L, Yuan F. Enhanced expression of aquaporin-4 aggravates brain edema after traumatic brain injury in rat. Shoudu Yike Daxue Xuebao. 2008;29(6):732–736. [Google Scholar]
- [38].Liu H, Yang M, Qiu GP, et al. Aquaporin 9 in rat brain after severe traumatic brain injury. Arq Neuropsiquiatr. 2012;70(3):214–220. doi: 10.1590/s0004-282x2012000300012. [DOI] [PubMed] [Google Scholar]
- [39].Nag S, Venugopalan R, Stewart DJ. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood-brain barrier breakdown. Acta Neuropathol. 2007;114(5):459–469. doi: 10.1007/s00401-007-0274-x. [DOI] [PubMed] [Google Scholar]
- [40].Avila MA, Sell SL, Hawkins BE, et al. Cerebrovascular connexin expression: effects of traumatic brain injury. J Neurotrauma. 2011;28(9):1803–1811. doi: 10.1089/neu.2011.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lin JY, Lin NY, Huang G. Expression and role of connexin-43 in brain tissue of hypoxic-ischemic neonatal rats. Zhongguo Xiandai Yisheng. 2007;45(8):18–20. [Google Scholar]
- [42].Zhang YM, Qi ST, Huang GL, et al. Blocking of gap junction communication affecting neuronal apoptosis in rabbit hypothalamus after explosive brain injury. Zhonghua Shenjing Yixue Zazhi. 2010;9(5):460–464. [Google Scholar]
- [43].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006 Sep 30; [Google Scholar]
- [44].Paxions G, Watson C. 6th ed. San Diego: Academic Press; 2005. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]