

Abstract
Even though many studies have identified roles of proteasomes in axonal degeneration, the molecular mechanisms by which axonal injury regulates proteasome activity are still unclear. In the present study, we found evidence indicating that extracellular calcium influx is an upstream regulator of proteasome activity during axonal degeneration in injured peripheral nerves. In degenerating axons, the increase in proteasome activity and the degradation of ubiquitinated proteins were significantly suppressed by extracellular calcium chelation. In addition, electron microscopic findings revealed selective inhibition of neurofilament degradation, but not microtubule depolymerization or mitochondrial swelling, by the inhibition of calpain and proteasomes. Taken together, our findings suggest that calcium increase and subsequent proteasome activation are an essential initiator of neurofilament degradation in Wallerian degeneration.
Keywords: neural regeneration, peripheral nerve injury, neurofilament degradation, sciatic nerve, calcium, calpain, mitochondria, microtubule depolymerization, axon, axon degeneration, neuroregeneration
INTRODUCTION
Following nerve injury, a catastrophic destruction of axons occurs in the distal side of an injured axon, and the degeneration is the result of several enzymatic processes that are distinct from cell death signaling pathways[1,2,3,4,5,6]. Energy failure in axons following nerve injury appears to result in the accumulation of intraaxonal sodium, and it subsequently leads to a rise of intraaxonal calcium levels through the activation of the reverse flow of the sodium/calcium exchanger[1,7,8]. One of the enzymes activated by extracellular calcium influx in axons is calpain, a calcium-dependent cysteine protease, and calpain breaks down axonal cytoskeletal structures such as neurofilaments[8,9,10,11,12,13,14,15]. Axonal degeneration encompasses the destruction of organelles, including mitochondria, as well as the destruction of axonal cytoskeletons. In axonal degeneration following nerve injury, rod-shaped mitochondria swell and break down[16,17], and this mitochondrial failure appears to be related to the energy disturbance observed in degenerating axons[16,17,18]. In contrast to the calcium-mediated destruction of neurofilament, we recently reported that microtubule depolymerization and mitochondrial swelling are not tightly regulated by extracellular calcium influx[17].
The ubiquitin-proteasome pathway has previously been implicated in axonal degeneration[19,20,21,22,23,24,25,26,27,28,29]. Proteasomes may de grade intracellular molecules that promote axonal survival such as AKT and nicotinamide mononucleotide adenylyltransferase following nerve injury[20,21], thereby inducing axonal degeneration, or proteasomes may directly regulate the destruction of cytoskeletal proteins[22,29]. Even though many studies have identified the roles of proteasomes in axonal degeneration, the molecular mechanisms by which axonal injury regulates protea-some activity are still unclear. In the present study, using sciatic nerve explant cultures[30,31,32], we tried to find evidence showing that extracellular calcium influx is an upstream regulator of proteasome activation and that proteasomes may not be related to microtubule depolymerization and mitochondrial swelling.
RESULTS
Inhibition of extracellular calcium influx and proteasomes significantly prevented neurofilament degradation in sciatic nerve explant cultures
We employed sciatic nerve explant cultures, which is a good ex vivo model for axonal degeneration[30,31,32], to determine the molecular mechanism of axonal degeneration. After 3 days of incubation (3 days in vitro, 3DIV), axonal degeneration was analyzed by neurofilament (high molecular weight) immunofluorescence staining and western blot analysis. In accordance with previous findings, an extracellular calcium chelator ethylene glycol tetraacetic acid (5 mmol/L), a calpain inhibitor (calpeptin, 50 μmol/L)[33] and a proteasome inhibitor (MG132; 20 μmol/L)[34] significantly protected against axonal degeneration (Figure 1A, B). As we previously reported[17], the repletion of energy with nicotinamide adenine dinucleotide, nicotinamide and methyl pyruvate also prevented axonal degeneration in sciatic nerve explant cultures (Figure 1A, B). Consistent with the results of immunofluorescence staining, western blot analysis showed that ethylene glycol tetraacetic acid and MG132 significantly suppressed neurofilament degradation (Figure 1C, D), indicating a role of the calcium/calpain pathway and proteasomes in neurofilament degradation in an axonal degeneration model.
Figure 1.
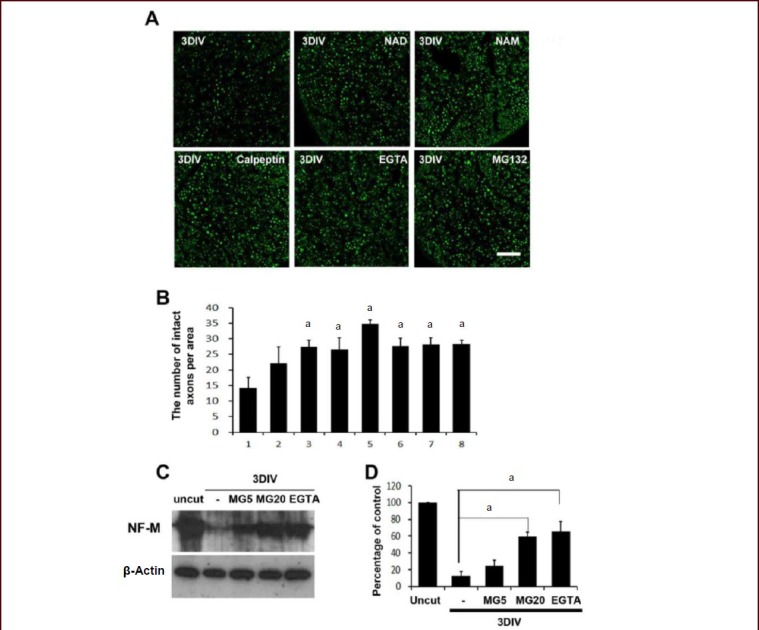
Extracellular calcium and proteasomes participated in neurofilament degradation in sciatic nerve explant cultures.
(A) Immunofluorescence staining against high molecular weight neurofilament (NF). Immunofluorescence microscopic images of cross-sections of sciatic nerve explants cultured for 3DIV were analyzed under a laser confocal microscope. Green fluorescence dots indicate neurofilament-positive axons. DIV: day in vitro. Scale bar: 100 μm. (B) Quantitative analysis of the number of high molecular weight NF in the sciatic nerve explant cultures. aP < 0.05, vs. vehicle-treated nerve controls. (n = 3; mean ± SD). 1: Vehicle; 2: nicotinamide adenine dinucleotide (NAD; 5 mmol/L); 3: nicotinamide (NAM; 20 mmol/L); 4: methyl pyruvate (20 mmol/L); 5: NAD (5 mmol/L) + methyl pyruvate (20 mmol/L); 6: ethylene glycol tetraacetic acid (EGTA, an extracellular calcium chelator; 5 mmol/L); 7: calpeptin (50 μmol/L; a calpain inhibitor); 8: MG132 (20 μmol/L; a proteasome inhibitor). (C) Western blot analysis showing the degradation of medium chain neurofilament (NF-M) in sciatic nerve explants cultured for 3DIV. MG5: 5 μmol/L of MG132; MG20: 20 μmol/L of MG132, EGTA (5 mmol/L). (n = 3; mean ± SD). (D) Quantitative analysis of NF-M immunoreactive bands. The intensity of bands was displayed as relative intensity to uncut nerve control. At least three independent experiments were performed for each condition. -: No treatment. aP < 0.05. Differences in the means between groups were statistically assessed using one-way analysis of variance followed by Bonferroni post hoc test.
Calcium-dependent activation of the proteasome and calpain
We next examined the role of calcium influx and calpain activation in proteasome activation in degenerating axons. First, calpain activity in the distal stump of an injured sciatic nerve was determined in vivo (Figure 2A). It was found that a sciatic nerve axotomy induced the calpain activation in a time-dependent manner. At 48 hours after axotomy, calpain activity was increased by ~3 fold compared to uncut controls. We also observed calpain activation in sciatic nerve explant cultures at 2 DIV (Figure 2B). As expected, the inclusion of calpeptin and extracellular calcium chelation by ethylene glycol tetraacetic acid in explant cultures completely suppressed calpain activity. This finding suggests that extracellular calcium influx regulates calpain activity during Wallerian degeneration.
Figure 2.
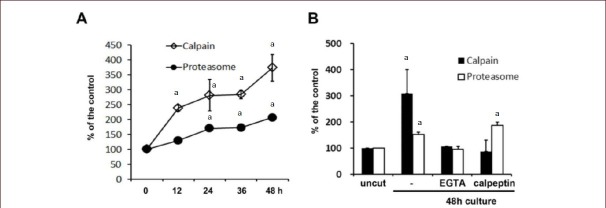
Calcium-induced activation of proteasome in the sciatic nerves following nerve injury.
(A) Following axotomy, the distal stump of the sciatic nerves was processed to measure calpain and proteasome activity. Each time point indicates the time after axotomy (n = 3; mean ± SD). aP < 0.05, vs. uncut control nerves (0 hour, 100 %). (B) Levels of calpain and proteasome activity in sciatic nerve explant cultures. Sciatic nerve explants were cultured for 2 days in the presence or absence of ethylene glycol tetraacetic acid (EGTA; 5 mmol/L) and MG132 (20 μmol/L), and the activities were measured. At least three independent experiments were performed for each condition. -: No treatment. aP < 0.05, vs. uncut control nerves. Differences in the means between groups were statistically assessed using one-way analysis of variance followed by Bonferroni post hoc test.
We studied the activity of 20S proteasomes[35] in degenerating axons (Figure 2A). Proteasome activity in the distal stump was increased within 12 hours following axotomy, and maximal activity was reached at 48 hours after axotomy. We next examined whether extracellular calcium influx is involved in proteasome activation using sciatic nerve explant cultures. The addition of ethylene glycol tetraacetic acid completely suppressed proteasome activation (Figure 2B). However, calpeptin did not inhibit proteasome activation, suggesting that calcium influx, but not calpain, is the primary regulator of proteasome activity.
Destruction of ubiquitinated proteins were inhibited by calcium chelation
Ubiquitinated proteins are degraded by proteasomes, and the inhibition of proteasomes with a proteasome inhibitor results in the accumulation of ubiquitinated proteins. Thus, extracellular calcium chelation may prevent the degradation of ubiquitinated proteins by inhibiting proteasomes. Using western blot analysis, we found that several ubiquitinated proteins were degraded in sciatic nerve explants, and the addition of ethylene glycol tetraacetic acid prevented the degradation of ubiquitinated proteins (Figure 3). Together, these findings further suggest that extracellular calcium influx is an upstream regulator of proteasomes.
Figure 3.
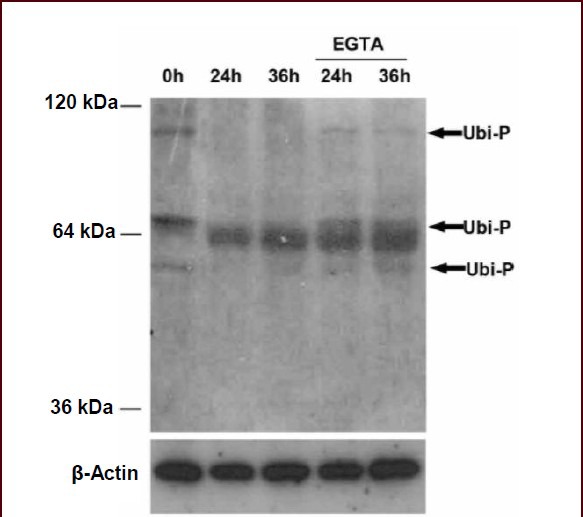
Calcium chelation suppressed the destruction of ubiquitinated proteins in degenerating axons.
Sciatic nerve explants were cultured for 24 and 36 hours in the presence or absence of ethylene glycol tetraacetic acid (EGTA; 5 mmol/L). Western blot analysis was performed to demonstrate ubiquitinated proteins (Ubi-P) in sciatic nerve explants using an antibody against ubiquitin. β-Actin: beta-actin.
Proteasomes were required for neurofilament degradation but not for energy depletion, microtubule depolymerization or mitochondrial swelling
It was recently reported that extracellular calcium chelation prevented neurofilament degradation but not energy depletion, microtubule depolymerization or mitochondrial swelling in degenerating axons[17]. Because proteasome activation is downstream of extracellular calcium influx in degenerating axons, proteasomes also may not play a role in energy depletion, microtubule depolymerization or mitochondrial swelling. We thus investigated the role of proteasomes in these processes using electron microscopy (Figure 4A). At 2DIV, sciatic nerves showed absolute axon degeneration and swollen mitochondria, as demonstrated by a decreased length index and increased mitochondrial diameter (Figure 4A–C, see Materials and Methods). Calpeptin (50 μmol/L) significantly suppressed axonal degeneration, however, calpain-protected axons contained swollen mitochondria (length index, 1.24 ± 0.28; diameter, 0.53 ± 0.15; Figure 4A–C) and did not show any microtubules when viewed using electron microscopy (Figure 4A). In explant cultures treated with MG132 (20 μmol/L) for 2 days, the length of mitochondria in protected axons was 1.39 ± 0.07, and the mean diameter was 0.43 ± 0.02 μm (Figure 4A–C). In addition, neurofilaments, but not microtubules, were well preserved in MG132-treated axons (Figure 4A), suggesting that both mitochondrial swelling and microtubule depolymerization still occur even after proteasome activity is suppressed.
Figure 4.

Calcium and the proteasome are late effectors in axonal degeneration.
(A) Representative ultrathin longitudinal cross-sections showing protection against axonal degeneration by MG132 (20 μmol/L) and calpeptin (50 μmol/L) in sciatic nerve explant cultures. Arrowheads: Microtubules. Scale bars: 0.2 μm. (B) Mean diameters (μm) of mitochondria in degenerating axons. (C) Mean length index (length/diameter) of mitochondria in degenerating axons. (D) The decrease of NAD and ATP levels in sciatic nerve explants cultured for 1 day could not be rescued by MG132. (B–D) Data were expressed as mean ± SD (n = 3). At least three independent experiments were performed for each condition. aP < 0.05, vs. uncut nerve controls (0 day). Differences in the means between groups were statistically assessed using one-way analysis of variance followed by Bonferroni post hoc test.
Energy failure, as demonstrated by the decrease of nicotinamide adenine dinucleotide and ATP levels in the sciatic nerve explant cultures, is known to be an early event that precedes microtubule depolymerization and mitochondrial swelling[17]. In this study, we measured nicotinamide adenine dinucleotide and ATP levels in degenerating sciatic nerves in the presence or absence of MG132 using sciatic nerve explant cultures (Figure 4D), and found that energy failure in degenerating axons was not rescued by MG132.
DISCUSSION
In the present study, we revealed for the first time that extracellular calcium influx affects proteasome activity in degenerating axons. This is in agreement with previous results showing that proteasome activity is regulated by calcium in neuronal and non-neuronal cells[36,37,38,39]. Our findings are novel because they show that calcium regulates the final steps of axonal degeneration via both calpain and proteasomes.
Axonal degeneration in rodent peripheral nerves following nerve injury shows the sequence of two morphologically distinct phases, a latency period and an execution period[1]. During the latency period, axonal cytoskeletons appear to be normal. However, we have recently reported that the major energy failure and microtubule depolymerization begin to occur in this period[17]. During the execution period, calcium influx from outside the axons is known to be a critical factor in the catastrophic destruction of axons. Our previous findings demonstrated that the inhibition of calcium influx could not prevent energy failure and microtubule depolymerization further supports a specific role of calcium influx during the execution period.
In the present study, we found evidence suggesting that proteasomes might also be a late effector, not an early initiator, of axonal degeneration. First, proteasome inhibition did not block energy depletion or microtubule depolymerization, two events that occur during the latency period. Second, neurofilament degradation was inhibited by proteasome inhibition. Finally, proteasomes were regulated by an extracellular calcium influx. Neurofilaments have previously been reported as a target of the ubiquitin-proteasome pathway and calpain[22,39,40], and thus, neurofilament degradation during axonal degeneration might also be performed by both calpain and proteasomes. It has previously been reported that microtubule depolymerization in degenerating cultured axons is delayed by proteasome inhibitors[19,21]. The reasons for the discrepancy between these findings and our results are currently unknown, although they may have resulted from different experimental conditions and analysis methods. For example, we used explant cultures, whereas they employed cultured primary neurons, and we used an electron microscopy to examine microtubule integrity whereas they used immunofluorescent light microscopy. It is possible that non-neuronal cells such as Schwann cells included in the ex vivo system have some effects on axonal degeneration. In addition, the explant culture system may require higher concentration of reagent than the concentration used in cell culture. However, we could not test this possibility because we have experienced that higher levels of MG132 induced non-specific toxicity on non-neuronal cells in the explant cultures. Further studies on how the ubiquitin-proteasome pathway regulates cytoskeletal degeneration at a molecular level will provide a better understanding of the molecular mechanisms of axonal degeneration.
Although calcium appeared to regulate proteasome activity in degenerating axons, we could not find the inhibitory effect of calpeptin on proteasome activation. This finding may indicate a calpain-independent activation of proteasomes in axonal degeneration. However, it seems too early to draw such a conclusion for the following reasons. First, there are more than ten calpains in eukaryotic cells that could not be suppressed by calpeptin[33]. Therefore, the potential role of calpeptin-insensitive calpains in proteasome activation cannot be excluded. Second, calpain-dependent proteasome activation in a proteolytic degradation has been reported[36,38]. It may be possible that the role of calpain in proteasome activations is cell-type specific or is dependent on local milieu in a cell. Further studies on the mechanistic relation between calpain and proteasomes are required.
The failure of axonal energy metabolism has been proposed as a key mechanism of axonal degeneration after injury[41,42]. The mechanisms by which axonal injury results in energy failure are still unknown. It was recently proposed that proteasomal destruction of nicotinamide mononucleotide adenylyltransferase, a nicotinamide adenine dinucleotide source, may underlie injury-induced axonal degeneration in cultured neurons[20]. However, it is still uncertain whether nicotinamide mononucleotide adenylyltransferase destruction is indeed responsible for energy failure in degenerating axons. We found that proteasome inhibition could not rescue nicotinamide adenine dinucleotide and adenosine triphosphate (ATP) reduction in sciatic nerve explant cultures, suggesting that the ubiquitin-proteasome pathway may not participate in energy failure in injured axons.
Because calcium buffering by intact mitochondria is important for calcium homeostasis in cells[42,43,44], swollen and dysfunctional mitochondria may participate in axonal degeneration by providing an intra-axonal calcium increase[16,17]. The activation of mitochondrial permeability transition pore is a mechanism of mitochondrial swelling in many pathological conditions and has been implicated in the loss of calcium buffering during axonal degeneration[42,43,44]. Several mitochondrial proteins including voltage-dependent anion channel, a component of mitochondrial permeability transition pore, are targets of the ubiquitin-proteasome pathway and proteasomes appear to implicate in mitochondrial swelling in a pathological condition[45,46,47,48,49]. However, in this study, we did not observe an absolute requirement of proteasomes in mitochondrial swelling during axonal degeneration in peripheral nerves. It was recently shown that mitochondrial swelling is an outcome but not a cause of energy failure in axonal degeneration[17], and energy failure as demonstrated by nicotinamide adenine dinucleotide and ATP reduction was not rescued by proteasome inhibition in this study. Therefore, the ubiquitin-proteasome pathway may not be a principal stimulator for mitochondrial degeneration in degenerating axons. This is in agreement with a recent study showing that axonal mitochondria may not be an initiating factor for axonal degeneration[50].
In conclusion, our results show a role of extracellular calcium influx in the regulation of proteasome-mediated protein degradation in axonal degeneration. This suggests that proteasomes are a calcium-dependent late effector of axonal degeneration.
MATERIALS AND METHODS
Design
A randomized, controlled ex vivo experiment.
Time and setting
This experiment was performed in the Department of Physiology, Dong-A University Medical School, South Korea from June 2011 to May 2013.
Materials
A total of 102 C57BL/6 female mice were obtained from Samtaco Inc, Daejeon, South Korea for providing sciatic nerve explants. Antibodies against tubulin and ubiquitin were obtained from Sigma (St. Louis, MO, USA). Polyclonal antibodies against high and medium chain neurofilament, calpeptin and MG132 were purchased from Millipore, Billerica, MA, USA. Other reagents were purchased from Sigma.
Methods
Sciatic nerve explant cultures
Sciatic nerve explant cultures were performed as previously reported[30]. Sciatic nerves were sectioned 5 mm proximal to the tibioperoneal bifurcation with a fine iris scissor (FST Inc., Foster City, CA, USA) under anesthesia by intraperitoneal injection of a mixture of 10% ketamine hydrochloride (Sanofi-Ceva, Dόsseldorf, Germany; 0.1 mL/100 g body weight) and Rompun (Bayer, Leverkusen, Germany; 0.05 mL/100 g body weight). Right side sciatic nerves were collected, and the connective tissues surrounding the nerves were carefully detached in ice-cold calcium/magnesium-free Hank's buffered solution under a stereomicroscope (SZX16, Olympus, Osaka, Japan). The sciatic nerves were then cut into small explants of about 3 mm in length. The explants were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 1% (v/v) heat-inactivated fetal bovine serum at 37°C with 5% CO2 for the indicated time. After culture, the nerve explants were prepared for further experiments. For uncut nerve controls, sciatic nerves (right side) of adult mice were removed and then used for an analysis within minutes.
Immunofluorescence staining
After sciatic nerve explant cultures or sciatic nerve axotomy, the sciatic nerve was fixed with 4% paraformaldehyde overnight. The sciatic nerves were cryoprotected in 20% sucrose, and frozen cross-sections (14 μm thick) were made with a cryostat (Frigocut, Leica, Germany), and processed for immunofluorescent staining. The slides were blocked with PBS containing 0.2% Triton X-100 and 2% bovine serum albumin for 1 hour. The sections were then incubated with a rabbit polyclonal antibody against high chain neurofilament (NF-M, 1:2 000) for 16 hours at 4°C and washed three times with PBS. Next, the slides were incubated with Alexa 488-conjugated goat anti-rabbit IgG (Amersham, Piscatway, NJ, USA;1:800) for 3 hours at room temperature. The sections were then washed three times with PBS, and coverslips were adhered to glass slides with a mounting medium and viewed under a laser confocal microscope (LSM510, Carl Zeiss, Germany). For the quantitative estimation of axonal degeneration, we counted the number of neurofilaments stained positively from two randomly selected areas (100 μm × 100 μm) of a sciatic nerve and six nerves were used for counting.
Calpain and proteasome activity assay
The quantitative measurement of calpain activity was performed using a Calpain Activity Assay Kit (BioVision, Mountain View, CA, USA). Sciatic nerves were homogenized in an extraction buffer (Qiagen, Valencia, CA, USA) with a TissueLyser system (Qiagen). The lysates were centrifuged at 8 000 × g, and the supernatants were used for enzymatic reactions. The calpain substrate was Ac-LLY-AFC, and the reaction product emited a yellow-green fluorescence measured at 505 nm with a spectrofluorometer (Molecular Devices, Sunnyvale, CA, USA).
For the proteasome assay, a 20S Proteasome Assay System (Enzo, Plymouth, PA, USA) was employed. Sciatic nerves were homogenized in an extraction buffer (Qiagen) with a TissueLyser system. The lysates were centrifuged at 8 000 × g, and the supernatant was collected. The 20S proteasome substrate was Suc-LLVY-AMC, and proteasome activity was determined by measuring the fluorescence of the reaction product at 460 nm.
Electron microscopic examination and morphometry of the mitochondria
The processing of sciatic nerves for electron microscopy has been previously reported[17]. Briefly, sciatic nerve explant cultures were fixed in 2% glutaraldehyde in 0.1 mol/L phosphate buffer (pH, 7.2). For semithin sections, 2 mm-long sciatic nerves were Epon-resin embedded, sectioned, stained with toluidine blue and examined with an Axiophot light microscope (Carl Zeiss, Germany). Ultrathin sections were examined with a Hitachi transmission electron microscope equipped with a digital camera. At least three independent experiments were performed for each condition, and about 100 randomly selected myelinated axons from three experiments were analyzed for cytoskeletal destruction and mitochondrial morphometry. In longitudinal sections, rod-shaped mitochondria are frequently found in axons, and we measured the length index of the mitochondria (length/diameter) to demonstrate mitochondrial swelling in degenerating axons using about 100 mitochondria. The mean length index and diameter of the mitochondria in normal axons were 6.92 and 0.21 μm, respectively (Figure 4A–C).
Western blot analysis
For western blot analysis, the distal stumps of the sciatic nerves were harvested and homogenized with a polytron homogenizer in modified radioimmune precipitation assay lysis buffer (150 mmol/L NaCl, 1% Nonidet P-40, 1 mmol/L ethylenediamine tetraacetic acid, 0.5% deoxycholic acid, 2 μg/mL aprotinin, 1 mmol/L phenylmethylsulfonyl fluoride, 5 mmol/L benzamidine, 1 mmol/L sodium orthovanadate, and 1 × protease inhibitor cocktail). The lysates were centrifuged at 8 000 × g for 10 minutes at 4°C, and the supernatant was collected. Protein (25–35 μg) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred onto a nitrocellulose membrane (Amersham). After blocking with 0.1% Tween-20 and 5% nonfat dry milk in Tris-buffered saline (TBS, 25 mmol/L Tris-HCl pH 7.5, 140 mmol/L NaCl) at room temperature for 1 hour, the membrane was incubated with a rabbit polyclonal antibody against medium chain neurofilament (1: 2 000; Sigma) or beta-actin (1:5 000; Sigma) in TBST containing 2% nonfat dry milk at 4°C overnight. After three washes with TBST for 15 minutes each, the membranes were incubated with a horseradish peroxidase-conjugated goat anti-rabbit IgG (1:3 000; Amersham) for 1 hour at room temperature. The signals were detected using the Enhanced Chemiluminescence System (Amersham).
Nicotinamide adenine dinucleotide and ATP measurements
Quantitative measurement of nicotinamide adenine dinucleotide and ATP concentration in the sciatic nerves was performed as previously reported[17]. Briefly, an EnzyChrom Assay Kit (BioAssay Systems, Hayward, CA, USA) was used for nicotinamide adenine dinucleotide measurement. After sciatic nerve explant culture, the sciatic nerves were homogenized in an extraction buffer (BioAssay Systems) and the supernatants were used for enzymatic reactions. The reaction products were chromogenic and were measured at 565 nm with a spectrofluorometer. For ATP measurement, Enliten ATP Assay System (Promega, San Luis Obispo, CA, USA) was used[17]. Sciatic nerve lysates were quick frozen with liquid nitrogen, and then boiled for 3 minutes at 95°C. The extracts were centrifuged at 8 000 × g for 20 minutes at 4°C, and the supernatant was collected. Luciferase reaction was performed with components of the assay system as Vendor's instruction.
Statistical analysis
Differences in the means between groups were statistically assessed using one-way analysis of variance followed by Bonferroni post hoc test (SPSS 12.0; IBM, Armonk, New York, NY, USA). Data were represented as mean ± SD. The differences were considered to be statistically significant at P < 0.05.
Footnotes
Funding: This research was supported by research funds from Dong-A University, South Korea.
Conflicts of interest: None declared.
Ethical approval: All procedures were approved by the Dong-A University Committee on animal research, which follows the guide for animal experiments established by the Korean Academy of Medical Sciences, South Korean.
(Reviewed by Zhong LR, Zhang N, Liu Q, Wang LS)
(Edited by Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Court FA, Coleman MP. Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 2012;35:364–372. doi: 10.1016/j.tins.2012.04.001. [DOI] [PubMed] [Google Scholar]
- [3].Yan T, Feng Y, Zhai Q. Axon degeneration: Mechanisms and implications of a distinct program from cell death. Neurochem Int. 2010;56:529–534. doi: 10.1016/j.neuint.2010.01.013. [DOI] [PubMed] [Google Scholar]
- [4].Martinelli P, Rugarli EI. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim Biophys Acta. 2010;1797:1–10. doi: 10.1016/j.bbabio.2009.07.013. [DOI] [PubMed] [Google Scholar]
- [5].Saxena S, Caroni P. Mechanisms of axon degeneration: from development to disease. Prog Neurobiol. 2007;83:174–191. doi: 10.1016/j.pneurobio.2007.07.007. [DOI] [PubMed] [Google Scholar]
- [6].Conforti L, Adalbert R, Coleman MP. Neuronal death: where does the end begin? Trends Neurosci. 2007;304:159–166. doi: 10.1016/j.tins.2007.02.004. [DOI] [PubMed] [Google Scholar]
- [7].George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Glass JD, Culver DG, Levey AI, et al. Very early activation of m-calpain in peripheral nerve during Wallerian degeneration. J Neurol Sci. 2002;196:9–20. doi: 10.1016/s0022-510x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- [9].Ma M. Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol Dis. 2013;60:61–79. doi: 10.1016/j.nbd.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ma M, Ferguson TA, Schoch KM, et al. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol Dis. 2013;56:34–46. doi: 10.1016/j.nbd.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kilinc D, Gallo G, Barbee KA. Mechanical membrane injury induces axonal beading through localized activation of calpain. Exp Neurol. 2009;219:553–561. doi: 10.1016/j.expneurol.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Touma E, Kato S, Fukui K, et al. Calpain-mediated cleavage of collapsin response mediator protein(CRMP)-2 during neurite degeneration in mice. Eur J Neurosci. 2007;26:3368–3381. doi: 10.1111/j.1460-9568.2007.05943.x. [DOI] [PubMed] [Google Scholar]
- [13].Huh JW, Franklin MA, Widing AG, et al. Regionally distinct patterns of calpain activation and traumatic axonal injury following contusive brain injury in immature rats. Dev Neurosci. 2006;285:466–476. doi: 10.1159/000094172. [DOI] [PubMed] [Google Scholar]
- [14].Thompson SN, Gibson TR, Thompson BM, et al. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006;201:253–265. doi: 10.1016/j.expneurol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- [15].Das A, Guyton MK, Smith A, et al. Calpain inhibitor attenuated optic nerve damage in acute optic neuritis in rats. J Neurochem. 2013;124:133–146. doi: 10.1111/jnc.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barrientos SA, Martinez NW, Yoo S, et al. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Park JY, Jang SY, Shin YK, et al. Mitochondrial swelling and microtubule depolymerization are associated with energy depletion in axon degeneration. Neuroscience. 2013;238:258–269. doi: 10.1016/j.neuroscience.2013.02.033. [DOI] [PubMed] [Google Scholar]
- [18].Avery MA, Rooney TM, Pandya JD, et al. WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhai Q, Wang J, Kim A, et al. Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron. 2003;39:217–225. doi: 10.1016/s0896-6273(03)00429-x. [DOI] [PubMed] [Google Scholar]
- [20].Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wakatsuki S, Saitoh F, Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Biol. 2011;13:1415–1423. doi: 10.1038/ncb2373. [DOI] [PubMed] [Google Scholar]
- [22].Gou JP, Leterrier JF. Possible involvement of ubiquitination in neurofilament degradation. Biochem Biophy Res Comm. 1995;217:529–538. doi: 10.1006/bbrc.1995.2808. [DOI] [PubMed] [Google Scholar]
- [23].Simonin Y, Ferrer-Alcon M, Ferri A, et al. The neuroprotective effects of the WldS gene are correlated with proteasome expression rather than apoptosis. Eur J Neurosci. 2007;25:2269–2274. doi: 10.1111/j.1460-9568.2007.05501.x. [DOI] [PubMed] [Google Scholar]
- [24].Hoopfer ED, McLaughlin T, Watts RJ, et al. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- [25].MacInnis BL, Campenot RB. of Wallerian degeneration and nerve growth factor withdrawal-induced pruning of axons of sympathetic neurons by the proteasome and the MEK/Erk pathway. Mol Cell Neurosci. 2005;28:430–439. doi: 10.1016/j.mcn.2004.10.003. [DOI] [PubMed] [Google Scholar]
- [26].Korhonen L, Lindholm D. The ubiquitin proteasome system in synaptic and axonal degeneration: a new twist to an old cycle. J Cell Biol. 2004;165:27–30. doi: 10.1083/jcb.200311091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ehlers MD. Deconstructing the axon: Wallerian degeneration and the ubiquitin-proteasome system. Trends Neurosci. 2004;27:3–6. doi: 10.1016/j.tins.2003.10.015. [DOI] [PubMed] [Google Scholar]
- [28].Watts RJ, Hoopfer ED, Luo L. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/s0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- [29].Shi K, Li J, Han K, Jiang H, et al. The degradation of kinesin-like calmodulin binding protein of D. salina (DsKCBP) is mediated by the ubiquitin-proteasome system. Mol Biol Rep. 2013;40:3113–3121. doi: 10.1007/s11033-012-2385-2. [DOI] [PubMed] [Google Scholar]
- [30].Jung J, Cai W, Lee HK, et al. Actin polymerization is essential for myelin sheath fragmentation during Wallerian degeneration. J Neurosci. 2011;31:2009–2015. doi: 10.1523/JNEUROSCI.4537-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shin YK, Jang SY, Park JY, et al. The Neuregulin-Rac-MKK7 pathway regulates antagonistic c-jun/Krox20 expression in Schwann cell dedifferentiation. Glia. 2013;61:892–904. doi: 10.1002/glia.22482. [DOI] [PubMed] [Google Scholar]
- [32].Lee HK, Shin YK, Jung J, et al. Proteasome inhibition suppresses Schwann cell dedifferentiation in vitro and in vivo. Glia. 2009;57:1825–1834. doi: 10.1002/glia.20894. [DOI] [PubMed] [Google Scholar]
- [33].Samantaray S, Ray SK, Banik NL. Calpain as a potential therapeutic target in Parkinson's disease. CNS Neurol Disord Drug Targets. 2008;7:305–312. doi: 10.2174/187152708784936680. [DOI] [PubMed] [Google Scholar]
- [34].Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- [35].Jung T, Grune T. The proteasome and the degradation of oxidized proteins: Part I-structure of proteasomes. Redox Biol. 2013;1:178–182. doi: 10.1016/j.redox.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Menconi MJ, Wei W, Yang H, et al. Treatment of cultured myotubes with the calcium ionophore A23187 increases proteasome activity via a CaMK II-caspase-calpain-dependent mechanism. Surgery. 2004;136:135–142. doi: 10.1016/j.surg.2004.03.014. [DOI] [PubMed] [Google Scholar]
- [37].Djakovic SN, Schwarz LA, Barylko B, et al. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nayak MK, Kumar K, Dash D. Regulation of proteasome activity in activated human platelets. Cell Calcium. 2011;49:226–232. doi: 10.1016/j.ceca.2011.02.005. [DOI] [PubMed] [Google Scholar]
- [39].Lopez-Picon FR, Kukko-Lukjanov TK, Holopainen IE. The calpain inhibitor MDL-28170 and the AMPA/KA receptor antagonist CNQX inhibit neurofilament degradation and enhance neuronal survival in kainic acid-treated hippocampal slice cultures. Eur J Neurosci. 2006;23:2686–2694. doi: 10.1111/j.1460-9568.2006.04793.x. [DOI] [PubMed] [Google Scholar]
- [40].Shaw G, Yang C, Zhang L, et al. Characterization of the bovine neurofilament NF-M protein and cDNA sequence, and identification of in vitro and in vivo calpain cleavage sites. Biochem Biophys Res Commun. 2004;325:619–625. doi: 10.1016/j.bbrc.2004.09.223. [DOI] [PubMed] [Google Scholar]
- [41].Wang J, Zhai Q, Chen Y, et al. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophy Acta. 2009;1787:1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chang KT, Niescier RF, Min KT. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci U S A. 2011;108:15456–15461. doi: 10.1073/pnas.1106862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Verkhratsky A, Fernyhough P. Mitochondrial malfunction and Ca2+ dyshomeostasis drive neuronal pathology in diabetes. Cell Calcium. 2008;44:112–122. doi: 10.1016/j.ceca.2007.11.010. [DOI] [PubMed] [Google Scholar]
- [45].Yoshii SR, Kishi C, Ishihara N, et al. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–19640. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- [47].Bragoszewski P, Gornicka A, Sztolsztener ME, et al. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol Cell Biol. 2013;33:2136–2148. doi: 10.1128/MCB.01579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Leboucher GP, Tsai YC, Yang M, et al. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. 2012;47:547–557. doi: 10.1016/j.molcel.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Clarke KJ, Adams AE, Manzke LH, et al. A role for ubiquitinylation and the cytosolic proteasome in turnover of mitochondrial uncoupling protein 1 (UCP1) Biochim Biophys Acta. 2012;1817:1759–1767. doi: 10.1016/j.bbabio.2012.03.035. [DOI] [PubMed] [Google Scholar]
- [50].Kitay BM, McCormack R, Wang Y, et al. Mislocalization of neuronal mitochondria reveals regulation of Wallerian degeneration and NMNAT/WLDS-mediated axon protection independent of axonal mitochondria. Human Mol Gen. 2013;22:1601–1614. doi: 10.1093/hmg/ddt009. [DOI] [PMC free article] [PubMed] [Google Scholar]