

Abstract
Accumulation of aberrant proteins and inclusion bodies are hallmarks in most neurodegenerative diseases. Consequently, these aggregates within neurons lead to toxic effects, overproduction of reactive oxygen species and oxidative stress. Autophagy is a significant intracellular mechanism that removes damaged organelles and misfolded proteins in order to maintain cell homeostasis. Excessive or insufficient autophagic activity in neurons leads to altered homeostasis and influences their survival rate, causing neurodegeneration. The review article provides an update of the role of autophagic process in representative chronic and acute neurodegenerative disorders.
Keywords: neural regeneration, reviews, oxidative stress, autophagy, autophagy-related genes, apoptosis, Parkinson's disease, amyotrophic lateral sclerosis, multiple sclerosis, acute and chronic neurode-generation, neuroregeneration
Research Highlights
-
(1)
There has been no consensus on the role of autophagy in the onset of neurodegenerative diseases, but autophagy is considered one of important issues in the field of neuroscience.
-
(2)
This review article summarizes the distinct steps of the autophagic process, correlates its orderly function with nerve cell homeostasis and analyzes the specific functional alterations observed in chronic and acute diseases of human central nervous system.
-
(3)
Data acquisition of autophagy in the onset of neurodegenerative diseases benefits for clinical physicians to develop new treatment strategies in neurodegenerative diseases.
INTRODUCTION
Catabolism is a process, which contributes to the recycling of the cellular material by degrading proteins or even whole organelles. During catabolic processes, a production of peroxides and free radicals can occur[1]. These represent a class of molecules that are derived from the metabolism of oxygen and exist inherently in all aerobic organisms. Antioxidant enzymes (superoxide dismutase, catalase and glutathione peroxidase) protect cells from the toxic effects of reactive oxygen species[1]. However, imbalanced process of production and consumption of reactive oxygen species leads to oxidative stress.
Cell homeostasis is disrupted by intracellular accumulation of aberrant proteins, inactive enzymes and damaged organelles. Eukaryotic cell response involves primarily two major systems for whole protein degradation: the proteasome system and autophagy; however, the latter has the ability to degrade entire organelles[2].
Autophagy (derived from Greek meaning “to eat oneself”) is a highly conserved process through the evolution of species, from eukaryotic microorganisms to humans[3]. Bulky cytoplasmic contents, organisms (bacteria, viruses) and soluble proteins are degraded by autophagy and reused for the synthesis of new molecules[4]. This process is generally induced by a change of environmental conditions, such as nutrient deprivation, oxidative stress and ultraviolet radiation. However, it has also been associated with normal procedures like development, differentiation and defence against pathogens. In spite of offering protection to cells, autophagy may also contribute to cell damage[5]. In this review, we collected data on molecular mechanisms of autophagy, and the role of autophagic process in acute and chronic neurodegeneration.
AUTOPHAGY AS A MECHANISM OF CELL HOMEOSTASIS
Three types of autophagy are currently identified: classic autophagy (macroautophagy), chaperone-mediated autophagy and microautophagy[6]. Macroautophagy becomes activated under stress conditions (nutrient deprivation, infection, toxins)[7]. It is the classic autophagic pathway in which a double membrane vesicle forms around cytosolic components and merges with lysosomes[8]. Chaperone-mediated autophagy (CMA) targets a certain group of proteins containing the pentapeptide KFERQ or similar sequences[9]. It does not necessitate vesicle formation, because all substrates form a complex with cytosolic chaperone heat shock protein 70, which binds to the lysosome-associated membrane protein type 2a[10,11]. The substrate/heat shock protein 70 complex enters the lysosome lumen by binding to the lysosomal heat shock protein 70, a second chaperone[12]. In microautophagy, the lysosome itself surrounds a targeted protein or organelle. When a peroxisome or a mitochondrion is engulfed, then the process is called pexophagy or mitophagy[13,14].
All the above processes maintain intracellular balance and can occur in every living cell. When cell homeostatic mechanisms fail to comply, death is inevitable. This leads to the induction of mitosis of neighbouring cells in order to maintain tissue integrity and function. In the adult brain tissue, this process of replenishment cannot occur, since neurons cannot divide. Neurons have to adjust all their intracellular processes in order to survive for decades. Thus, proper autophagy regulation is essential for neuronal survival and its malfunction has been entailed in neurodegenerative diseases.
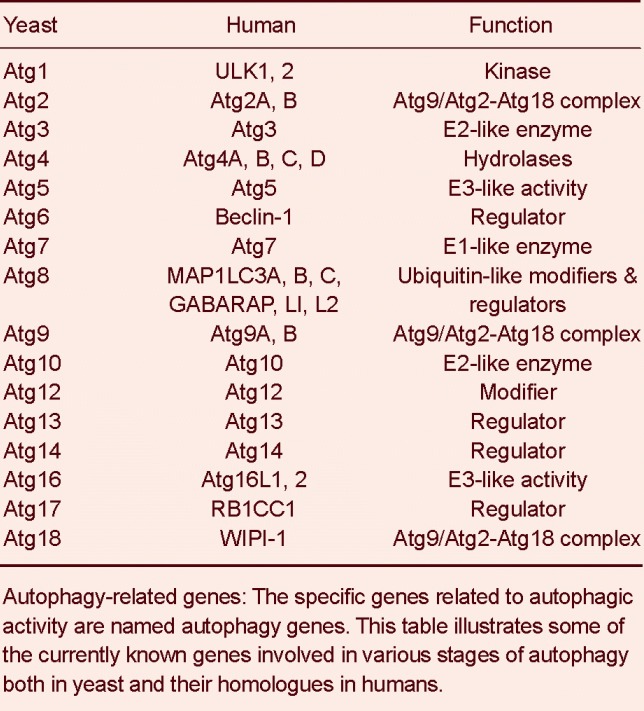
There is molecular machinery responsible for autophagy. Specialized molecules, such as kinases, phosphatases, enzymes that can bind and hydrolyze guanosine triphosphate (GTPases) participate in this process, all encoded by autophagy-related (Atg) genes (Table 1)[21].
Table 1.
Yeast and human autophagy-related genes
INDUCTION OF AUTOPHAGY
There are common autophagy signaling pathways in mammals and other organisms such as yeast[15]. A phosphatidylinositol kinase related serine/threonine protein kinase TOR (target of rapamycin) plays a crucial part in the induction of autophagy. Mammalian target of rapamycin (mTOR) in its phosphorylated form (activated mammalian target of rapamycin) acts as a negative regulator of autophagy by hyperphosphorylation of Atg13[15]. This hyperphosphorylation prevents Atg13 from undergoing an interaction with Atg1, a serine/threonine protein kinase, which forms a complex necessary for the induction of autophagy. Other components of the Atg1 complex are Vac8, Atg11, Atg20, Atg24 and Atg17[16]. In particular, Akt and MAPK signaling triggers mammalian target of rapamycin, whereas AMPK and p53 signaling suppresses mammalian target of rapamycin activity. Another signaling pathway which mediates in the initiation of autophagy involves Vps34 phosphatidylinositol 3-kinase complex (PI3K). Vacuolar protein sorting (Vps34) interacts with Vps15, a membrane associated serine/threonine kinase that regulates the kinase activity of Vps34. Vps34 is primarily involved in vacuolar protein targeting and forms a complex with Vps15-Vps38-Atg6[17]. At the same time, Vps34 has been found to be associated with Vps15-Atg6-Atg14 on pre-autophagosomal membranes. The process includes multiple steps (Figure 1).
Figure 1.
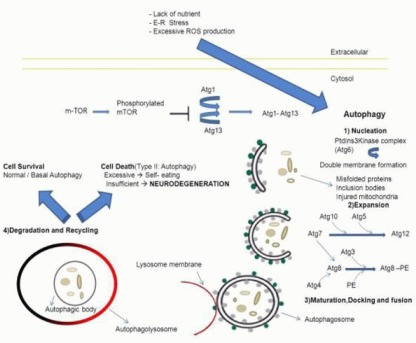
Overview and distinct steps of autophagy.
Extracellular stimuli (lack of nutrient, ER stress, excessive ROS production) can trigger the autophagic process. A serine/threonine kinase, mTOR, plays a central role in the inhibition of autophagy by blocking the interaction between Atg1 and Atg13. Atg1–Atg13 complex formation leads to the induction of Autophagy.
The first step (Nucleation) begins with a double membrane that engulfs cargo (misfolded proteins, inclusion bodies or injured mitochondria). It is mediated by a class III PtdIns3Kinase, which includes Atg6. The second step (Expansion) implicates two ubiquitin-like systems that lead to the elongation of the double membrane. In the first system Atg7 interacts with Atg10 and Atg5 in order to activate Atg12. In the second system Atg4 cleaves Atg8, which is activated by Atg7, moves to Atg3 and conjugates with PE. The form Atg8-PE is transferred to the autophagosome membrane allowing the transition to the next steps.
In these steps (maturation, docking and fusion) autophagosome formation is completed (double membrane vesicle), its outer membrane fuses with the lysosomal membrane (single membrane vesicle) and the autophagolysosome is formed. The inner membrane of the autophagosome includes the cargo and it is called autophagic body. In the last two steps (Degradation and Recycling) the cargo is degraded by lysosomal enzymes and the resulting small molecules (peptides, amino acids, etc) are recycled. The autophagic process may lead to cell survival (normal activity), cell death (excessive activity) or neurodegeneration (insufficient degradation rates).
ER: Endoplasmic reticulum; ROS: reactive oxygen species; mTOR: mammalian target of rapamycin; Atg: autophagy related gene; PtdIns3Kinase: phosphatidylinositol 3-kinase PE: phosphatidylethanolamine.
The first step in the autophagy process is nucleation. In particular, an autophagosomal membrane engulfs the targeted cargo (e.g. misfolded proteins, inclusion bodies, injured mitochondria) within a double membrane forming a vesicle called autophagosome. This phase is mediated by a class III phosphatidylinositol 3-kinase complex, which includes Atg6 (called Beclin-1 in mammals)[18]. Next comes the expansion phase. The elongation of vesicles is executed by following steps that include two systems which accept conjugation in the same way as ubiquitin. In the first system, Atg7 (E1-like protein) activates Atg12, binds with Atg10 (E2-like protein) and conjugates with Atg5[19]. In the second step, a cysteine protease (Atg4) acts on Atg8 (LC3 in mammals), which then is activated by Atg7 and moves to Atg3. Last, Atg8 makes a bond with the lipid phosphatidylethanolamine (PE)[20].
In the maturation, docking and fusion steps that follow, the autophagosomes bind to and fuse with lysosomes (autophagolysosomes). Their outer membranes fuse with each other and the inner membranes with their constituents are released into the lysosome/autolysosome lumen. The final steps of the autophagy process are degradation and recycling where the cargo breaking down is performed by lysosomal hydrolases and other proteolytic enzymes. After degradation, free amino acids, nucleotides, fatty acids and other components are released into the cytosol and get recycled.
AUTOPHAGY AND APOPTOSIS
There is a controversial role of autophagy in cell survival and cell death. Observations of autophagy process in dying cells indicate that this is a form of autophagic cell death. This represents type 2 programmed cell death and it is morphologically different from apoptosis (type 1 cell death)[22]. Autophagic cell death is mediated by a large number of autophagosomes in the cytoplasm, whereas apoptosis is mediated by caspases[23,24].
Under normal conditions, autophagy and apoptosis become inactive by inhibiting each other[25]. Under cellular stress, mitochondria release cytochrome C leading to apoptosis, while endoplasmic reticulum stress or altered metabolism (nutrient deprivation) can trigger autophagy. Specifically, during nutrient deficiency, autophagy functions as a pro-survival mechanism, but overelevated autophagy results in autophagic cell death. Some pro-apoptotic signals, such as tumor necrosis factor (TNF), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas-associated protein with death domain (FADD) can also induce autophagy. On the other hand, pro-survival signals suppress autophagy through another pathway[26]. Experiments and studies suggest that autophagy precedes apoptosis.
Atg5 proteolysis, transcriptional factor p53[27] and the Bcl-2 protein family play an important role in the interplay between apoptosis and autophagy. Beclin 1 regulates autophagy by interacting with the BH3 domain of Bcl-2 proteins. Bcl-2 functions primarily as an anti-apoptotic molecule, but can also act as an anti-autophagic mediator in the endoplasmic reticulum. The latter role is performed by inhibiting the participation of Beclin 1 in the nucleation of autophagosomes[28]. Moreover, caspase-3, -7 and -8 cleave Beclin 1 and produce N-terminal and C-terminal fragments which lose their ability to induce autophagy. C-terminal fragments translocate within mitochondria and make cells prone to apoptotic signals[29].
Atg5 is another link between autophagy and apoptosis. It promotes autophagy associated cell death by interacting with FADD protein, a molecule that promotes apoptosis[30]. Calpain cleaves Atg5 in the N-terminus and the fragment is translocated to mitochondria, where it interacts with Bcl-xL leading to apoptosis via cytochrome c release and caspase activation[31].
Concurrent apoptotic and autophagic features are observed in dying neurons in neonatal hypoxia-ischemia[32]. Thorough investigation of the apoptotic and autophagic molecular mechanisms will lead to deeper understanding of their interplay and may reveal ways regarding their manipulation in neurodegenerative diseases.
AUTOPHAGY IN NEURONAL CELLS AND NEURODEGENERATION
Autophagic pathways have been reported to exist in neurons. In neuronal culture models, some characteristics of neuronal macroautophagy have been identified, thereby suggesting that macroautophagy in neurons may provide a protective mechanism[33]. However, autophagosomes may hardly be detected in healthy neurons under nutrient rich conditions[34,35]. Interestingly, mammalian target of rapamycin has been known to regulate post-synaptic long-term potentiation or long-term depression, suggesting that autophagic regulation may be essential for synaptic plasticity[36]. On the other hand, the inhibition of mammalian target of rapamycin kinase activity in glial cells results in anti-inflammatory actions, whereas it plays an important role in the regulation of oligodendrocyte development and myelination process, as well as in several neuronal functions[37].
The pathogenesis of neurodegenerative diseases involves a gradual loss of neuronal cells. Altered activity of proteolytic systems and accumulation of protein aggregates are basic characteristics of neurodegenerative disorders. The clearance of aggregate-prone proteins is very important in post-mitotic cells, like neurons, because they cannot be diluted by cell division. Possible toxic effects of these aggregates include: blockage of axonal transport (occupation of intracellular space) and transcriptional dysregulation (inactivation of transcriptional factors)[38]. Thus, non-dividing neural cells demand a protein quality-control process executed by autophagy in order to maintain homeostasis throughout the whole cell (body, dendrites, axon)[39]. However, there are a number of questions yet to be answered with regard to the role of autophagy in neurodegenerative process. Understanding of the biological effects and the mechanisms underlying autophagy in neurons and glia might be helpful in seeking effective new treatments for neurodegenerative diseases[40]. The effect of autophagy in neurons during disease can be divided in two classes: chronic and acute condition response. Studies in chronic central nervous system diseases such as Alzheimer's disease, Parkinson's disease and Huntington's disease, Amyotrophic lateral sclerosis and Multiple sclerosis have shown that altered autophagic activity may be implicated in their pathogenesis. Autophagy is also a contributor in acute central nervous system diseases, such as vascular episodes, hypoxia/ischemia, brain injury and experimental pharmacological brain injury.
Chronic central nervous system diseases
Alzheimer's disease
Main clinical manifestations include selective memory impairment and degenerative dementia. These are associated with neuronal loss, silk-like extracellular senile plaques, neurofibrillary tangles and threads that accumulate within the gray matter. Two proteins are involved in the onset and progress of brain cell damage in Alzheimer's disease: tau protein and amyloid beta-peptide (Aβ).
The normal tau protein is involved in microtubule stabilization and vesicle transport along the axis. When the protein is hyperphosphorylated, it loses its affinity for microtubules and accumulates in neurons forming toxic neurofibrillary tangles[41]. Excessive amounts of tau are to be removed from the autophagolysosome system, aiming to prevent neurodegeneration[42].
The amyloid beta-peptide is the major protein constituent of senile plaques and results from the enzymatic degradation of a larger protein, amyloid precursor protein (APP)[43]. APP is a membrane protein expressed primarily in neuronal synapses. When APP is degraded by α-and γ-secretase, it produces peptide p3, whereas when it is degraded by β- and γ-secretase, it results in the production of amyloid beta-peptide. This peptide is normally degraded in the lysosomes.
In Alzheimer's disease, though not in normal brain cells, autophagosomes are large in numbers thus providing evidence of activation of autophagy with damaged autophagosome-lysosome fusion[44]. Other observations include increased mitophagy. The lack of function in Alzheimer's disease is due to impaired fusion between lysosomes and autophagosomes filled with amyloid beta-peptide[45]. The gene that encodes presenilin-1 (part of the gamma-secretase complex) is implicated in the majority of inherited forms of Alzheimer's disease. Its mutations deteriorate lysosome function and promote the accumulation of amyloid beta-peptide, resulting in neuronal loss. Recent observations also show a reduced expression of Beclin 1 gene leading to impaired autophagy in neurons, amyloid beta-peptide deposition and consequent neurodegeneration[46].
Parkinson's disease
The disease is characterized by progressive loss of dopamine-producing brain cells in the substantia nigra. The main characteristic feature is the presence of Lewy bodies: round, eosinophilic, intracytoplasmic inclusions in the nuclei of neurons[47]. Further studies have shown that these bodies consist of an aggregated and insoluble form of α-synuclein. This cytosolic protein exists in abundancy in the brain and is mainly expressed in presynaptic membranes[48]. It has the ability to bind with membrane phospholipids and participates in membrane-related procedures in presynaptic termini[49]. There are two missense mutations in α-synuclein, A53T and A30P that lead to early-onset Parkinson's disease[50].
α-Synuclein recycling is performed by the proteasome system[51], CMA and macroautophagy. The proteasome degrades only soluble forms of the protein. A percentage of intracellular α-synuclein is normally degraded through CMA. Aberrant forms of alpha-synuclein (oxidized or phosphorylated) modify the CMA pathway, having loose affinity for the CMA receptor on the lysosomal membrane. Instead of entering for degradation, they accumulate in the cytoplasm. Moreover, missense mutations of α-synuclein or dopamine mediated modifications result in even greater impairment of CMA[52]. Autophagy/mitophagy appears to be impaired, autophagosome-like structures accumulate and the cells are prone to apoptosis and necrosis[53].
Further data reveal mutations in PINK 1 (PTEN-induced putative kinase 1) gene, which promotes basal autophagy and normally controls mitochondrial balance acting on Parkin protein. The latter promotes autophagy, but if PINK 1 gene carries a mutation, altered Parkin activity leads to autophagy dysregulation and mitochondrial imbalance. PINK 1 kinase 1 interplays with Beclin1. A mutation in this gene provokes damaged interaction, thus resulting in ineffective or insufficient autophagic activity[54]. This provides more evidence about the role of autophagy in neurodegenerative diseases.
Huntington's disease
This disease is caused by expansion of the CAG trinucleotide repeats in the Huntington's disease gene which encodes huntingtin (polyglutamine – polyQ disorder). This protein is present in a large number of tissues and it is associated with vesicle and microtubule function[55]. Mutations make huntingtin aggregation-prone and toxic. Huntingtin polyglutamine residues cannot act as a substrate for proteasome degradation and macroautophagy acts as a compensatory mechanism. A variety of mechanisms may contribute to neurodegeneration[56], such as: excitotoxicity, mitochondrial dysfunction and oxidative stress, promotion of apoptosis and/or autophagy, dysfunction of neuronal interaction and circuits.
Observations include impaired sorting/degradation of autophagosomes, insufficient levels of autophagy and a resulting accumulation of highly ubiquitinated aggregates of huntingtin (htt) in the endosomal-lysosomal organelles[57]. Recent investigations of eight polymorphisms concerning five Atg genes (Atg3, Atg5, Atg7, Atg16L1 and Beclin-1) showed two significant findings in Atg7 and Atg16. Specifically, Atg7 polymorphism that encodes alanine for valine (V471A), has been linked to earlier disease onset (4 years earlier)[58].
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease characterized by early degeneration of upper and lower motor neurons resulting in inevitable paralysis. All forms of the disease, sporadic and familial, affect the same neurons displaying similar pathological features (muscle weakness, atrophy, spasticity). About 20% of familial forms of amyotrophic lateral sclerosis are caused by mutations in the gene coding for peroxide dismutase (SOD1)[59]. Mutations cause defective protein folding, mitochondrial dysfunction, oxidative stress, inflammation and excitotoxicity. Amyotrophic lateral sclerosis neuronal aggregates are composed of intermediate filaments and insoluble forms of proteins (such as SOD1). Aggregate formation leads to disturbance of proteasome activity, axial transport, chaperone protein function and damage of cell organelles such as Golgi, endoplasmic reticulum and mitochondria[60]. Aggregates consisting of TAR DNA-binding protein and intraneuronal protein A are also present in sporadic and familial forms of amyotrophic lateral sclerosis[61].
The above aggregates are cleared by autophagy and the proteasome mechanism[62]. In experimental models of amyotrophic lateral sclerosis, inhibition of autophagy exacerbates the viability of motor neurons, whereas its stimulation prevents degeneration[63,64].
Multiple sclerosis
This is the most common autoimmune inflammatory demyelinating disease of the central nervous system. Multiple sclerosis is characterized by multifocal areas of demyelination with loss of oligodendrocytes, astroglial scarring and axonal injury. Observations showed that a population of T-cells and activated microglia attack myelin proteins, causing chronic neurodegeneration. Other findings include blood brain barrier disruption, increased T and B lymphocyte levels in demyelinated areas and characteristic oligoclonal IgM and IgG bands in cerebrospinal fluid electrophoresis[65].
It is found that molecules which participate in the autophagy process interfere with T-cell balance[66,67,68]. Generally, it is proved that there is correlation between autophagy and autoimmune-mediated demyelination. In particular, Atg5 gene function has been reported to modulate T-cell survival and proliferation. An experimental mouse model has shown that Atg5 mRNA is raised in T cells, overexpressed in multiple sclerosis brain tissue and that clinical severity corresponds with expression of Atg5 in peripheral blood[69]. Evidence from studies in Atg-5 deficient mice reported that Atg-5 deletion has strong immunological and neurological effects: impaired T cell function and survival and neurodegeneration with presence of intracellular inclusions in neurons[67,68,70]. These data indicate that Atg5 relates to extended T-cell survival, which are associated with autoimmunity in multiple sclerosis. Furthermore, B cell survival is linked to Atg5 expression[71]. Thus, mutations in Atg5 may determine the severity and age of onset of multiple sclerosis in humans. Whether increased Atg5 expression is specific to MS or is just another feature of autoimmune diseases remains to be clarified.
Neuronal homeostasis is also affected in multiple sclerosis. The hypersecretion of glutamate (from lymphocytes, microglia and macrophages) intoxicates neurons by increasing intracellular calcium which in turn causes mitochondrial damage, release of reactive oxygen species and stimulates caspases and calpains. These two protein families lead to autophagic activity disruption[72]. Defective mitochondria, not subjectable to mitophagy, accumulate within axons rendering them sensitive to energy deficits and prone to degeneration[73].
Acute central nervous system diseases
Increased levels of autophagic process and elevated numbers of autophagosomes are found in acute central nervous system diseases such as stroke, hypoxia/ischemia, brain injury and experimental brain trauma[74,75].
In brain hypoxia-ischemia, the main mediator in neuronal death is excitotoxicity (a phenomenon caused by hyperactivation of glutamate receptors)[76]. Induction of autophagy has been demonstrated in various models of acute and chronic excitotoxicity. The hypoxia-induced oxidative stress is characterized by overproduction of reactive oxygen species within mitochondria and the neuronal cell appears to adapt by increasing mitochondrial autophagy (mitophagy)[77]. Studies in brain trauma showed overexpression of Beclin 1, large numbers of autophagosomes and increased expression of lysosomal proteases[74,75,76,77,78].
Experiments involving mouse brain injury and consequent administration of rapamycin revealed improved neurological function. In conclusion, autophagy, triggered by rapamycin, acts as a compensatory mechanism in order to protect brain tissue[79,80]. However, in other cases of acute central nervous system disorders (pharmacological induction of injury and lack of trophic support) both autophagy and apoptosis are involved in cell death process. Autophagy appears to be deleterious and its inhibition reduces cell death[81]. In traumatic brain injury in particular, autophagy pathway has been reported to be involved in the underlying pathology. Inhibition of this pathway may help attenuate traumatic damage and functional outcome deficits[82]. Similarly, the inhibition of mammalian target of rapamycin signaling using rapamycin during the acute phase of spinal cord injury produces neuroprotective effects and reduces secondary damage at lesion sites[83]. Interestingly enough, methylprednisolone treatment which is a common strategy following spinal cord injury, has been reported to inhibit the enhanced autophagy expression in neurons soon after experimental contusion injury, thereby protecting neurons from cell death[84]. Moreover, the clinical significance of targeting autophagy in promoting neurodegeneration has recently been emphasized by the finding that pretreatment with tetracycline may inhibit autophagy in the ischemic stroke brain and then suppress the inflammatory process via inhibiting the activation of nuclear factor kappa B pathway[85]. Last but not least, there is increasing evidence that neuroprotection may be achieved by modulating autophagy in retinal ganglion cells under glaucoma-related stressing conditions[86]. Nevertheless as far as acute central nervous system disease is concerned, a very intriguing field of interest is whether inhibition or promotion of autophagy will promote neuroprotection in acute central nervous system injuries and whether an early or delayed intervention is needed.
Conclusion
Autophagy is a promising field of study with many aspects remaining uninvestigated. The appropriate rate of autophagy and its interaction with apoptosis are key factors for cell homeostasis and survival. Autophagy dysregulation impedes normal cell function and may contribute in neurodegenerative disorders pathways when neuronal cells are concerned. Future research will provide further data about this astonishing homeostatic mechanism which allows neuronal cells to survive for decades. Deeper understanding of implicated molecular and intracellular components of autophagy may contribute to direct pharmacologic manipulation in order to prevent neurodegeneration.
Footnotes
Conflicts of interest: None declared.
(Reviewed by Choi JI, Finana IT, Salmina AB)
(Edited by Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Butler D, Bahr BA. Oxidative stress and lysosomes: CNS-related consequences and implications for lysosomal enhancement strategies and induction of autophagy. Antioxid Redox Signal. 2006;8:185–196. doi: 10.1089/ars.2006.8.185. [DOI] [PubMed] [Google Scholar]
- [2].Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- [4].Lee HK, Marzella L. Regulation of intracellular protein degradation with special reference to lysosomes: role in cell physiology and pathology. Int Rev Exp Pathol. 1994;35:39–147. [PubMed] [Google Scholar]
- [5].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dice J. Austin: Landes Bioscience; 2000. Lysosomal Pathways of Protein Degradation. [Google Scholar]
- [8].Dunn WA., Jr Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- [10].Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- [11].Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- [12].Bandyopadhyay U, Kaushik S, Varticovski L, et al. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–5763. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sakai Y, Oku M, van der Klei IJ, et al. Pexophagy: autophagic degradation of peroxisomes. Biochim Biophys Acta. 2006;1763:1767–1775. doi: 10.1016/j.bbamcr.2006.08.023. [DOI] [PubMed] [Google Scholar]
- [14].Tolkovsky AM. Mitophagy. Biochim Biophys Acta. 2009;1793:1508–1515. doi: 10.1016/j.bbamcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- [15].Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- [16].Abeliovich H, Zhang C, Klionsky DJ, et al. Chemical genetic analysis of Apg1 reveals a non-kinase role in the induction of autophagy. Mol Biol Cell. 2003;14:477–490. doi: 10.1091/mbc.E02-07-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sohal RS, Ku HH, Agarwal S, et al. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech Ageing Dev. 1994;74:121–133. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- [18].Bossy B, Perkins G, Bossy-Wetzel E. Clearing the brain's cobwebs: the role of autophagy in neuroprotection. Curr Neuropharmacol. 2008;6:97–101. doi: 10.2174/157015908784533897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859–864. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- [21].Behrends C, Sowa ME, Harper JW, et al. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- [23].Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- [24].Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- [25].González-Polo RA, Boya P, Pauleau AL, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- [26].Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feng Z, Zhang H, Levine AJ, et al. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- [29].Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- [30].Pyo JO, Jang MH, Kwon YK, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formationand cell death. J Biol Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- [31].Yousefi S, Simon HU. Autophagy in cancer and chemotherapy. Results Probl Cell Differ. 2009;49:183–190. doi: 10.1007/400_2008_25. [DOI] [PubMed] [Google Scholar]
- [32].Ginet V, Puyal J, Clarke PG, et al. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol. 2009;175:1962–1974. doi: 10.2353/ajpath.2009.090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- [34].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nixon RA, Cataldo AM, Mathews PM. The endosomal-lysosomal system of neurons in Alzheimer's disease pathogenesis: a review. Neurochem Res. 2000;25:1161–1172. doi: 10.1023/a:1007675508413. [DOI] [PubMed] [Google Scholar]
- [36].Lee JA. Neuronal autophagy: a housekeeper or a fighter in neuronal cell survival? Exp Neurobiol. 2012;21:1–8. doi: 10.5607/en.2012.21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dello Russo C, Lisi L, Feinstein DL, et al. mTOR kinase, a key player in the regulation of glial functions: Relevance for the therapy of multiple sclerosis. Glia. doi: 10.1002/glia.22433. in press. [DOI] [PubMed] [Google Scholar]
- [38].Ravikumar B, Rubinsztein DC. Can autophagy protect against neurodegeneration caused by aggregate-prone proteins. Neuroreport? 2004;15:2443–2445. doi: 10.1097/00001756-200411150-00001. [DOI] [PubMed] [Google Scholar]
- [39].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- [40].Rami A. Review: autophagy in neurodegeneration: firefighter and/or incendiarist? Neuropathol Appl Neurobiol. 2009;35:449–461. doi: 10.1111/j.1365-2990.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- [41].Mocanu MM, Nissen A, Eckermann K, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hamano T, Gendron TF, Causevic E, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–1130. doi: 10.1111/j.1460-9568.2008.06084.x. [DOI] [PubMed] [Google Scholar]
- [43].Tiraboschi P, Hansen LA, Thal LJ, et al. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–1989. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- [44].Nixon RA, Wegiel J, Cuervo AM, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- [45].Ling D, Salvaterra PM. A central role for autophagy in Alzheimer-type neurodegeneration. Autophagy. 2009;5:738–740. doi: 10.4161/auto.5.5.8626. [DOI] [PubMed] [Google Scholar]
- [46].Pickford F, Masliah E, Wyss-Coray T, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lewy FH. Paralysis Agitans. In: Lewandowsky M, editor. Handbuch der Neurologie. Berlin: Springer-Verlag; 1912. [Google Scholar]
- [48].Jenco JM, Rawlingson A, Morris AJ, et al. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry. 1998;37:4901–4909. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- [49].Abeliovich A, Schmitz Y, Fariñas I, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- [50].Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- [51].Betarbet R, Sherer TB, Greenamyre JT. Ubiquitin-proteasome system and Parkinson's diseases. Exp Neurol. 2005;191(Suppl 1):S17–27. doi: 10.1016/j.expneurol.2004.08.021. [DOI] [PubMed] [Google Scholar]
- [52].Cuervo AM, Stefanis L, Fredenburg R, et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- [53].Stefanis L. Caspase-dependent and -independent neuronal death: two distinct pathways to neuronal injury. Neuroscientist. 2005;11:50–62. doi: 10.1177/1073858404271087. [DOI] [PubMed] [Google Scholar]
- [54].Michiorri S, Gelmetti V, Giarda E, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010;17:962–974. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- [55].Hoffner G, Kahlem P, Djian P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: relevance to Huntington's disease. J Cell Sci. 2002;115:941–948. doi: 10.1242/jcs.115.5.941. [DOI] [PubMed] [Google Scholar]
- [56].Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- [57].Sapp E, Schwarz C, Chase K, et al. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- [58].Metzger S, Saukko M, Van Che H, et al. Age at onset in Huntington's disease is modified by the autophagy pathway: implication of the V471A polymorphism in Atg7. Hum Genet. 2010;128:453–459. doi: 10.1007/s00439-010-0873-9. [DOI] [PubMed] [Google Scholar]
- [59].Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- [60].Tummala H, Jung C, Tiwari A, et al. Inhibition of chaperone activity is a shared property of several Cu, Zn-superoxide dismutase mutants that cause amyotrophic lateral sclerosis. J Biol Chem. 2005;280:17725–17731. doi: 10.1074/jbc.M501705200. [DOI] [PubMed] [Google Scholar]
- [61].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- [62].Wang X, Fan H, Ying Z, et al. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010;469:112–116. doi: 10.1016/j.neulet.2009.11.055. [DOI] [PubMed] [Google Scholar]
- [63].Madeo F, Eisenberg T, Kroemer G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009;23:2253–2259. doi: 10.1101/gad.1858009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pasquali L, Longone P, Isidoro C, et al. Autophagy, lithium, and amyotrophic lateral sclerosis. Muscle Nerve. 2009;40:173–194. doi: 10.1002/mus.21423. [DOI] [PubMed] [Google Scholar]
- [65].Freedman MS, Thompson EJ, Deisenhammer F, et al. Recommended standard of cerebrospinal fluid analysis in the diagnosis of multiple sclerosis: a consensus statement. Arch Neurol. 2005;62:865–870. doi: 10.1001/archneur.62.6.865. [DOI] [PubMed] [Google Scholar]
- [66].Li C, Capan E, Zhao Y, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- [67].Pua HH, Dzhagalov I, Chuck M, et al. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pua HH, He YW. Maintaining T lymphocyte homeostasis: another duty of autophagy. Autophagy. 2007;3:266–267. doi: 10.4161/auto.3908. [DOI] [PubMed] [Google Scholar]
- [69].Alirezaei M, Fox HS, Flynn CT, et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy. 2009;5:152–158. doi: 10.4161/auto.5.2.7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- [71].Miller BC, Zhao Z, Stephenson LM, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- [72].Manev H, Favaron M, Guidotti A, et al. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36:106–112. [PubMed] [Google Scholar]
- [73].Mahad D, Ziabreva I, Lassmann H, et al. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131:1722–1735. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Diskin T, Tal-Or P, Erlich S, et al. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma. 2005;22:750–762. doi: 10.1089/neu.2005.22.750. [DOI] [PubMed] [Google Scholar]
- [75].Liu XS, Chopp M, Zhang XG, et al. profiles and electrophysiology of doublecortin-expressing cells in the subventricular zone after ischemic stroke. J Cereb Blood Flow Metab. 2009;29:297–307. doi: 10.1038/jcbfm.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Borsello T, Croquelois K, Hornung JP, et al. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–485. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- [77].Azad MB, Chen Y, Henson ES, et al. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. 2008;4:195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wen YD, Sheng R, Zhang LS, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- [79].Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- [80].Erlich S, Alexandrovich A, Shohami E, et al. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- [81].Kunchithapautham K, Rohrer B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy. 2007;3:433–441. doi: 10.4161/auto.4294. [DOI] [PubMed] [Google Scholar]
- [82].Luo CL, Li BX, Li QQ, et al. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience. 2011;184:54–63. doi: 10.1016/j.neuroscience.2011.03.021. [DOI] [PubMed] [Google Scholar]
- [83].Kanno H, Ozawa H, Sekiguchi A, et al. The role of mTOR signaling pathway in spinal cord injury. Cell Cycle. 2012;11:3175–3179. doi: 10.4161/cc.21262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chen HC, Fong TH, Lee AW, et al. Autophagy is activated in injured neurons and inhibited by methylprednisolone after experimental spinal cord injury. Spine (Phila Pa 1976) 2012;37:470–475. doi: 10.1097/BRS.0b013e318221e859. [DOI] [PubMed] [Google Scholar]
- [85].Jiang Y, Zhu J, Wu L, et al. Tetracycline Inhibits Local Inflammation Induced by Cerebral Ischemia via Modulating Autophagy. PLoS One. 2012;7:e48672. doi: 10.1371/journal.pone.0048672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Russo R, Berliocchi L, Adornetto A, et al. In search of new targets for retinal neuroprotection: is there a role for autophagy? Curr Opin Pharmacol. doi: 10.1016/j.coph.2012.09.004. in press. [DOI] [PubMed] [Google Scholar]