

Abstract
Metformin may reduce food intake and body weight, but the anorexigenic effects of metformin are still poorly understood. In this study, Sprague-Dawley rats were administered a single intracere-broventricular dose of metformin and compound C, in a broader attempt to investigate the regula-tory effects of metformin on food intake and to explore the possible mechanism. Results showed that central administration of metformin significantly reduced food intake and body weight gain, par-ticularly after 4 hours. A reduction of neuropeptide Y expression and induction of AMP-activated protein kinase phosphorylation in the hypothalamus were also observed 4 hours after metformin administration, which could be reversed by compound C, a commonly-used antagonist of AMP-activated protein kinase. Furthermore, metformin also improved lipid metabolism by reducing plasma low-density lipoprotein. Our findings suggest that under normal physiological conditions, central regulation of appetite by metformin is related to a decrease in neuropeptide Y gene expres-sion, and that the activation of AMP-activated protein kinase may simply be a response to the anorexigenic effect of metformin.
Keywords: neural regeneration, metformin, food intake, body weight gain, hypothalamus, AMP-activated protein kinase, neuropeptide Y, grants-supported paper, neuroregeneration
Research Highlights
-
(1)
Under normal physiological conditions, an intracerebroventricular approach was applied to in-vestigate the anorexigenic effects of metformin in vivo in the central nervous system.
-
(2)
Under normal physiological conditions, metformin inhibited neuropeptide Y gene expression, suggesting that the central regulation of appetite by metformin is related to hypothalamic neuropeptide Y signaling pathway. Metformin activated AMP-activated protein kinase phosphorylation, which may simply be a response to the anorexigenic effects of metformin.
INTRODUCTION
People who are overweight or obese are at a markedly higher risk of developing metabolic syndrome[1,2] and cardiovascular disease[2] compared with people with normal body weight. Metformin (one of the biguanides) is widely used in the treatment of type 2 diabetes mellitus, and is linked to promoting a broad range of health benefits by lowering the risks associated with the metabolic syndrome[3,4,5]. Although its molecular target is still to be identified, metformin is known to decrease intestinal absorption of glucose[6], improve hyperglycemia primarily through its suppression of hepatic glucose production[7,8], and increase peripheral insulin sensitivity[9,10,11]. In addition, metformin has been shown to reduce food intake and body weight gain in high-fat diet-fed mice[12,13], obese diabetic rats[14] and normotensive patients[15,16]. Such effects of metformin have also been observed in non-diabetic mammals[17,18,19]. For instance, prolonged treatment with metformin decreased the body weight and food intake of female outbred SHR mice[20]. A recent study revealed that intracerebroventricular injections of metformin induced anorexia in rats[21]. Nevertheless, the precise mechanism underlying the role of metformin in regulating feeding behavior and energy balance is not yet known.
AMP-activated protein kinase is a major sensor of cellular energy charge and an important regulator of food intake by responding to hormonal and nutrient signals in the hypothalamus[22,23]. Metformin can activate AMP-activated protein kinase in tissues such as skeletal muscle, liver, and heart[24,25,26,27,28]. The drug is also used in the central nervous system as an agonist of AMP-activated protein kinase[29,30,31]. However, the effect of metformin on AMP-activated protein kinase in the central nervous system and how this contributes to whole body energy homeostasis remains controversial. It has been observed that, in contrast to its effect on AMP-activated protein kinase in the periphery, metformin inhibits the activation of AMP-activated protein kinase in response to low glucose in cultured hypothalamic neurons[32]. Intracerebroventricular administration of metformin may produce an anorexigenic effect by preventing ghrelin-induced activation of AMP-activated protein kinase[18]. In contrast, metformin activates AMP-activated protein kinase in both a neuronal culture system and in mouse brain[33]. These discrepancies may be explained by a differential regulation of AMP-activated protein kinase activation that depends on the tissue, degree of stimulation, and condition of activation[34]. Some researchers have also proposed that the mechanism of AMP-activated protein kinase activation by metformin is different from that of the existing AMP-activated protein kinase activating agent, 5-aminoimidazole-4-car- boxamide riboside[35].
In summary, the roles of metformin in the central nervous system are still poorly understood. In this study, we applied compound C, an antagonist of AMP-activated protein kinase, in a broader attempt to verify the correlation between metformin and AMP-activated protein kinase, to investigate the central effects of metformin, and to explore the possible mechanism of metformin in the hypothalamus. We also investigated the expression status of several appetite-related genes, including neuropeptide Y, proopiomelanocortin, neuromedin U and neuromedin U receptor 2. Prior to experiments, rats were fasted for 12–14 hours, because metformin administration may have no effect on food intake in normally fed rats. We also applied an intracerebroventricular approach for metformin administration because a peripheral approach may be impeded by peripheral metabolism and/or the blood-brain barrier. A possible central interaction responsible for metformin-induced feeding changes in the rats has been suggested.
RESULTS
Quantitative analysis of experimental animals
A total of 55 rats were included in this study. Only animals that showed progressive weight gain after intracerebroventricular surgery were used. The general health status, including abnormal temperature, vomiting, diarrhea and polyuria, was observed during the experiment to exclude the presence of illness and/or malaise in the animals. Eleven rats were excluded from the study because of abnormal temperature (three rats), vomiting (two rats), diarrhea (four rats), or polyuria (two rats). The remaining 44 rats were included in the final results. Of these, 20 rats were used to evaluate the effect of metformin on feeding behavior and biochemical parameters, and 24 rats were used to evaluate the role of AMP-activated protein kinase and the expression status of several appetite-related genes. For experiments concerning the effect of metformin on feeding behavior and biochemical parameters, rats were randomly assigned to two groups (n = 10 per group): a metformin group and a control group. For each rat, metformin or saline was injected into the third ventricle. For the role of AMP-activated protein kinase and the expression status of several appetite-related genes, rats were divided into three groups (n = 8 per group): a control group, a metformin group, and a metformin + compound C group. Three hours after a single intracerebroventricular administration of metformin or saline, the rats from the metformin + compound C group received an intracerebroventricular dose of compound C, while the other two groups received dimethyl sulfoxide.
Metformin decreased food intake in rats
Three days before administration, there were no significant differences in food intake, water consumption, or body weight between the two groups of rats (data not shown). The cumulative food intake in metformin-treated rats reduced by 43%, 56% and 54% at 6, 8 and 12 hours post-treatment, respectively, and the most obvious anorexigenic effect, 85% lower than the control, was observed from 2–4 hours (Figure 1A); this difference was no longer present 24 hours after intracerebroventricular administration (data not shown). Measurement of food intake at 2-hour intervals showed that rats in the metformin group consumed much less food than those in the control group from 2–4 hours (P < 0.01) and from 4–6 hours (P < 0.05; Figure 1B). Such an anorexic role was also evident in rats in the metformin group from 6–8 hours, although this difference did not show statistical significance (Figure 1B). There was no significant difference in water consumption between the two groups (data not shown).
Figure 1.
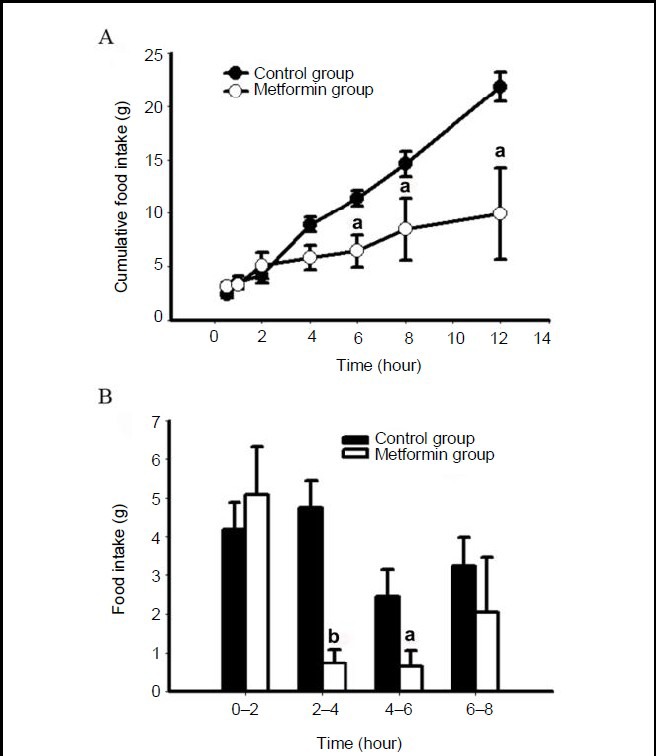
Metformin decreases food intake in rats.
After a single intracerebroventricular administration of metformin or saline, food intake was measured at 0.5, 1, 2, 4, 6, 8 and 12 hours.
(A) Cumulative food intake of the rats. (B) Food intake of the rats for every 2 hour period. Data are expressed as mean ± SEM. There were five rats in each group. Differences between the groups were analyzed using two-tailed t-tests. aP < 0.05, bP < 0.01, vs. control group.
Meanwhile, a significant difference was found between the two groups of rats in cumulative change of body weight from 4–12 hours post-administration (Figure 2A). Notably, during the periods from 2–4 hours and from 4–6 hours (P < 0.05), rats in the metformin group lost weight (Figure 2B).
Figure 2.
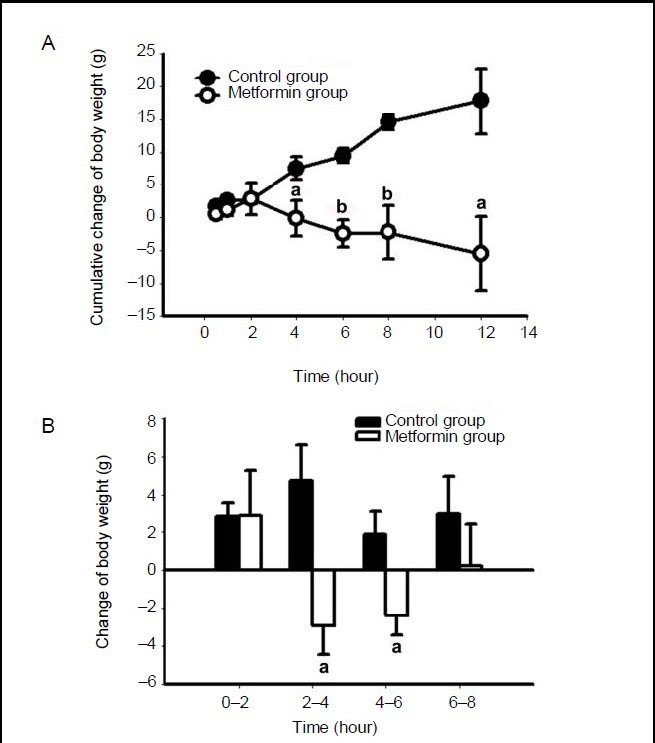
Metformin decreases body weight gain in rats.
After a single intracerebroventricular administration of metformin or saline, body weight was measured at 0.5, 1, 2, 4, 6, 8 and 12 hours.
(A) Cumulative body weight change of the rats. (B) Body weight change of the rats for every 2 hour period. Data are expressed as mean ± SEM. There were five rats in each group. Differences between the groups were analyzed using two-tailed t-tests. aP < 0.05, bP < 0.01, vs. control group.
Effect of metformin on biochemical parameters in rats
Five rats in each group were sacrificed 4 hours after a single intracerebroventricular administration of metformin or saline. While no differences were found for other measured biochemical parameters between the two groups of rats, plasma low-density lipoprotein was significantly lower in the metformin-treated rats than those in the control group (P < 0.05; Figure 3). No differences in the weights of liver, spleen, heart, kidney, perirenal fat or testes were found (data not shown).
Figure 3.
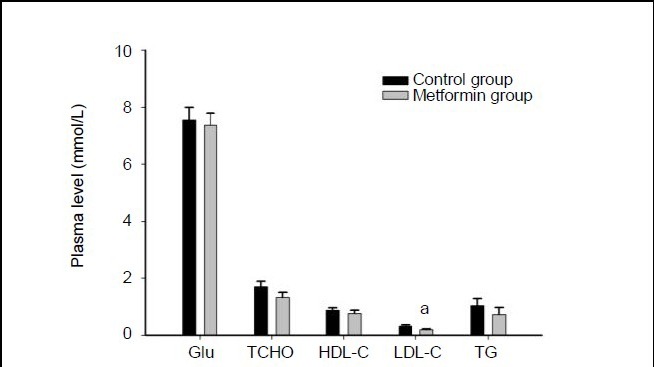
Biochemical characteristics after administration of metformin.
Rats were sacrificed 4 hours after a single intracerebroventricular administration of metformin or saline. The plasma glucose (Glu), total cholesterol (TCHO), high-density lipoprotein-cholesterol (HDL-C), low-density lipoprotein-cholesterol (LDL-C), and triglycerides (TG) levels were analyzed using a 7020 Automatic Biochemistry Analyzer. Data are expressed as mean ± SEM. There were five rats in each group. Differences between the groups were analyzed using two-tailed t-tests. aP < 0.05, vs. control group.
Metformin activated AMP-activated protein kinase phosphorylation in the hypothalamus
To explore the underlying association of the hypothalamic effect of metformin with feeding behavior and energy balance, 3 hours after a single intracerebroventricular administration of metformin or saline, the rats from the metformin + compound C group were administered with an intracerebroventricular dose of compound C, while the other two groups received dimethyl sulfoxide. One hour later, rats were sacrificed and western blot assay was used to analyze hypothalamic AMP-activated protein kinase phosphorylation levels. The results showed that central administration of metformin significantly increased hypothalamic AMP-activated protein kinase phosphorylation levels compared with the control group, and that AMP-activated protein kinase activation by metformin was blocked by compound C, an inhibitor of AMP-activated protein kinase phosphorylation (Figure 4).
Figure 4.
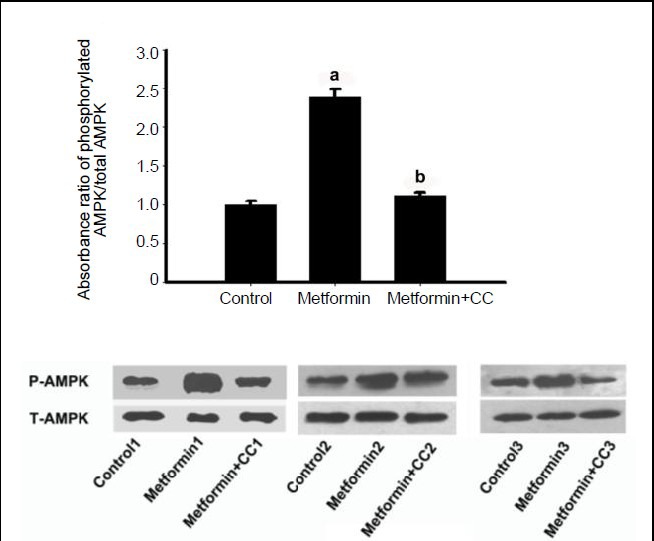
Effects of metformin and compound C on AMP-activated protein kinase (AMPK) phosphorylation in the hypothalamus.
Three hours after a single intracerebroventricular administration of metformin or saline, rats from the metformin + compound C (CC) group received an intracerebroventricular dose of compound C, while the other two groups were given an equal volume of dimethyl sulfoxide. One hour later, rats were sacrificed and western blot assay was used to analyze hypothalamic AMP-activated protein kinase phosphorylation (P-AMPK) levels. Data are expressed as mean ± SEM. There were eight rats in each group. Differences between the groups were analyzed using analysis of variance and least significant difference tests. aP < 0.001, vs. control group; bP < 0.001, vs. metformin group.
Metformin down-regulated neuropeptide Y expression in hypothalamus
The hypothalamus represents a key regulatory network in the central nervous system, maintaining a balance at the whole body level between energy input and output. To examine the interactions of the metformin-mediated anorexigenic effect with other appetite stimulating/inhibit- ing signals in the hypothalamus, we examined the transcriptional responses of hypothalamic neuropeptide Y, proopiomelanocortin, neuromedin U and neuromedin U receptor 2 to metformin treatment in the rats. The results of quantitative real-time PCR showed that the PCR efficiency of the four genes was similar (Figure 5). Hypothalamic expression of neuropeptide Y was significantly lower in rats in the metformin group compared with those in the control group, and the down-regulation of gene expression by metformin was inhibited by compound C (Figure 6), suggesting that metformin's regulation of neuropeptide Y may be related to AMP-activated protein kinase. Metformin had no effect on the hypothalamic expression of proopiomelanocortin, neuromedin U or neuromedin U receptor 2 (Figure 7).
Figure 5.
Real-time PCR plots of neuropeptide Y, proopiomelanocortin, neuromedin U and neuromedin U receptor 2 gene expression.
Hypothalamic expression of neuropeptide Y, proopiomelanocortin, neuromedin U and neuromedin U receptor 2 were profiled in the cDNA samples. The shapes of the plots were similar, showing similar PCR efficiency.
Figure 6.
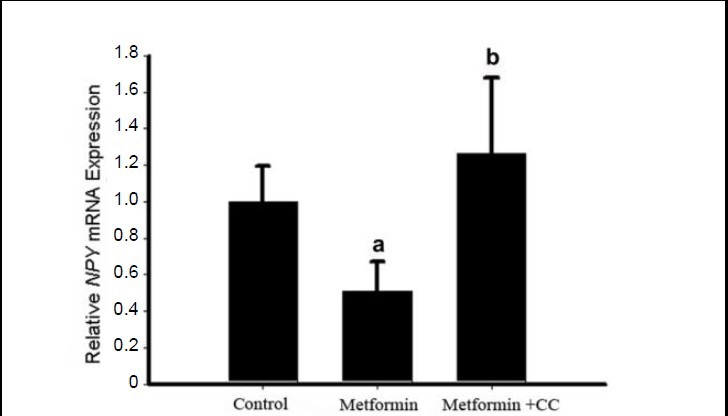
Effects of metformin and compound C on neuropeptide Y (NPY) expression in the hypothalamus.
Three hours after a single intracerebroventricular administration of metformin or saline, rats from the metformin + compound C (CC) group received an intracerebroventricular dose of compound C, while the other two groups received an equal volume of dimethyl sulfoxide. One hour later, rats were sacrificed and hypothalamic expression of neuropeptide Y was determined using real-time PCR. Relative neuropeptide Y mRNA expression levels were calculated as the ratio of the neuropeptide Y mRNA expression level to the GAPDH expression level in the same sample. Data are expressed as mean ± SEM. There were eight rats in each group. Differences between the groups were analyzed using analysis of variance and least significant difference tests. aP < 0.05, vs. control group; bP < 0.05, vs. metformin group.
Figure 7.
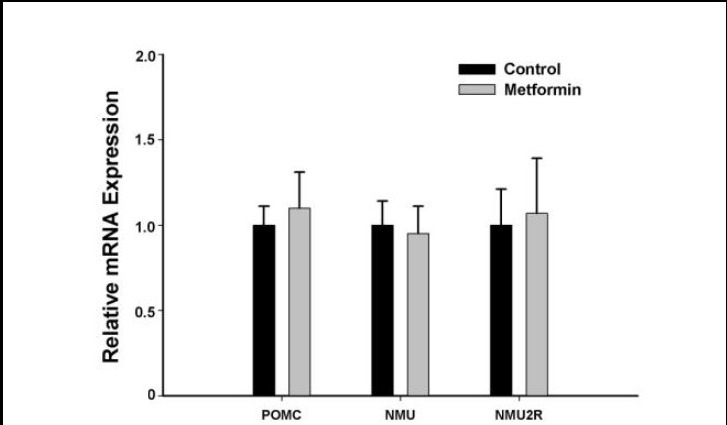
Effects of metformin on proopiomelanocortin, neuromedin U and neuromedin U receptor 2 mRNA expression in the hypothalamus.
Rats were sacrificed 4 hours after a single intracerebroventricular administration of metformin or saline. Hypothalamic expression of proopiomelanocortin (POMC), neuromedin U (NMU) and neuromedin U receptor 2 (NMU2R) was determined using real-time PCR. Relative mRNA expression levels were calculated as the ratio of proopiomelanocortin, neuromedin U and neuromedin U receptor 2 mRNA expression level to the GAPDH expression level in the same sample. Data are expressed as mean ± SEM. There were eight rats in each group. Differences between the groups were analyzed using two-tailed t-tests. Metformin had no effect on the hypothalamic expression of proopiomelanocortin, neuromedin U or neuromedin U receptor 2.
DISCUSSION
In addition to its glucose-lowering effect, metformin also has a role in regulating energy balance, presuming via an AMP-activated protein kinase cascade pathway in endothelial cells, skeletal muscle and liver[12,27,36,37]. Given that whole body energy homeostasis is critically regulated by multiple signaling pathways in the central nervous system as well as between the central nervous system and the periphery, whether and how metformin also exerts its energy-modulating role in the central nervous system is still uncertain. In this study, we took the approach of intracerebroventricular administration to investigate the direct effects of metformin on food intake, and to explore the possible mechanism of metformin in the rat hypothalamus.
Metformin can inhibit weight gain and reduce appetite[13,38,39]. It can even induce weight loss[40,41], preferentially in adipose tissue[42]. Approximately half of drug-naive type 2 diabetic cases displayed significant weight loss with metformin compared with baseline or comparator drugs[15]. Decreased visceral adipose tissue mass and increased glucose uptake were also reported in type 2 diabetic patients after metformin treatment[43]. In this study, metformin achieved its maximum effect on food intake and body weight from 2–6 hours after intracerebroventricular administration. Interestingly, at 12 hours after administration, the amount of food consumed by the metformin-treated rats was 11.87 g less than control group, but their weight gain was 23.21 g lower than the control group. This evidence suggests that metformin had stronger central inhibitory effects on weight gain than on food intake. The short-term effects of metformin on body weight might be relevant in the setting of concomitant insulin use[44]. Metformin acts by improving the action of insulin, particularly in the liver[45]. However, whether the effect of metformin on body weight in the central nervous system is similar to that in the periphery is unclear. Together, these findings suggest that the central nervous system participates, at least partially, in the action of metformin in regulating food intake and body weight.
Metformin can modestly improve lipid metabolism in patients with metabolic syndrome and diabetes via reducing the levels of low-density lipoprotein-cholesterol, total cholesterol and triglycerides, and by increasing the level of high-density lipoprotein-cholesterol[46]. In this study, the level of plasma low-density lipoprotein-cholesterol was significantly decreased in metformin-treated rats, thus confirming the benefits of central metformin on blood lipids. We also measured the weights of viscera, as well as hepatic and renal function; no hypoglycemia or other undesirable effects were found after intracerebroventricular administration of metformin.
To explore the underlying association of the hypothalamic effect of metformin with feeding behavior and energy balance, we analyzed the activation status of AMP-activated protein kinase in the hypothalamus of rats. Our results showed that the level of phosphorylated AMP-activated protein kinase was significantly increased in response to exposure to metformin in the hypothalamus of rats, and that activation of AMP-activated protein kinase can be suppressed by compound C, an AMP-activated protein kinase inhibitor. It is well recognized that AMP-activated protein kinase is a major sensor of cellular energy charge and an important regulator of food intake by responding to hormonal and nutrient signals in the hypothalamus. The activation of AMP-activated protein kinase can lead to an increase in food intake and body weight while inhibition of AMP-activated protein kinase activity can result in a decrease in these parameters in the hypothalamus[47]. Seemingly these data are not consistent with what we observed in this study. Given that AMP-activated protein kinase could be activated in hypothalamic neurons under conditions of diet restriction[22], it is not surprising that the increased AMP-activated protein kinase phosphorylation after intracerebroventricular dosing of metformin in this study may be merely attributed to the anorexigenic effect of metformin, similar to the effect of diet restriction. Nevertheless, the precise mechanism, particularly in the central nervous system, by which metformin activates AMP-activated protein kinase remains unclear. The data from Chau-Van's work, which was probably the first study on the effect of metformin on neuronal AMP-activated protein kinase, demonstrated that metformin can block AMP-activated protein kinase activation under conditions of low glucose[32]. An in vivo study also found an inhibitory role of intracerebroventricularly-admini- stered metformin on ghrelin-induced AMP-activated protein kinase activation[18]. Another in vivo study showed that oral administration of lower-dose leptin can decrease the phosphorylation level of AMP-activated protein kinase, while metformin alone does not change the level of phosphorylated AMP-activated protein kinase in the hypothalamus of rats fed standard chow[48].
These results are not in line with the findings of the present study. These discrepancies might be explained as follows. First, different experiment systems were used in Chau-Van's[32] and our studies, and from both a biological and a physiological point of view it is not always possible to make simple comparisons between data derived from in vitro and in vivo experiments. Our observation of the AMP-activated protein kinase response to metformin was based on an intracerebroventricular administration approach that allowed us to obtain more direct and physiologically relevant evidence toward the argument on whether phosphorylation of AMP-activated protein kinase is activated or inhibited by metformin in the hypothalamus. Second, the administration route and dosing duration of the drug are different between our studies and others. Stevanovic et al[18] examined hypothalamic phosphorylated AMP-activated protein kinase after 1 hour of intracerebroventricular metformin administration and found that metformin blocked ghrelin-induced AMP-activated protein kinase activation, whereas Kim et al[48] detected hypothalamic AMP-activated protein kinase after 4 weeks of oral metformin administration. The unchanged hypothalamic AMP-activated protein kinase activity observed by Kim et al may merely reflect a return to normal physiological steady-state of this kinase following chronic treatment with metformin, and the integrated function of AMP-activated protein kinase in mediating diverse metabolic signaling between the periphery and central nervous system. Collectively, the results from the present study and others could be supportive of the emerging concept that AMP-activated protein kinase activation may have differential regulating functions that depend on the tissue, degree of stimulation and conditions of activation[34]. Our findings suggest that under physiological conditions, a single intracerebroventricular dose of metformin can regulate feeding responses and energy expenditure, and these changes might further activate AMP-activated protein kinase in the hypothalamus. Beyond a master sensor that is activated with more extreme changes in energy levels[34], AMP-activated protein kinase could also respond to transient physiological fluctuation. Activation of hypothalamic AMP-activated protein kinase by metformin was also evident in the work of Chen et al[33]. These authors showed that in neuronal culture systems, and in mouse brain, the phosphorylation of AMP-activated protein kinase and acetyl CoA carboxylase were both induced by metformin treatment, suggesting a similar effect of the drug in vitro and in vivo[33]. Our finding is certainly not able to rule out the possibility that the consequences of hypothalamic AMP-activated protein kinase activation in response to metformin under pathological conditions may differ from those under normal physiological conditions. Indeed, elevated hypothalamic AMP-activated protein kinase activity was evident in streptozocin-induced diabetic rats[49]. It will thus be interesting to elucidate the outcome of AMP-activated protein kinase alterations in the central nervous system if metformin, a widely used anti-diabetic drug, is used in such a diabetic model.
The hypothalamus represents a key regulating network in the central nervous system, maintaining a balance at the whole body level between energy input and output[50,51,52,53,54,55,56,57]. To examine the interactions of a metformin-mediated anorexigenic effect with other appetite stimulating/inhibiting signals in the hypothalamus, we examined the transcriptional responses of hypothalamic neuropeptide Y, proopiomelanocortin, neuromedin U and neuromedin U receptor 2 to metformin treatment in rats. Neuropeptide Y is a 36 amino acid peptide which is involved in many physiological activities in the brain, primarily increasing food intake and decreasing physical activity[50,51,52]. In some animal models of obesity, such as diet-induced obesity, leptin-deficient ob/ob mice, Zucker fatty fa/fa rats, melanocortin receptor 4-knockout mice, brown adipose tissue-deficient mice and tubby mice, the expression levels of neuropeptide Y are elevated[53,54,55,56,57]. Our results showed that the expression level of neuropeptide Y was significantly reduced, while expression of the remaining genes remained unchanged. These findings suggest that the central regulation of appetite by metformin may be related to hypothalamic neuropeptide Y signaling pathway. Moreover, the inhibition of neuropeptide Y expression by central metformin administration was found to be abolished by an AMP-activated protein kinase antagonist, compound C, further indicating that metformin-mediated alterations in hypothalamic neuropeptide Y expression toward the appetite regulation may be AMP-activated protein kinase-related, although not exclusively so. While opposing regulation of neuronal AMP-activated protein kinase by metformin is evident in Chau-Van's in vitro[32] and our in vivo studies, the expression levels of neuropeptide Y and proopiomelanocortin from the current study are partially consistent with that from Chau-Van's work, in which metformin inhibited low glucose-stimulated, but not unstimulated, neuropeptide Y expression while no change was found in proopiomelanocortin expression[32]. It has been also reported that hypothalamic neuropeptide Y gene expression was not inhibited after oral metformin treatment for 12 days in genetically obese Zucker rats[58]. Again, the differences in experimental models chosen or the administration route and dosing duration of metformin used might to a certain extent account for the discrepancies between our current data and other results[32,58].
In summary, our results demonstrate that central metformin can regulate food intake and locomotor activity, and that under normal physiological conditions metformin can activate AMP-activated protein kinase phosphorylation, although this may simply be a response to the anorexigenic effects of metformin. Although the central interaction of metformin with AMP-activated protein kinase in coordinating feeding behavior and peripheral energy expenditure is not yet entirely clear, the anorexigenic role of the drug in the hypothalamus might be related to neuropeptide Y signaling, which is, at least partially, modulated by AMP-activated protein kinase.
MATERIALS AND METHODS
Design
A randomized, controlled experiment.
Time and setting
Experiments were performed at the Key Laboratory of Brain Functional Genomics, East China Normal University, Shanghai, China from December 2011 to July 2012.
Materials
Animals
Male Sprague-Dawley rats weighing 130 ± 10 g were purchased from Slac Animal Center (Shanghai, China, license No. SCXK (Hu) 2012-0002). Rats were housed in the specific pathogen-free animal facility with controlled temperature (22–24°C), humidity (40–70%) and light/ dark regime (lights on between 7:00 and 19:00). Standard laboratory food and water were available ad libitum before the experiments. The experimental protocols for care and use of experimental animals described in the present study were performed in accordance with NIH guidelines and approved by the Animals Ethical Committee of the East China Normal University in China.
Drugs
Metformin tablets (Shanghai Pharmaceutical Co., Ltd., Shanghai, China) were dissolved in sterile saline (40 mg/mL). Compound C powder (Sigma, Shanghai, China) was dissolved in dimethyl sulfoxide.
Methods
Intracerebroventricular surgery and metformin administration
For intracerebroventricular surgery, rats were intraperitoneally anaesthetized with sodium pentobarbital (70 mg/kg; Sigma) and positioned on a Stereotaxic apparatus (Stoelting Co., Wood Dale, IL, USA). Three stainless steel screws were anchored to the skull and a chronic 9 mm sterile cannula was implanted into the third ventricle. The stereotaxic coordinates were: anterior-posterior to 0.8 mm, dorsoventral, 6.5 mm from the bregma. At the end of the surgery, dental cement was used to fix the cannula and a 10 mm plunger was inserted into the cannula in case of blockage. The location of the cannula in the third ventricle was confirmed by trypan blue staining. After surgery, animals were allowed to recover for 1 week before the administration procedure, by which time they were accustomed to handling on a daily basis. Their food intake, body weight and water intake were measured daily during that week. The rats were fasted for 12–14 hours before the administration procedure. For each rat, 200 μg metformin (in 5 μL saline) or saline of the same volume was slowly injected (over 2–3 minutes) through the cannula using a micro-injector. Food intake, water consumption and body weight were measured at 0.5, 1, 2, 4, 6, 8 and 12 hours after administration.
Biochemical analysis
Rats were deeply anaesthetized with pentobarbital (50 mg/kg body weight) and rapidly decapitated 4 hours after administration of metformin or saline. Plasma glucose, total cholesterol, high-density lipoprotein, low- density lipoprotein, and triglycerides were analyzed using a 7020 Automatic Biochemistry Analyzer (Hitachi, Tokyo, Japan). The weights of the liver, spleen, heart, kidney, perirenal fat and testes were also recorded.
Western blot assay
Three hours after metformin administration, rats from the metformin + compound C group received an intracerebroventricular 10 μL dose of compound C (100 nmol/L), and rats from the control group and the metformin group were given an equal volume of dimethyl sulfoxide. 1 hour later, all rats were deeply anaesthetized with injected pentobarbital (50 mg/kg body weight) and rapidly decapitated and the hypothalamus was harvested and stored at –80°C.
Hypothalamic tissue was homogenized in lysis buffer and 20 μg samples of lysate were separated by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose filter membranes. Blots were then blocked and incubated overnight at 4°C with rabbit monoclonal T172-phosphorylated AMP-activated protein kinase antibody and rabbit monoclonal total AMP-activated protein kinase antibody (1:1 000; Cell Signaling Technology Inc., Shanghai, China). Blots were developed using anti-rabbit secondary antibody (1:5 000; Kang Chen Bio-tech Inc, Shanghai, China) for 1 hour at 37°C, and visualized using an electrogenerated chemiluminescence system (Millipore, Shanghai, China).
Quantitative real-time PCR
Hypothalamic expression of neuropeptide Y, proopiome- lanocortin, neuromedin U and neuromedin U receptor 2 were determined using real-time PCR. Briefly, total RNA was extracted from the hypothalamus using Trizol (Invitrogen, Shanghai, China) and cDNA was generated using M-MLV reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Specific primers of the genes of interest were used as previously reported[59,60,61] and synthesized by Invitrogen (Shanghai, China). GAPDH (Invitrogen) was used as an endogenous reference.
Statistical analysis
Differences between the groups were analyzed using two-tailed t-tests or analysis of variance and least significant difference tests using SPSS 13.0 software (SPSS, Chicago, IL, USA). Data are presented as mean ± SEM. Statistical significance was accepted as P < 0.05.
Footnotes
Funding: This work was supported by grants from the National Natural Science Foundation of China, No. 31171019 and No. 31200820; the Opening Project of Shanghai Key Laboratory of Brain Functional Genomics and Key Laboratory of Brain Functional Genomics (East China Normal University) of the Ministry of Education.
Conflicts of interest: None declared.
Ethical approval: This study was approved by the Animal Ethics Committee of the East China Normal University, China.
(Reviewed by Barnett M, Qiu YM, Liang GB)
(Edited by Mu WJ, Yang Y, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- 1.Svacina S. Treatment of obese diabetics. Adv Exp Med Biol. 2012;77:459–464. [PubMed] [Google Scholar]
- 2.Aballay LR, Eynard AR, Diaz Mdel P, et al. Overweight and obesity: a review of their relationship to metabolic syndrome, cardiovascular disease, and cancer in South America. Nutr Rev. 2013;7:168–179. doi: 10.1111/j.1753-4887.2012.00533.x. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg RB, Mather K. Targeting the consequences of the metabolic syndrome in the Diabetes Prevention Program. Arterioscler Thromb Vasc Biol. 2012;32:2077–2090. doi: 10.1161/ATVBAHA.111.241893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutcheson R, Rocic P. The metabolic syndrome, oxidative stress, environment, and cardiovascular disease: the great exploration. Exp Diabetes Res 2012. 2012 doi: 10.1155/2012/271028. 271028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viollet B, Guigas B, Sanz Garcia N, et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGill JB. Pharmacotherapy in type 2 diabetes: a functional schema for drug classification. Curr Diabetes Rev. 2012;8:257–267. doi: 10.2174/157339912800840541. [DOI] [PubMed] [Google Scholar]
- 7.Viollet B, Foretz M. Revisiting the mechanisms of metformin action in the liver. Ann Endocrinol (Paris) 2013;74(2):123–129. doi: 10.1016/j.ando.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Al-Abri SA, Hayashi S, Thoren KL, et al. Metformin overdose-induced hypoglycemia in the absence of other antidiabetic drugs. Clin Toxicol (Phila) doi: 10.3109/15563650.2013.784774. in press. [DOI] [PubMed] [Google Scholar]
- 9.Carvajal R, Rosas C, Kohan K, et al. Metformin augments the levels of molecules that regulate the expression of the insulin-dependent glucose transporter GLUT4 in the endometria of hyperinsulinemic PCOS patients. Hum Reprod. doi: 10.1093/humrep/det116. in press. [DOI] [PubMed] [Google Scholar]
- 10.Lu J, Ji J, Meng H, et al. The protective effect and underlying mechanism of metformin on neointima formation in fructose-induced insulin resistant rats. Cardiovasc Diabetol. 2013;12:58. doi: 10.1186/1475-2840-12-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ibanez L, Diaz M, Sebastiani G, et al. Oral contraception vs insulin sensitization for 18 months in nonobese adolescents with androgen excess: posttreatment differences in c-reactive protein, intima-media thickness, visceral adiposity, insulin sensitivity, and menstrual regularity. J Clin Endocrinol Metab. 2013;98(5):E902–907. doi: 10.1210/jc.2013-1041. [DOI] [PubMed] [Google Scholar]
- 12.Matsui Y, Hirasawa Y, Sugiura T, et al. Metformin reduces body weight gain and improves glucose intolerance in high-fat diet-fed C57BL/6J mice. Biol Pharm Bull. 2010;33:963–970. doi: 10.1248/bpb.33.963. [DOI] [PubMed] [Google Scholar]
- 13.Hou M, Venier N, Sugar L, et al. Protective effect of metformin in CD1 mice placed on a high carbohydrate-high fat diet. Biochem Biophys Res Commun. 2010;397:537–542. doi: 10.1016/j.bbrc.2010.05.152. [DOI] [PubMed] [Google Scholar]
- 14.Lv WS, Wen JP, Li L, et al. The effect of metformin on food intake and its potential role in hypothalamic regulation in obese diabetic rats. Brain Res. 2012;1444:11–19. doi: 10.1016/j.brainres.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 15.Paolisso G, Amato L, Eccellente R, et al. Effect of metformin on food intake in obese subjects. Eur J Clin Invest. 1998;28:441–446. doi: 10.1046/j.1365-2362.1998.00304.x. [DOI] [PubMed] [Google Scholar]
- 16.Yener S, Comlekci A, Akinci B, et al. Soluble CD40 ligand, plasminogen activator inhibitor-1 and thrombin-activatable fibrinolysis inhibitor-1-antigen in normotensive type 2 diabetic subjects without diabetic complications. Effects of metformin and rosiglitazone. Med Princ Pract. 2009;18:266–271. doi: 10.1159/000215722. [DOI] [PubMed] [Google Scholar]
- 17.Smith DL, Jr, Elam CF, Jr, Mattison JA, et al. Metformin supplementation and life span in Fischer-344 rats. J Gerontol A Biol Sci Med Sci. 2010;65:468–474. doi: 10.1093/gerona/glq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevanovic D, Janjetovic K, Misirkic M, et al. Intracerebroventricular administration of metformin inhibits ghrelin-induced hypothalamic AMP-kinase signalling and food intake. Neuroendocrinology. 2012;96:24–31. doi: 10.1159/000333963. [DOI] [PubMed] [Google Scholar]
- 19.Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS One. 2010;5:e8758. doi: 10.1371/journal.pone.0008758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anisimov VN, Berstein LM, Egormin PA, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7:2769–2773. doi: 10.4161/cc.7.17.6625. [DOI] [PubMed] [Google Scholar]
- 21.Lee CK, Choi YJ, Park SY, et al. Intracerebroventricular injection of metformin induces anorexia in rats. Diabetes Metab J. 2012;36:293–299. doi: 10.4093/dmj.2012.36.4.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minokoshi Y, Alquier T, Furukawa N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 23.McCrimmon RJ, Shaw M, Fan X, et al. Key role for AMP- activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes. 2008;57:444–450. doi: 10.2337/db07-0837. [DOI] [PubMed] [Google Scholar]
- 24.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suwa M, Egashira T, Nakano H, et al. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol. 2006;101:1685–1692. doi: 10.1152/japplphysiol.00255.2006. [DOI] [PubMed] [Google Scholar]
- 26.Kim YD, Park KG, Lee YS, et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase- dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57:306–314. doi: 10.2337/db07-0381. [DOI] [PubMed] [Google Scholar]
- 27.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin JT, Troke JJ, Kimura N, et al. A novel cardioprotective agent in cardiac transplantation: metformin activation of AMP-activated protein kinase decreases acute ischemia-reperfusion injury and chronic rejection. Yale J Biol Med. 2011;84:423–432. [PMC free article] [PubMed] [Google Scholar]
- 29.Amato S, Liu X, Zheng B, et al. AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science. 2011;332:247–251. doi: 10.1126/science.1201678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams T, Courchet J, Viollet B, et al. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc Natl Acad Sci U S A. 2011;108:5849–5854. doi: 10.1073/pnas.1013660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roland AV, Moenter SM. Prenatal androgenization of female mice programs an increase in firing activity of gonadotropin-releasing hormone (GnRH) neurons that is reversed by metformin treatment in adulthood. Endocrinology. 2011;152:618–628. doi: 10.1210/en.2010-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chau-Van C, Gamba M, Salvi R, et al. Metformin inhibits adenosine 5’-monophosphate-activated kinase activation and prevents increases in neuropeptide Y expression in cultured hypothalamic neurons. Endocrinology. 2007;148:507–511. doi: 10.1210/en.2006-1237. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y, Zhou K, Wang R, et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J Physiol. 2006;574:85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hawley SA, Gadalla AE, Olsen GS, et al. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 36.Musi N, Hirshman MF, Nygren J, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 37.Ghatak SB, Dhamecha PS, Bhadada SV, et al. Investigation of the potential effects of metformin on atherothrombotic risk factors in hyperlipidemic rats. Eur J Pharmacol. 2011;659:213–223. doi: 10.1016/j.ejphar.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 38.Nakano M, Inui A. Metformin and incretin-based therapies up-regulate central and peripheral Adenosine monophosphate-activated protein affecting appetite and metabolism. Indian J Endocrinol Metab. 2012;16:S529–531. doi: 10.4103/2230-8210.105567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Werneke U, Taylor D, Sanders TA. Behavioral interventions for antipsychotic induced appetite changes. Curr Psychiatry Rep. 2013;15:347. doi: 10.1007/s11920-012-0347-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glueck CJ, Fontaine RN, Wang P, et al. Metformin reduces weight, centripetal obesity, insulin, leptin, and low-density lipoprotein cholesterol in nondiabetic, morbidly obese subjects with body mass index greater than 30. Metabolism. 2001;50:856–861. doi: 10.1053/meta.2001.24192. [DOI] [PubMed] [Google Scholar]
- 41.Komori T, Yoshida F, Nakamura J, et al. Metformin ameliorates treatment of obese type 2 diabetic patients with mental retardation; its effects on eating behavior and serum leptin levels. Exp Clin Endocrinol Diabetes. 2004;112:422–428. doi: 10.1055/s-2004-821187. [DOI] [PubMed] [Google Scholar]
- 42.Stumvoll M, Nurjhan N, Perriello G, et al. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333:550–554. doi: 10.1056/NEJM199508313330903. [DOI] [PubMed] [Google Scholar]
- 43.Virtanen KA, Hallsten K, Parkkola R, et al. Differential effects of rosiglitazone and metformin on adipose tissue distribution and glucose uptake in type 2 diabetic subjects. Diabetes. 2003;52:283–290. doi: 10.2337/diabetes.52.2.283. [DOI] [PubMed] [Google Scholar]
- 44.Golay A. Metformin and body weight. Int J Obes (Lond) 2008;32:61–72. doi: 10.1038/sj.ijo.0803695. [DOI] [PubMed] [Google Scholar]
- 45.Giannarelli R, Aragona M, Coppelli A, et al. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003;29:6S28–35. doi: 10.1016/s1262-3636(03)72785-2. [DOI] [PubMed] [Google Scholar]
- 46.Lawrence JM, Reid J, Taylor GJ, et al. Favorable effects of pioglitazone and metformin compared with gliclazide on lipoprotein subfractions in overweight patients with early type 2 diabetes. Diabetes Care. 2004;27:41–46. doi: 10.2337/diacare.27.1.41. [DOI] [PubMed] [Google Scholar]
- 47.Xue B, Kahn BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. J Physiol. 2006;574:73–83. doi: 10.1113/jphysiol.2006.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim YW, Kim JY, Park YH, et al. Metformin restores leptin sensitivity in high-fat-fed obese rats with leptin resistance. Diabetes. 2006;55:716–724. doi: 10.2337/diabetes.55.03.06.db05-0917. [DOI] [PubMed] [Google Scholar]
- 49.Namkoong C, Kim MS, Jang PG, et al. Enhanced hypothalamic AMP-activated protein kinase activity contributes to hyperphagia in diabetic rats. Diabetes. 2005;54:63–68. doi: 10.2337/diabetes.54.1.63. [DOI] [PubMed] [Google Scholar]
- 50.Gilpin NW. Neuropeptide Y (NPY) in the extended amygdala is recruited during the transition to alcohol dependence. Neuropeptides. 2012;46:253–259. doi: 10.1016/j.npep.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirsch D, Zukowska Z. NPY and stress 30 years later: the peripheral view. Cell Mol Neurobiol. 2012;32:645–659. doi: 10.1007/s10571-011-9793-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mercer RE, Chee MJ, Colmers WF. The role of NPY in hypothalamic mediated food intake. Front Neuroendocrinol. 2011;32:398–415. doi: 10.1016/j.yfrne.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 53.Makimura H, Mizuno TM, Roberts J, et al. Adrenalectomy reverses obese phenotype and restores hypothalamic melanocortin tone in leptin-deficient ob/ob mice. Diabetes. 2000;49:1917–1923. doi: 10.2337/diabetes.49.11.1917. [DOI] [PubMed] [Google Scholar]
- 54.Dryden S, Pickavance L, Frankish HM, et al. Increased neuropeptide Y secretion in the hypothalamic paraventricular nucleus of obese (fa/fa) Zucker rats. Brain Res. 1995;690:185–188. doi: 10.1016/0006-8993(95)00628-4. [DOI] [PubMed] [Google Scholar]
- 55.Marsh DJ, Hollopeter G, Huszar D, et al. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 56.Tritos NA, Elmquist JK, Mastaitis JW, et al. Characterization of expression of hypothalamic appetite-regulating peptides in obese hyperleptinemic brown adipose tissue-deficient (uncoupling protein-promoter-driven diphtheria toxin A) mice. Endocrinology. 1998;139:4634–4641. doi: 10.1210/endo.139.11.6308. [DOI] [PubMed] [Google Scholar]
- 57.Guan XM, Yu H, Van der Ploeg LH. Evidence of altered hypothalamic pro-opiomelanocortin/neuropeptide Y mRNA expression in tubby mice. Brain Res Mol Brain Res. 1998;59:273–279. doi: 10.1016/s0169-328x(98)00150-8. [DOI] [PubMed] [Google Scholar]
- 58.Rouru J, Pesonen U, Koulu M, et al. Anorectic effect of metformin in obese Zucker rats: lack of evidence for the involvement of neuropeptide Y. Eur J Pharmacol. 1995;273:99–106. doi: 10.1016/0014-2999(94)00669-x. [DOI] [PubMed] [Google Scholar]
- 59.Sindelar DK, Ste Marie L, Miura GI, et al. Neuropeptide Y is required for hyperphagic feeding in response to neuroglucopenia. Endocrinology. 2004;145:3363–3368. doi: 10.1210/en.2003-1727. [DOI] [PubMed] [Google Scholar]
- 60.Nogueiras R, Tovar S, Mitchell SE, et al. Negative energy balance and leptin regulate neuromedin-U expression in the rat pars tuberalis. J Endocrinol. 2006;190:545–553. doi: 10.1677/joe.1.06577. [DOI] [PubMed] [Google Scholar]
- 61.Gartlon J, Szekeres P, Pullen M, et al. Localisation of NMU1R and NMU2R in human and rat central nervous system and effects of neuromedin-U following central administration in rats. Psychopharmacology (Berl) 2004;177:1–14. doi: 10.1007/s00213-004-1918-3. [DOI] [PubMed] [Google Scholar]