

Abstract
After intraperitoneal injection of 20 mg/kg lead acetate, rats received 8 weeks of treadmill exercise (15–22 m/min, 25–64 minutes) and/or treadmill exercise at 1.6 km/h until exhaustion. The markers related to neurotoxicity were measured by enzyme-linked immunosorbent assay method. 8 weeks of treadmill exercise significantly increased brain-derived neurotrophic factor level in the hippocampus (P = 0.04) and plasma level of total antioxidant capacity of rats exposed to lead acetate (P < 0.001), and significantly decreased plasma level of malondialdehyde (P < 0.001). Acute exercise only decreased the hippocampal malondialdehyde level (P = 0.09) and increased brain-derived neurotrophic factor level in the hippocampus (P = 0.66). Acute exercise also enhanced the total antioxidant capacity in rats exposed to lead acetate, insignificantly (P = 0.99). These findings suggest that chronic treadmill exercise can significantly decrease neurotoxicity and alleviate oxidative stress in rats exposed to lead acetate. However, acute endurance exercise was not associated with these beneficial effects.
Keywords: neural regeneration, neurorehabilitation, long-term exercise training, endurance exercise, treadmill, lead poisoning, neurotoxicity, oxidative stress, hippocampus, brain-derived neurotrophic factor, neuroregeneration
Research Highlights
(1) We investigated the effects of acute endurance exercise and chronic exercise at low intensity on lead acetate-induced neurotoxicity and oxidative stress.
(2) Acute endurance exercise was associated with lower induction of brain-derived neurotrophic factor in the hippocampus of rats exposed to lead acetate compared with chronic exercise.
(3) Chronic exercise increased total antioxidant capacity, but acute exercise had no effect.
(4) Chronic exercise greatly decreased the plasma level of malondialdehyde, while acute endurance exercise had no such effect.
(5) Exercise training can improve brain resistance to neurodegenerative diseases induced by air pollutants.
INTRODUCTION
Human exposure to environmental lead is a public health problem of global proportion[1]. It is recognized as a dangerous neurotoxin, even at low levels of exposure[2], and time-series analyses show that risks are likely to increase with longer exposures[3]. The central nervous system has been recognized as a primary target for lead-induced toxicity. There is evidence showing that lead readily crosses the blood brain barrier and, in adult rats, enters the brain with rapid kinetics[4] and induces several brain alterations leading to encephalopathy, cellular damage, and reduced axonal and dendritic development. In addition, lead exposure during development impairs neuronal function[2], and potentially induces oxidative stress[5] in different brain regions, particularly in the hippocampus and cerebral cortex, which have been found to be more vulnerable to lead-induced neurotoxicity[6,7]. Evidence is accumulating that supports the role of oxidative stress in the pathophysiology of lead toxicity[5].
A previous study has shown that molecules typically implicated in synaptic plasticity such as brain-derived neurotrophic factor, are affected by cellular energy metabolism[8]. New findings indicate that the interaction between oxidative stress and brain-derived neurotrophic factor can affect neuronal plasticity. These studies indicate that elevated reactive oxygen species level decreases brain-derived neurotrophic factor-mediated synaptic plasticity[8]. The influence of environmental factors on the brain is manifested by their ability to promote adaptive changes using principles in common with hormesis[8].
There is accumulating evidence that exercise can promote brain health and function by protecting neurons and improving neuronal plasticity[9]. The capacity of physical activity to maintain and compensate for deterioration of neural function is becoming increasingly recognized[10]. Exercise may promote antioxidant defenses that could attenuate central nervous system vulnerability to neuronal degeneration[11]. In addition, it has been suggested that physical exercise modulates cognitive functions through various signaling mechanisms that lead to brain-derived neurotrophic factor up-regulation, especially in the hippocampus, a major hub for learning and memory[12,13,14]. Brain-derived neurotrophic factor is important for cell survival in neurogenesis studies and has been thought to play an important role in antidepressant action[14]. These observations, together with evidence that it is a powerful modifier of neuronal excitability and synaptic transmission[15,16,17], suggest that brain-derived neurotrophic factor is a crucial effecter of experience-dependent plasticity[18].
How an exercise-induced elevation of selected trophic factors can modulate crucial aspects of neural cellular plasticity, however, remains unknown. Previous studies in animals have demonstrated that acute and chronic exercise lead to an increase in brain-derived neurotrophic factor in various brain regions[11]. However, exercise-induced increases in brain-derived neurotrophic factorare transient[19]. Thus, prolonged reduction in serum brain-derived neurotrophic factor in patients with affective disorder is most likely associated with other factors, in addition to whatever transient effect one sees after physical exertion[20,21].
In contrast, there have been many reports showing that exercise causes oxidative stress, free radical generation, increases in oxidative damage biomarkers such as thiobarbituric acid reactive substances, effects on mitochondrial function, and decreases in levels of antioxidants in the brain[22]. However, Navarro et al[23] found lower thiobarbituric acid reactive substance levels in brain samples from mice after 24-week running exercise period, and Liu et al[22] demonstrated that there was a significant decrease in all brain homogenate and mitochondrial malondialdehyde levels and no change in brain DNA damage indicators after an 8-week running exercise period. The decrease in mitochondrial malondialdehyde levels was interpreted as a favorable effect of chronic exercise. Thus far, the evidence for tissue oxidative stress and damage due to exercise remains incomplete because of the complexity of exercise models[22].
A standing question for planning the design of studies using the therapeutic potential of exercise is whether exercise provided for a short period can have the same benefit as long-term exercise and also whether chronic and acute exercise show different effects on oxidative stress in the brain. To resolve these questions, in this study, we investigated the separate and combined effects of chronic and acute treadmill running protocols on hippocampal brain-derived neurotrophic factor levels in rats exposed to lead acetate. Additionally, we examined thiobarbituric acid reactive substances, as an indicator of lipid peroxidation, and total antioxidant capacity levels to assess the effects of two types of exercise on lead-induced neurotoxicity.
RESULTS
Quantitative analysis of animals
Forty rats were equally randomized into five groups: a lead acetate group (animals received an intraperitoneal injection of lead acetate), a chronic exercise training group (in addition to lead acetate exposure, rats were subjected to progressive running exercise, 5 successive days per week for a total of 8 weeks), a acute endurance exercise group (in addition to lead acetate exposure, rats were made to run on the treadmill until exhaustion), a chronic exercise training + acute endurance exercise group, and a sham group (rats received water and ethyl oleate administration). All 40 rats were suitable for final analysis.
Neurotoxicity of lead acetate
As shown in Table 1, after administration of 20 mg/kg lead acetate, body mass and brain mass decreased in the lead acetate group compared with the sham group. There was no significant effect of training protocols on brain mass/body mass ratio in the sham group compared with the lead acetate group. Additionally, administration of lead at a concentration of 20 mg/kg for 8 weeks resulted in a non-significant decrease in hippocampal brain-derived neurotrophic factor level (14%) and a significant decrease in plasma total antioxidant capacity (27%), compared with the sham group. In contrast, administration of lead acetate (20 mg/kg) was associated with an increase in hippocampal and plasma level of mitochondrial malondialdehyde, by 59% and 72%, respectively, compared with the sham group (Figures 1–4).
Table 1.
Effects of 8-week training and acute exercise protocols on body mass and brain mass in rats chronically exposed to lead acetate
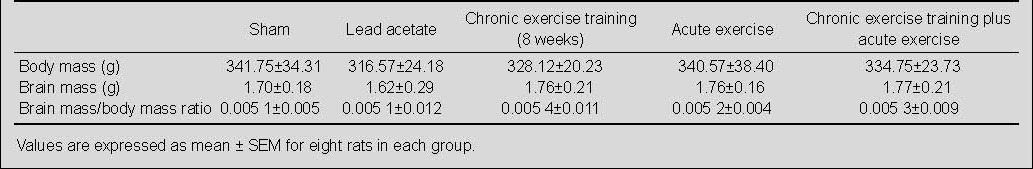
Figure 1.
Effects of chronic and acute treadmill exercise on hippocampal brain-derived neurotrophic factor (BDNF) levels in rats exposed to lead acetate.
The data in the left diagram are expressed as the percentage of hippocampal BDNF levels in lead acetate, chronic or/and acute exercise groups relative to the sham group. The data in the right diagram are expressed as the percentage of hippocampal BDNF levels in chronic or/and acute exercise, and sham groups relative to the lead acetate group. aP < 0.05 (one-way analysis of variance).
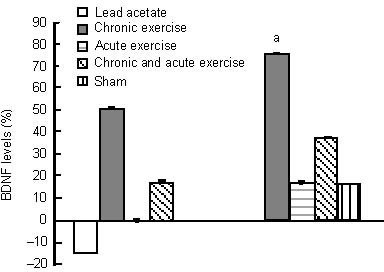
Figure 4.
Effects of chronic and acute treadmill exercise on plasma malondialdehyde (MDA) in rats exposed to lead acetate.
The data in the left diagram are expressed as the percentage of plasma MDA content in lead acetate, chronic or/and acute exercise groups relative to the sham group. The data in the right diagram are expressed as the percentage of plasma MDA content in chronic or/and acute exercise, and sham groups relative to the lead acetate group. aP < 0.05 (one-way analysis of variance).
Neuroprotective effects of chronic exercise on rats exposed to lead acetate
A period of 8 weeks of exercise significantly increased hippocampal brain-derived neurotrophic factor levels (76%) compared with the lead acetate group, and (51%) compared with the sham group (not significant), and was also associated with a significant increase in plasma total antioxidant capacity level (47% and 7%, respectively), compared with the lead acetate and sham groups (Figures 1 and 2). Chronic exercise training decreased the malondialdehyde level in the hippocampus (17%, not significant), and significantly in the plasma (60%) compared with the lead acetate group. Whereas, chronic exercise training increased hippocampal malondialdehyde level (32%) and also reduced plasma malondialdehyde level (31%) compared with the sham group (not significant; Figures 3 and 4).
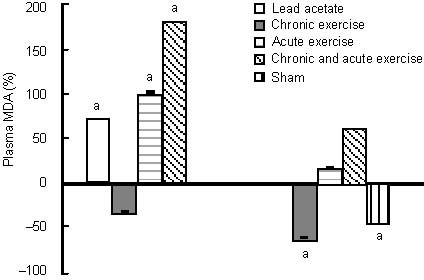
Figure 2.
Effects of chronic and acute treadmill exercise on plasma total antioxidant capacity (TAC) in rats exposed to lead acetate.
The data in the left diagram are expressed as the percentage of plasma TAC levels in lead acetate, chronic or/and acute exercise groups relative to the sham group. The data in the right diagram are expressed as the percentage of plasma TAC levels in the chronic or/and acute exercise, and sham groups relative to the lead acetate group. aP < 0.05 (one-way analysis of variance).
Figure 3.
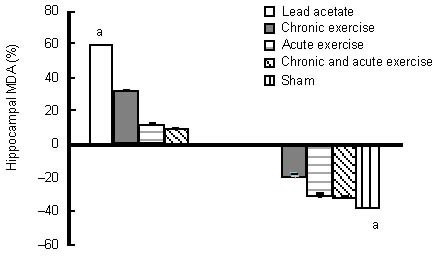
Effects of chronic and acute treadmill exercise on hippocampal malondialdehyde (MDA) content in rats exposed to lead acetate.
The data in the left diagram are expressed as the percentage of hippocampal MDA content in lead acetate, chronic or/and acute exercise groups relative to the sham group. The data in the right diagram are expressed as the percentage of hippocampal MDA content in chronic or/and acute exercise, and sham groups relative to the lead acetate group. aP < 0.05 (one-way analysis of variance).
Effect of acute endurance exercise on rats exposed to lead acetate
Brain-derived neurotrophic factor levels in the hippocampus increased non-significantly after acute endurance exercise alone and combined with chronic exercise (17% and 38%, respectively) compared with the lead acetate group, and with acute exercise combined with chronic exercise (18%) compared with the sham group, while there was no change after acute exercise compared with the sham group (Figure 1). Both acute exercise protocols reduced plasma total antioxidant capacity significantly (26% and 38%) compared with the sham group, however, only the acute exercise combined with chronic exercise group had a significant decrease in antioxidant level (total antioxidant capacity) compared with the lead acetate group. Acute exercise increased total antioxidant capacity in plasma insignificantly (1.5%) (Figure 2).
Acute endurance exercise alone and combined with chronic exercise was associated with a non-significant decrease in hippocampal malondialdehyde content (30% and 31%, respectively), compared with the lead acetate group, but an increase in plasma malondialdehyde (17% and 62%, respectively) (Figures 3, 4). Acute exercise combined with chronic exercise significantly increased plasma malondialdehyde level. Acute endurance exercise alone and combined with chronic exercise increased hippocampal malondialdehyde content non-significantly (12% and 9%, respectively) and plasma malondialdehyde, significantly (101% and 178%, respectively) compared with the sham group (Figures 3, 4).
DISCUSSION
Chronic exercise and acute exercise are two totally different exercise paradigms. Our results indicate that a single bout of acute exercise or acute exercise with previous experience of 8-week moderate exercise training leads to a lowered induction of hippocampal brain-derived neurotrophic factor in rats exposed to lead acetate compared with chronic exercise.
In addition to enhancing hippocampal brain-derived neurotrophic factor, chronic and acute exercise increased total antioxidant capacity levels, while acute exercise combined with chronic exercise decreased plasma total antioxidant capacity levels. In contrast, hippocampal malondialdehyde level was decreased after three types of exercise training whereas only chronic exercise reduced plasma malondialdehyde level in rats exposed to lead acetate.
A previous study has shown that chronic exposure to lead, a pervasive environmental pollutant, impairs synaptic plasticity in the form of long-term potentiation and cognitive function in experimental animals[1]. Several mechanisms including cholinergic dysfunction, glutamate receptor alterations, impaired antioxidant defense enzymes in the brain, and enhanced oxidative stress have been suggested to be causative factors of lead neurotoxicity[24]. There may be two independent sources of lead-induced oxidative damage; the first is the pro-oxidative effect of δ-aminolevulinic acid (δ-ALA), and the second is connected with the direct effect of lead on membrane lipids[5]. The depletion of glutathione and protein bound sulfhydryl groups, and changes in the activity of various antioxidant enzymes indicative of lipid peroxidation have been implicated in lead-induced oxidative tissue damage[5]. Lipid peroxidation appears to be markedly enhanced in the brain of lead-treated rats[25], which concurs with our findings of decreased production of total antioxidant capacity and increased malondialdehyde.
The ability of specific aspects of lifestyle, such as exercise and other factors, to modulate mental function is becoming increasingly recognized. In the present study, rats were subjected to both acute and chronic exercise protocols and were chronically exposed to lead acetate. Theoretically, exercise may cause oxidative stress in the brain. First, exercise enhances brain dopamine synthesis. Dopamine may form reactive oxygen species through either dopamine metabolism by monoamine oxidase or autoxidation. Second, exercise leads to increased serum glucocorticoid levels. Corticosterone increases the toxicity of oxygen radical generators, the basal level of reactive oxygen species, and may alter antioxidant enzyme activities in the brain[7].
The results suggest a positive correlation between physical fitness and cognitive function[26]. Moreover, acute exercise leads to improvements in performance of a hippocampal-based cognitive task, which may be related to a concomitant increase in circulating brain-derived neurotrophic factor concentration[22]. Although, chronic voluntary physical activity has been shown to enhance hippocampal brain-derived neurotrophic factor expression in animals exposed to lead acetate, It has also been shown that minimally stressful low-intensity exercise results in hippocampal activation and brain-derived neurotrophic factor expression; lending support to the idea that mild exercise can yield greater benefits in hippocampal function compared with more strenuous forms of exercise[22]. In this study, it is interesting to note that repetitive exercise increased brain-derived neurotrophic factor protein levels to a greater extent than did a single bout of exercise[19]. This observation suggests that upregulation of brain-derived neurotrophic factor expression depends on exercise intensity or duration. However, it has been shown that both acute moderate and severe exercise increases brain-derived neurotrophic factor protein content in the hippocampus[22]. Acute submaximal aerobic exercise is an external stimulus, which can stimulate the brain-derived neurotrophic factor gene to increase brain-derived neurotrophic factor protein levels by stimulating cholinergic neurons via the septo- hippocampal axis[11]. Rojas Vegaa et al[9] reported that exercise to exhaustion induced an elevation of brain-derived neurotrophic factor peaking at cessation of exercise, followed by a fast return to baseline level post-exercise.
It is now understood that the action of exercise on the brain-derived neurotrophic factor system modulates the function of intracellular signaling systems such as calcium-calmodulin kinase II and mitogen-activated protein kinase, with endpoint effects on the production and function of cAMP response element binding protein[27]. Enhanced learning after chronic activity wheel running has also been accompanied by decreases in extracellular amyloid plaques and proteolytic fragments of amyloid precursor protein in a transgenic mouse model of Alzheimer's disease[27].
In the present report, we also measured indices of oxidative stress, thiobarbituric acid reactive substances as a marker of lipid peroxidation and total antioxidant capacity as an indicator of overall antioxidant capacity of the serum. It is well known that different forms of exercise result in different levels of oxidative stress. Studies also support the idea that chronic exercise has dual effects: chronic exercise results in oxidant formation and oxidative stress and, perhaps as a consequence, induces antioxidant enzymes and antioxidant synthesis that minimize the effects of oxidants[22]. Changes due to exercise are subtle, suggesting that there are active stress-strain relationships between oxidant formation and scavenging during exercise[22]. Davies et al[28] showed that exhaustive exercise causes an increase in free radicals and thiobarbituric acid reactive substances, suggesting that endurance training induces free radical damage. The rate of free radical or oxidant generation in biological tissue is closely related to oxygen consumption indicating a possible beneficial effect of chronic exercise on brain function. Exhausting aerobic exercise increases oxygen demand 10–15 fold compared with rest, resulting in increased oxygen consumption, increased oxygen cell uptake, and increased flow in the electron transport chain[29]. Previously, we have shown that both acute and chronic exercise decreases malondialdehyde level in the hippocampus, indicating a possible beneficial effect of chronic exercise[22]. Acute exercise increases serum malondialdehyde level, which supports the hypothesis of the involvement of oxidative stress in tissues by acute exercise. In contrast to results reported by Liu and colleagues[22], we showed that effect of acute exercise on malondialdehyde level was greater compared with chronic exercise.
However, the initiation of oxidative damage can be reversed by the stimulation of antioxidants, by maintaining an adequate concentration of intracellular antioxidants, and by endogenous repair systems[22]. Antioxidant status varied in direction and magnitude according to different kinds of exercise[22], and there was a significant difference between the acutely exercised and chronically exercised rats. In our study, chronic exercise caused a significant increase in the levels of plasma total antioxidant capacity, although, acute endurance exercise alone and combined with chronic exercise did not have such effects and caused little increase or a significant decrease in serum total antioxidant capacity levels. An increase in the levels of antioxidants leads to the scavenging of excess reactive oxygen species and thereby may contribute to a decrease in brain oxidative damage[22].
The most important effect of exercise on the body is the adaptation process. As a stressor, a single bout of exercise has the capability to induce adaptation. Chronic stressors could be very dangerous since the resting period, which is obligatory for recovery and an efficient stress response, is missing[30]. The effects of acute exercise on oxidative stress have been reported to be dependent on the type of exercise, which strongly implies that chronic exercise with different characteristics might provoke different adaptations in the redox system[29,30]. Results of our study revealed that 8 weeks of endurance training before acute exercise cannot improve oxidative/antioxidative balance, and can even exacerbate the trend caused by acute exercise. Overtraining or excessive exercise, at the other end point of the hormesis curve, enhances the risk of disease and jeopardizes health. Exercise can increase the generation of reactive oxygen species[29,30] likely through mechanisms such as acidosis, catecholamine oxidation, and xanthine oxidase activation[29]. This is especially true for single bouts of exercise[30]. Oxidative damage to lipids, proteins, and DNA as a consequence of increased reactive oxygen species levels have been reported following single bouts of exercise[30]. There is ample evidence to suggest that regular exercise increases the activity of antioxidant enzymes, and increased levels of reactive oxygen species appear to be necessary during the exercise session[30]. In addition to the first line of antioxidant enzymes, the second line, the oxidative damage repair systems, are important to minimize the dangerous effects of reactive oxygen species. It is our view that reactive oxygen species-generating effects of exercise are very important, because this process can initiate adaptive processes, which result in lower base levels of reactive oxygen species, increased activity of antioxidant and damage repair enzymes, and lower levels of oxidative damage. This reactive oxygen species mediated adaptation could be a mechanism by which exercise decreases the incidence of reactive oxygen species-associated diseases, including Alzheimer's disease[30].
In summary, our results demonstrated that the brain is responsive to both acute and chronic exercise. Overall, exercise increases brain health in addition to body health, and represents an exciting lifestyle intervention technique to improve brain plasticity, function, and resistance to neurodegenerative diseases induced by air pollutants. Although, it could be concluded from this study that chronic exercise produced benefits more reliably, the different effect of the two training protocols may be partly due to the high oxygen load imposed during acute exercise bouts. Further studies are needed to clarify other mechanisms involved in this process.
MATERIALS AND METHODS
Design
A randomized, controlled animal experiment.
Time and setting
This study was performed at the Cell Biology Laboratory, Faculty of Animal Science, Mazandaran University, Iran from November 2009 to September 2010.
Materials
The experimental protocol was performed according to guidelines regarding Care and Use of Animals, issued by the American Physiological Society. Forty male Wistar rats, 8 weeks of age, with an initial body weight of 240 ± 20 g, were obtained from the Center of Laboratory Animals at the Pasture Institute of Iran. Rats were raised separately in standard cages (20 cm × 15 cm × 15 cm) made of polycarbonate at the Pasture Institute of Iran, in a large air-conditioned room with a controlled temperature of 22 ± 2°C, 12-hour light-dark cycle, and humidity of 50 ± 5%. The pollutant standard index was in an acceptable range as determined by the Iranian Meteorological Organization. Rats were fed with a standard rat chow (provided by the Pars Institute for Animals and Poultry) with a daily regimen of 10/100 g body weight for each rat. Water was available ad libitum.
Methods
Exercise protocol
After familiarization with the laboratory environment and running on the rodent treadmill (KN-73, Natsume Ltd., Japan). Rats in the lead acetate group received an intraperitoneal injection of lead acetate[31] at a concentration of 20 mg/kg in water solution, 3 alternate days weekly for a total of 8 weeks. Rats in the chronic exercise training group, in addition to lead acetate exposure, as in the lead acetate group, were subjected to progressive running exercise for 15–22 m/min for 25–64 minutes, 5 successive days per week for a total of 8 weeks. This protocol started with a running speed of 15 m/min for 25 minutes in the first week, and then gradually increased according to training protocol over 8 weeks to a running speed of 22 m/min for 64 minutes in the last week. Rats in the acute endurance exercise group, in addition to lead acetate exposure, as in the lead acetate group, were made to run on the treadmill at 1.6 km/h until exhaustion, which was defined as the animal touching the electrified grid at the rear of the treadmill 5 times in 2 minutes and then receiving lead acetate administration. The running time varied between 20 and 110 minutes. Rats in the chronic exercise training + acute endurance exercise group received a physical training protocol similar to that in chronic exercise training group, i.e., after lead acetate exposure, they were subjected to an acute exercise protocol immediately before death. Rats in sham group received water and ethyl oleate administration.
Lead acetate (Sigma, St. Louis, MO, USA) was solubilized in Milli-Q water. Because a previously reported lead dosing regimen[31] was shown to cause oxidative stress, a 20 mg/kg lead acetate dose was selected. A mild shock (0.75 mA, 500 ms duration, 0.5 Hz) was delivered through electrified stationary wire loops to motivate the rats to continuously walk on the moving belt (consisting of stationary wire loops), and thus avoid foot shock. The wire loops were activated during all exercise sessions, and an experimenter monitored all treadmill sessions. Rats quickly learned to stay on the belt and avoid shock, except one rat, which would not stay on the moving belt, and thus was quickly removed from the exercise group.
Sample preparation
At 12–14 hours after the last dose of treatment, rats in the acute exercise alone and combined with chronic exercise groups were subjected to the acute training protocol. After 12–14 hours of overnight fastening, rats in all groups were decapitated under anesthesia administered by an intraperitoneal injection of ketamine and xylazine according to the animal guidelines formulated by Tehran Institute of Endocrine and Metabolism of Shahid Beheshti University in Tehran.
Blood samples obtained from the heart were first centrifuged at 3 000 r/min for 15 minutes within 30 minutes after collection, and then stored at –80°C before assay and serum were separated for biochemical estimations of total antioxidant capacity and mitochondrial malondialdehyde. The brains were rapidly removed and the two hemispheres were separated along the midline according to Paxinos and Watson[32]. The hippocampus from each hemisphere was then micro-dissected and frozen in liquid nitrogen and subsequently stored at –80°C for future analysis.
Hippocampal brain-derived neurotrophic factor assay
For protein extraction, the hippocampus was homogenized in a lysis buffer (18 mL/mg tissue) containing 137 mM NaCl, 20 mM Tris-HCl (pH 8.0), 1% NP40, 10% glycerol, 1 mM phenylmethyl sulfonyl fluoride, leupeptin (1mg/mL), sodium vanadate (0.5 mM), and 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (100 mg/mL)[33]. Samples were then centrifuged for 3 minutes (14 000 r/min, 3 minutes, 4°C) and supernatant was collected and stored at –80°C. The brain-derived neurotrophic factorprotein level was determined using the brain-derived neurotrophic factorELISA kit (Demeditec Diagnostics GmbH, Kiel, Germany) according to the manufacturer's recommendations. Supernatant was diluted in sample buffer and incubated on 96-well flat-bottom plates, previously coated with buffer containing anti-brain-derived neurotrophic factormonoclonal antibody. After blocking, plates were incubated with anti-human brain-derived neurotrophic factor polyclonal antibody for 2 hours and horseradish peroxidase for 1 hour. Then a color reaction with tetraethyl benzedrine was quantified in a plate reader (State Fax-2100, Awareness Technology, USA) at 450 nm. The standard brain-derived neurotrophic factorcurve ranged from 0 to 500 pg/mL[33].
Hippocampal and plasma malondialdehyde assay
Lipid peroxidation levels in the tissue homogenate were measured with a thiobarbituric acid reaction using the method of Ohkawa et al[34]. Sample homogenates (1 mL) were incubated at 37°C in an oscillating water bath for 1 hour. At the end of the incubation period, 0.5 mL of butylated hydroxytoluene (0.5 mg/mL in absolute ethanol) and 1 mL of trichloroacetic acid (25%) were added. The tubes were sealed and heated for 10 minutes in a boiling water bath to release malondialdehyde (the end product of lipid peroxidation) from proteins. To avoid adsorption of malondialdehyde to insoluble proteins, the samples were cooled to 4°C and centrifuged at 2 000 × g for 20 minutes. Following centrifugation, 2 mL of the protein free supernatant was removed from each tube and 0.5 mL of thiobarbituric acid (0.33%) was added to this fraction. All tubes were heated for 1 hour at 95°C in a water bath. After cooling, the thiobarbituric acid-malondialdehyde complexes were extracted with 2 mL of butanol. The light absorbance was read at 532 nm on a spectrophotometer (Jenway6505 UV/VIS, Dunmow, Essex, UK) and malondialdehyde levels were determined from a standard curve that was generated from 1,1,1,3 tetramethoxypropan. Results are represented as nmol/mg tissue[35]. Additionally, plasma level of malondialdehyde was measured by the thiobarbituric acid method as described above[34]. The results are represented as nmol/mL.
Plasma total antioxidant capacity assay
Plasma total antioxidant capacity was measured using a commercially available kit (Randox Laboratories, Crumlin, UK) as previously described by Erel et al[36]. In this method, the most potent radical, hydroxyl radical, is produced. First, a ferrous ion solution was mixed with hydrogen peroxide. The sequentially produced radicals such as brown colored dianisidinyl radical cation, produced by the hydroxyl radical, were potent radicals. The antioxidative effect of the sample against the potent free radical was then measured. The assay had excellent precision, which was lower than 3%. The results are expressed in μmol/mL.
Statistical analysis
Statistical analysis was performed using SPSS version 16.0 for Windows (SPSS, Chicago, IL, USA). Results are expressed as mean ± SEM. Data for brain-derived neurotrophic factor and oxidant/antioxidant markers were normally distributed after log transformation. One-way analysis of variance followed by post hoc multiple comparison tests was used to compare differences among the groups. The differences were considered statistically significant at P < 0.05.
Acknowledgments
We would like to thank the Department of Sport Physiology, College of Physical Education and Sport Sciences, University of Mazandaran for their cooperation during experimentation.
Footnotes
Conflicts of interest: None declared.
Ethical approval: All experiments were approved by the Animal Ethics Committee of the University of Mazandaran, Iran.
(Edited by Yang CM, Olex-Zarychta D/Song LP)
REFERENCES
- [1].Verina T, Rohde CA, Guilarte TR. Environmental lead exposure during early life alters granule cell neurogenesis and morphology in the hippocampus of young adult rats. Neuroscience. 2007;145(3):1037–1047. doi: 10.1016/j.neuroscience.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sansar W, Ahboucha S, Gamrani H. Chronic lead intoxication affects glial and neural systems and induces hypoactivity in adult rat. Acta Histochem. 2011;113(6):601–607. doi: 10.1016/j.acthis.2010.06.005. [DOI] [PubMed] [Google Scholar]
- [3].Makri A, Stilianakis NI. Vulnerability to air pollution health effects. Int J Hyg Environ Health. 2008;211(3-4):326–336. doi: 10.1016/j.ijheh.2007.06.005. [DOI] [PubMed] [Google Scholar]
- [4].García-Arenas G, Ramírez-Amaya V, Balderas I, et al. Cognitive deficits in adult rats by lead intoxication are related with regional specific inhibition of cNOS. Behav Brain Res. 2004;149(1):49–59. doi: 10.1016/s0166-4328(03)00195-5. [DOI] [PubMed] [Google Scholar]
- [5].Ahamed M, Siddiqui MK. Low level lead exposure and oxidative stress: current opinions. Clin Chim Acta. 2007;383(1-2):57–64. doi: 10.1016/j.cca.2007.04.024. [DOI] [PubMed] [Google Scholar]
- [6].Prasanthi RP, Devi CB, Basha DC, et al. Calcium and zinc supplementation protects lead (Pb)-induced perturbations in antioxidant enzymes and lipid peroxidation in developing mouse brain. Int J Dev Neurosci. 2010;28(2):161–167. doi: 10.1016/j.ijdevneu.2009.12.002. [DOI] [PubMed] [Google Scholar]
- [7].Aksu I, Topcu A, Camsari UM, et al. Effect of acute and chronic exercise on oxidant-antioxidant equilibrium in rat hippocampus, prefrontal cortex and striatum. Neurosci Lett. 2009;452(3):281–285. doi: 10.1016/j.neulet.2008.09.029. [DOI] [PubMed] [Google Scholar]
- [8].Gomez-Pinilla F. The influences of diet and exercise on mental health through hormesis. Ageing Res Rev. 2008;7(1):49–62. doi: 10.1016/j.arr.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rojas Vega S, Strüder HK, Vera Wahrmann B, et al. Acute BDNF and cortisol response to low intensity exercise and following ramp incremental exercise to exhaustion in humans. Brain Res. 2006;1121(1):59–65. doi: 10.1016/j.brainres.2006.08.105. [DOI] [PubMed] [Google Scholar]
- [10].Kempermann G, van Praag H, Gage FH. Activity-dependent regulation of neuronal plasticity and self repair. Prog Brain Res. 2000;127:35–48. doi: 10.1016/s0079-6123(00)27004-0. [DOI] [PubMed] [Google Scholar]
- [11].Panaree B, Daengwilai N, Chantana M, et al. Effect of acute submaximal aerobic exercise on serum brain derived neurotrophic factor protein and verbal short term memory in Thai male sedentary non-medical students. Thai J Physio Sci. 2008;21(2):63–67. [Google Scholar]
- [12].Vaynman S, Ying Z, Gomez-Pinilla F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci. 2004;20(10):2580–2590. doi: 10.1111/j.1460-9568.2004.03720.x. [DOI] [PubMed] [Google Scholar]
- [13].Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25(6):295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- [14].Sairanen M, Lucas G, Ernfors P, et al. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci. 2005;25(5):1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Causing CG, Gloster A, Aloyz R, et al. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18(2):257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- [16].Kafitz KW, Rose CR, Thoenen H, et al. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401(6756):918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- [17].Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8(1):1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- [18].Gómez-Pinilla F, Ying Z, Roy RR, et al. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol. 2002;88(5):2187–2195. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- [19].Huang AM, Jen CJ, Chen HF, et al. Compulsive exercise acutely upregulates rat hippocampal brain-derived neurotrophic factor. J Neural Transm. 2006;113(7):803–811. doi: 10.1007/s00702-005-0359-4. [DOI] [PubMed] [Google Scholar]
- [20].Burger J, Gochfeld M. Effects of lead and exercise on endurance and learning in young herring gulls. Ecotoxicol Environ Saf. 2004;57(2):136–144. doi: 10.1016/S0147-6513(03)00035-6. [DOI] [PubMed] [Google Scholar]
- [21].Wolff EA, 3rd, Putnam FW, Post RM. Motor activity and affective illness. The relationship of amplitude and temporal distribution to changes in affective state. Arch Gen Psychiatry. 1985;42(3):288–294. doi: 10.1001/archpsyc.1985.01790260086010. [DOI] [PubMed] [Google Scholar]
- [22].Liu J, Yeo HC, Overvik-Douki E, et al. Chronically and acutely exercised rats: biomarkers of oxidative stress and endogenous antioxidants. J Appl Physiol. 2000;89(1):21–28. doi: 10.1152/jappl.2000.89.1.21. [DOI] [PubMed] [Google Scholar]
- [23].Navarro A, Gomez C, López-Cepero JM, et al. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol. 2004;286(3):R505–511. doi: 10.1152/ajpregu.00208.2003. [DOI] [PubMed] [Google Scholar]
- [24].Shukla PK, Khanna VK, Khan MY, et al. Protective effect of curcumin against lead neurotoxicity in rat. Hum Exp Toxicol. 2003;22(12):653–658. doi: 10.1191/0960327103ht411oa. [DOI] [PubMed] [Google Scholar]
- [25].Antonio-García MT, Massó-Gonzalez EL. Toxic effects of perinatal lead exposure on the brain of rats: involvement of oxidative stress and the beneficial role of antioxidants. Food Chem Toxicol. 2008;46(6):2089–2095. doi: 10.1016/j.fct.2008.01.053. [DOI] [PubMed] [Google Scholar]
- [26].Wood RH, Reyes-Alvarez R, Maraj B, et al. Physical fitness, cognitive function, and health-related quality of life in older adults. JAPA J. 1999;7:217–230. [Google Scholar]
- [27].Dishman RK, Berthoud HR, Booth FW, et al. Neurobiology of exercise. Obesity (Silver Spring) 2006;14(3):345–356. doi: 10.1038/oby.2006.46. [DOI] [PubMed] [Google Scholar]
- [28].Davies KJ, Quintanilha AT, Brooks GA, et al. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun. 1982;107(4):1198–1205. doi: 10.1016/s0006-291x(82)80124-1. [DOI] [PubMed] [Google Scholar]
- [29].Kostaropoulos IA, Nikolaidis MG, Jamurtas AZ, et al. Comparison of the blood redox status between long-distance and short-distance runners. Physiol Res. 2006;55(6):611–616. doi: 10.33549/physiolres.930898. [DOI] [PubMed] [Google Scholar]
- [30].Radak Z, Chung HY, Koltai E, et al. Exercise, oxidative stress and hormesis. Ageing Res Rev. 2008;7(1):34–42. doi: 10.1016/j.arr.2007.04.004. [DOI] [PubMed] [Google Scholar]
- [31].Daniel S, Limson JL, Dairam A, et al. Through metal binding, curcumin protects against lead- and cadmium-induced lipid peroxidation in rat brain homogenates and against lead-induced tissue damage in rat brain. J Inorg Biochem. 2004;98(2):266–275. doi: 10.1016/j.jinorgbio.2003.10.014. [DOI] [PubMed] [Google Scholar]
- [32].Paxinos G, Watson C. 4th ed. San Diego: Academic Press; 1998. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]
- [33].Adlard PA, Perreau VM, Cotman CW. The exercise- induced expression of BDNF within the hippocampus varies across life-span. Neurobiol Aging. 2005;26(4):511–520. doi: 10.1016/j.neurobiolaging.2004.05.006. [DOI] [PubMed] [Google Scholar]
- [34].Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95(2):351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- [35].Ataie A, Sabetkasaei M, Haghparast A, et al. An investigation of the neuroprotective effects of Curcumin in a model of Homocysteine - induced oxidative stress in the rat's brain. DARU. 2010;18(2):128–136. [PMC free article] [PubMed] [Google Scholar]
- [36].Erel O. A novel automated direct measurement method for total antioxidant capacity using a new generation, more stable ABTS radical cation. Clin Biochem. 2004;37(4):277–285. doi: 10.1016/j.clinbiochem.2003.11.015. [DOI] [PubMed] [Google Scholar]