

Abstract
Neuroacanthocytosis is an autosomal recessive or dominant inherited disease characterized by widespread, non-specific nervous system symptoms, or spiculated “acanthocytic” red blood cells. The clinical manifestations typically involve chorea and dystonia, or a range of other movement disorders. Psychiatric and cognitive symptoms may also be present. The two core neuroacanthocytosis syndromes, in which acanthocytosis is atypical, are autosomal recessive chorea-acanthocytosis and X-linked McLeod syndrome. Acanthocytes are found in a smaller proportion of patients with Huntington's disease-like 2 and pantothenate kinase-associated neurodegeneration. Because the clinical manifestations are diverse and complicated, in this review we present features of inheritance, age of onset, neuroimaging and laboratory findings, as well as the spectrum of central and peripheral neurological abnormalities and extraneuronal involvement to help distinguish the four specific syndromes.
Keywords: neural regeneration, neurodegenerative disease, reviews, neuroacanthocytosis, chorea-acanthocytosis, pantothenate kinase-associated neurodegeneration, Huntington's disease-like 2, McLeod syndrome, clinical manifestations, features of inheritance, extrapyramidal disease, photographs-containing paper, neuroregeneration
Research Highlights
Features of inheritance, age of onset, neuroimaging, laboratory findings, and the spectrum of central and peripheral neurological abnormalities and extraneuronal involvement of neuroacanthocytosis are reviewed to help guide research and instruct clinical diagnosis.
INTRODUCTION
Neuroacanthocytosis is a rare and formerly neglected group of neurodegenerative disorders. The clinical manifestations are associated with widespread non-specific nervous system symptoms or acanthocytosis[1]. Since there are considerable phenotypic overlaps, it is easy to misdiagnose. The core symptom is basal ganglia degeneration, especially ataxia caused by striatum degeneration[1].
Historically, neuroacanthocytosis were termed as chorea-acanthocytosis and McLeod syndrome. Presently, pantothenate kinase-associated neurodegeneration and Huntington's disease-like 2 are also categorized into this group[2,3,4]. Here, neuroacanthocytosis, its clinical features and its potential molecular basis are reviewed to facilitate diagnosis and study of this disease.
CHOREA-ACANTHOCYTOSIS
Chorea-acanthocytosis is an autosomal recessive disease, except for a few reported to be dominantly inherited[5]. Most patients experience the onset approximately at 20 years old; the mean age of onset is 35 years old. The incidence among the juvenile and the senile is rare. The course of the disease is about 15–30 years. The main clinical manifestations are involuntary movements of the facial muscles, tongue and throat, including pronunciation, dysarthria, tongue and lip biting. Other symptoms can be paroxysmal dyspnea and belching[6], as well as nystagmus, especially vertical nystagmus[7]. Almost half of the patients may suffer grand mal epilepsy, and one-third have epilepsy as the first symptom. At the early stages, there could be signs of personality changes, difficulty in concentrating and forced psychosis[8], and decreased myodynamia and muscular atrophy.
Imaging and pathological features
The lesions mainly exist in the basal ganglia, especially the caudate nucleus and putamen (Figure 1). The magnetic resonance volume determination of the caudate nucleus objectively serves as an imaging diagnostic tool of chorea-acanthocytosis[9]. Recent studies suggest that the chorea-acanthocytosis are various: an autosomal dominant inheritance, and brain MRI presents cortical atrophy[10]. The latest report of one autosomal dominant autopsy case from Japan found neuronal loss and astrocyte proliferation throughout the brain, which was significantly different from that of the recessive form of inheritance, in which the basal ganglia is damaged[11]. Histological examination revealed neuronal loss and proliferation of astrocytes, especially in the caudate nucleus[12]. Detection by 133Xe inhalation or injection of 99Tc marked 6 methyl malonyl-based amine oxime revealed the blood flow of the frontal lobe to be decreased[13], which provides a theoretical basis for the decline of cognitive function. A monozygotic twin study of chorea-acanthocytosis using magnetic resonance diffusion tensor fluorodeoxyglucose-positron emission tomography and 123I-β-CIT-single photon emission computed tomography showed caudate nucleus atrophy. In this twin study, the diffusion coefficient of the bilateral striatum increased significantly, glucose use of the bilateral striatum decreased, and the dopamine transporter protein binding rate of the striatum and the hypothalamus in the midbrain 5-hydroxytryptamine transporter was reduced to a normal range[14]. The signs of hypothalamic endocrine disorders include hypothyroidism and abnormal growth hormone secretion[15].
Figure 1.
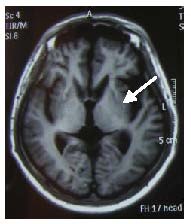
MRI finding showing atrophy of the caudate/putamen (arrow) from a patient with chorea-acanthocytosis.
The main peripheral nervous pathological manifestation of chorea-acanthocytosis is chronic axonal neuropathy. Tissue biopsies have shown that large myelinated nerve fibers were mainly injured and secondary proliferation was discernible, while primary demyelination and remyelination were rare[12]. Ultrastructural examination revealed that many neurofilaments were gathered around swelling axons[12] (Figure 2). The nerve conduction velocity was usually normal[12]. For no more than half of the patients, sensory action potentials decreased[12]. Inflammation may be involved in the pathogenesis of axonal neuropathy; however, the mechanism is not yet clear.
Figure 2.
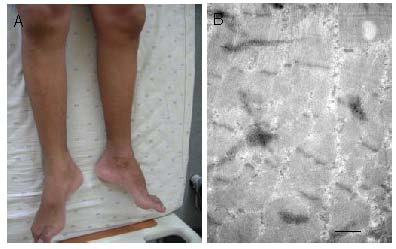
Symmetrical amyotrophy of the legs (A) and an electron microscope image (B; scale bar: 500 nm, cross section) of the gastrocnemius from a patient with chorea-acanthocytosis.
In the 1980s, chorea-acanthocytosis was defined as familial amyotrophic chorea with acanthocytosis[16]. CT suggests selective symmetrical amyotrophy[17]. Electromyographic examination has shown partial denervation of the proximal and distal muscles[17]. Biopsies may present neurogenic myotrophy, with small clusters of muscle fibers, and occasional necrotic fibers. Recently, a case of chorea-acanthocytosis with cardiomyopathy has been demonstrated[18].
Genetic research
In recent years, chorea-acanthocytosis gene mutations in different geographical regions have been reported. There is now a consensus of studies that the chorea-acanthocytosis locus is mapped to chromosome 9q21 within a 6-cM interval in the VPS13A gene[19]. Gene mutation loci and causes of mutation are various. So far, 92 different mutations have been found distributed throughout the VPS13A gene[20]. Most of the gene mutations result in loss of function, thus it is difficult to infer a relationship between genes and phenotypes. In recent years, the correspondence between certain gene mutation and the resulting clinical manifestations has led to new research ideas. CHAC expression was detected in tissues with erythroid precursors, skeletal muscles, and in the brain, where equal levels of transcript were found in all locations[21] (Figure 3).
Figure 3.
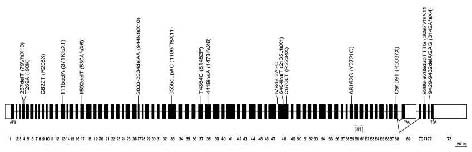
Schematic representation of the CHAC gene.
Thick bars represent exons (white blocks UTR regions); thin lines (not scaled) represent introns. The two splicing variants are shown. The approximate position of all presently reported mutations in CHAC is indicated and the resulting changes in the protein sequence (parenthesis). All mutations, except where indicated (brackets), were reported by Rampoldi et al[21].
The protein product of the CHAC gene is chorein[22], consisting of four proteins with major sequence similarities in the NH2- and COOH-terminal regions. Subcellular analysis has revealed high levels of chorein in brain microsomes and synaptic fragments[23].
Molecular pathology
Little is known about the function of chorein. Amino acid sequence analysis failed to identify its conserved domains, motifs or identifiable structural features. Furthermore, the presence of transmembrane domains could not be unequivocally determined using several prediction programs[1]. Nevertheless, the strong conservation of the terminal domains suggests that they are important for biological function of the protein[1]. Some hints to understanding chorein's function come from functional studies of its homologues. Among these, Vps13 of S. cerevisiae and TipC of Dictyostelium are known proteins. In Dictyostelium, TipC probably plays a role in the signaling pathway responsible for directing morphogenesis beyond the aggregated state[24]. In S. cerevisiae, VPS mutants show abnormal intracellular trafficking with mislocalization of vacuolar hydrolases to the cell surface[25]. At least 40 complementation groups have been reported so far. In particular, the chorein homologue Vps13 is required for proper trafficking of Kex2p, and to a lesser extent, Vps10 and Ste13, between the trans-Golgi network[26] and the pre-vacuolar compartment, which correspond to the multivesicular body in animal cells. Chorein might control one or more steps in the cycling of proteins through the trans-Golgi network to cytorhyctes, lysosomes and the plasma membrane in the early and late stages[27]. Functional loss of chorein protein may lead to bridging proteins and GABRG2 compensatory up-regulation[28]. Of course, functional experiments are required to assess the biological function of chorein in mammalian systems.
Acanthocytosis
Under normal circumstances, the acanthocytes should not be present in the peripheral blood. Pathological conditions are confirmed when more than 3% of crenated forms appear in the peripheral blood to avoid false positives caused by experimental artifacts or echinocytes[12]. Sometimes the search for acanthocytes as diagnostic criteria can be misleading, not only because of the different sensitivities of different methods used to detect them but also because they can appear during the late stage of the disease[29]. The percentage of acanthocytes in the blood of chorea-acanthocytosis patients is highly variable, usually between 5% and 50%, and does not seem to be correlated with the severity of the disease[1] (Figure 4).
Figure 4.

Acanthocytosis in the peripheral blood from a patient with the chorea-acanthocytosis (scanning electron microscope, scale bar in A: 1 μm; in B: 10 μm).
Presence of erythrocytes with thorn-like protrusions was originally called acanthocytosis.
The mechanism of acanthocyte formation in chorea-acanthocytosis is unknown. Studies of the lipid components of patient erythrocyte membranes have been somewhat discordant[1]. No abnormalities are generally reported in European patients[12]. On the other hand, in Japanese patients a decrease in linoleic acid (C18:2) and stearic acid (C18:0), and an increase in palmitic acid (C16:0) and docosahexaenoic acid (C22:6) were found[30]. It has been suggested that covalently bound fatty acids influence the shape of erythrocytes via the modulation of the interaction of membrane proteins with the plasma membrane. There are several indications of functional and probably structural modification of membrane proteins, in particular the anion transporter band 3. An increase in the phosphorylation of erythrocyte membrane proteins, particularly band 3 and spectrin-subunit, have been reported[31,32]. Anion transport, in most of the cases, is increased, but was found to be reduced in one case[33]. Erythrocytes from chorea-acanthocytosis patients exhibit immunological changes indicative of cellular aging/transporter damage, and some of their membrane proteins, such as band 3 and 2.1 (ankyrin), self-digest faster than normal protein[34]. The latter evidence could be due to either conformational change at the junction of band 3 to spectrin with band 2.1 and 4.2, or up-regulation of protease activity[34]. Interestingly, antibodies to neural tissues that appear to have band 3 specificity have been found in chorea-acanthocytosis patient sera. Band 3 protein in mammalian brain has a role in anion transport, ankyrin binding, and generation of senescent cell antigen[31]. The possibility that band 3 abnormalities have a role in chorea-acanthocytosis pathogenesis is a fascinating avenue for further investigation.
MCLEOD SYNDROME
McLeod syndrome, a rare X-linked disorder, was first described in 1961 and named propositus. It is characterized by choreic movements, peripheral neuropathy with areflexia, myopathy, cardiomyopathy (about two-thirds of patients), elevated serum creatine kinase levels and a permanent hemolytic state[35]. At the late stage, the majority of patients present with cognitive dysfunction and psychological disturbances[36]. Onset ranges between 25 years and 60 years of age. Survival is approximately 7–51 years and the mean age of death is 53 years of age (31–69 years)[35,37]. Approximately one-third of McLeod syndrome patients present with a choreatic movement disorder[35,37]. During the disease course, the majority of patients develop chorea or other involuntary movements, such as facial dyskinesia, dysarthria, and involuntary vocalizations[38]. Compared with patients with chorea-acanthocytosis, few McLeod syndrome patients develop lip or tongue biting, dysphagia, dystonia or extrapyramidal symptoms.
Imaging and pathological features
Neuropathological examination has revealed significant neuronal loss and glial cell proliferation in the caudate nucleus and putamen[39]. Fully automated magnetic resonance subcortical segmentation measurement studies of striatum volume have shown that[40] the caudate nucleus and putamen volume decrease continuously during 7 years of follow-up visits. Positron emission tomography analysis has revealed indications of absent metabolism in the basal ganglia and hypometabolism in the frontal and parietal cortex[41]. A fast multiple spin-echo imaging study[42] found that in patients with cognitive dysfunction, the N-acetyl aspartate/creatine or choline ratio in the frontal lobe, temporal lobe and insular lobe is abnormal. Nervous dysfunction may be due to damage or obvious structural changes of the basal ganglia-thalamus-cortex circuit loop. Interestingly, reduced 18F-labeled deoxyglucose uptake in the caudate nucleus was observed in the absence of clinical signs of cerebral involvement, movement disorder and unimpaired D2 receptor binding, indicating that abnormalities of glucose metabolism precede the onset of McLeod syndrome[43].
Areflexia is a common feature, suggesting involvement of the peripheral nervous system that is generally subclinical and less significant than in chorea-acanthocytosis in almost all the patients[38]. There is no significant neuropathologic difference compared with the former condition[38]. About 30% of patients feel decreased foot vibration sensation[44]. Only a few patients feel abnormal symptoms. In addition to the symptoms described thus far, some uncommon features have been reported, such as sleep behavior and autonomic nervous system abnormalities[35].
Muscle weakness and myoatrophy have been reported in about 65% of patients, especially during the late stage of the disease[44]. Only a small number of patients develop severe muscle weakness. Changes in biopsy specimens of muscular atrophy are various and usually consist of type 2 fiber atrophy, increased variability of the fiber size and central nucleation[41,45]. However, in some rare cases, severe muscle weakness and atrophy, and muscle pathology similar to muscular dystrophy, have been reported[46]. Myopathic changes in biopsies and elevated levels of muscle creatine kinase are not due to involvement of dystrophin, as demonstrated at the DNA and protein levels[47,48]. Immunohistochemical staining of XK protein in the lesioned muscles showed[49] positive staining within the sarcoplasmic reticulum in type 2 muscle fiber cells, while the control group only displayed weak background staining without specific labeling, indicating that the physiological XK expression deficiency is related to type 2 fiber atrophy, and suggesting that the XK protein is one of the key factors for maintenance of normal muscle structure and function. Muscle CT has revealed in some cases a selective and asymmetrical atrophy, which has been suggested as a factor in differential diagnosis between McLeod syndrome and chorea-acanthocytosis[50]. Signs of heart involvement have been found in two-thirds of the patients analyzed[41]. Cardiac disease, such as congestive cardiomyopathy, dilated cardiomyopathy, atrial fibrillation, and tachyarrhythmia, generally progresses from the fifth decade onwards[38,51]. Myocardial tissue presents with non-specific local muscle cell hypertrophy, mild variation of muscle fiber size, and patchy interstitial fibrosis[52]. Mean follow-up of 15 years of nine patients revealed that eight of them developed muscle weakness and myotrophy with elevated skeletal muscle creatine kinase levels, approximately 300–3 000 U/L (usually < 4 000 U/L; clinically, upper normal limit of creatine kinase is 173 U/L), and two of them were disabled because of lower limb weakness[52]. The muscle tissues of all patients were abnormal. Only four patients showed an obvious non-specific myopathic change. Movement and sensory nerve examinations showed that neurogenic myasthenia and atrophy were not the primary cause of myopathy[52].
Genetic research
The McLeod locus is localized on chromosome Xp21.1. The XK gene is organized in three exons with significant homology to the Ced-8 gene of C. elegans, which controls the apoptosis cycle[53]. With the increase of case studies, 35 different mutations have been reported to date[54]. They seem to be evenly distributed throughout the XK gene sequence and the complete XK mRNA has not yet been detected. Correlation is difficult between the type of mutation and the variations of the clinical phenotype, mainly because almost all the changes lead to loss of XK function.
Molecular pathology
Kell is the third most important blood group system after ABO and rhesus, comprising at least 23 different erythrocyte antigens. Expression of Kell antigen is dependent on the presence of the “precursor substance” XK, a 37-kDa membrane protein expressing antigen Kx[55]. The erythrocyte phenotype of McLeod syndrome can only be tested through special laboratory examination. The function of XK, Kell, and their complex has been only partly elucidated[1], and the molecular mechanisms that lead to McLeod syndrome pathogenesis are still unknown. Kell, an endothelin-processing enzyme, is the only M13 family member known to be covalently linked by a disulphide bond to another membrane-associated protein. Endothelins and their G protein coupled receptor ETB are present in brain dopaminergic areas such as the striatum, and activation of these receptors causes dopamine release[56]. Thus, a role of endothelins as basal ganglia neurotransmitters might be relevant to the pathogenesis of McLeod syndrome. Kell and XK may function as a complex. However, there are indications that in skeletal muscle, Kell and XK do not co-localize[49]; and XK expression appears within the intervals of neurons, while Kell expression occurs on red blood cells in cerebral blood vessels[57,58], suggesting the possibility of their independent functions in these tissues. The grouped atrophy in McLeod myopathy involves mainly type 2 fibers. The predominant XK expression in type 2 fibers in normal muscle suggests that the XK protein represents a crucial factor for the maintenance of normal muscle structure and function[1]. XK might have a role in transport, which can be inferred from its amino acid sequence, and in preserving the structural integrity of the membrane, as suggested on the basis of the clinical features of McLeod syndrome, i.e. acanthocytosis and elevated levels of serum creatine kinase[1].
Acanthocytosis
Acanthocytes account for approximately 3–40% of peripheral blood. In McLeod syndrome, acanthocytes are reported to be similar to those for chorea-acanthocytosis[59], suggesting some weakness regarding membrane tension. McLeod red blood cells show decreased deformability[60], partly because of decreased cell surface area and partly because of an increased intrinsic membrane mechanical stability in this syndrome. These abnormalities may account for the reduced red blood cell survival observed in McLeod syndrome. Decreased deformability and increased stability depend on skeletal proteins and their association with integral transmembrane proteins. These changes in McLeod erythrocytes are likely due to a combination of a lack of XK protein and dramatic decrease of transmembrane Kell protein, which is associated with the membrane skeleton[61,62]. Thus, in McLeod syndrome a defect in XK and Kell is likely to lead to changes in the organization of the membrane skeletal network responsible for membrane structural changes.
HUNTINGTON'S DISEASE-LIKE 2
Some cases share clinical and pathological characteristics with Huntington's disease, but the gene mutation for this disease has not yet been found. Therefore, they are named Huntington's disease-like 2[63], which is an autosomal dominant condition due to a trinucleotide repeat expansion in the junctophilin-3 gene on chromosome 16q[64]. In addition, 1% of Huntington's disease gene mutation-negative patients are Huntington's disease-like 2[65]. So far, all the affected families are of African ancestry, with the highest prevalence among black South Africans[66]. Some African Caribbean familial cases are reported to be of North American origin. Other families have been found in the United States and Brazil. Age of onset of most patients is 30–50 years old, with the general course of 10–20 years. The initial signs of the disease are personality changes or psychiatric symptoms[67]. Parkinson's syndrome and dystonia are common, especially in late stage. There are not any peripheral nervous or muscular disorders or epilepsy. Individual clinical manifestations vary widely. About 10% of the Huntington's disease-like 2 patients have accompanying acanthocytosis[64].
Imaging and pathological features
The brain MRI findings are similar to those of Huntington's disease: marked atrophy of the caudate nucleus and the cortex, and mild atrophy of the ventricular system[68] (Figure 5). Under the microscope, severe neuronal degeneration, reactive astrocytosis and vacuolization of the nerve fibers can be seen[69]. The caudate nucleus is not completely involved and microscopic changes are more evident in the dorsal region than the ventral region[69]. Putamen degeneration is milder than in the caudate nucleus, but the dorsal and ventral sides are the same. Lewy bodies, amyloid beta protein or neurofibrillary tangles were not found. An immunohistochemical study found 1C2 staining, nuclear aggregation, and anti-ubiquitin antibody, but not anti-Huntington antibody[2].
Figure 5.
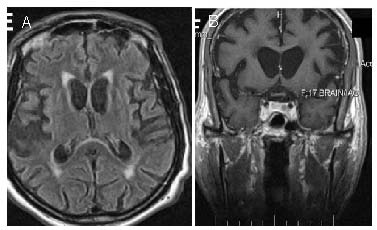
Axial fluid attenuated inversion recovery (A) and coronal T1-weighted (B) images show atrophy of the caudate nucleus and the fronto-temporal cortex in an Huntington's disease-like 2 patient.
Genetic research
Huntington's disease-like 2 is due to the CAG/CTG selective amplification of junctophilin-3 gene exon 2A fragment in chromosome 16q24.3[70]. The age of onset is inversely proportional to the size of the amplification fragment[71]. Normal amplification range is 6–27, and 41–59 copies may give rise to the disease[72]. The youngest case was an individual 21 years of age (56 copies) and the oldest was a 60-year-old male (46 copies)[71]. In middle-aged adults (30–50 years old) the symptoms are typical. An induced-expression Huntington's disease-like 2 mouse model suggested that overlapping polyglutamine mediates the pathogenesis of the disease[73]. Immunohistochemical analyses of muscle function and tissue morphology of knockout mice support the hypothesis that junctophilin-3 helps to link membrane voltage sensors and the ion channels of the brain, especially calcium channels[73]. At ages 1.5 to 3 months after junctophilin-3 knockout, the mice showed balance disturbances, without abnormal behavior, neuropathology, or electrophysiological damage. After 18 months, the junctophilin-3 knockout mice had abnormally slow movement, and ultimately lost weight and died.
Molecular pathology
Factors affecting gene mutation are unknown. The variation of junctophilin-3 exon 2A fragments repeat transcription, resulting in damage to transcription or alteration in protein levels. In myotonia, repetitive CUG causes toxic effects at the transcriptional level[74]. At the protein level, multi-poly-alanine repeated cohesion causes cytotoxic damage in cultured mouse Purkinje cells[75]. The cytorhyctes in Huntington's disease-like 2 nucleus perhaps are formed because of extended poly-alanine or repetitive poly-leucine, which can be detected by the 1C2 antibody[76]. A repeated amplified sequence may change expression of the junctophilin-3 gene, which can affect the flow of calcium ions, leading to excitotoxicity and cell death[77,78,79]. Other potential mechanisms may be related to metabolic disturbance. The latest study demonstrated that[80] cytoplasmic dynein plays a major role in lipid metabolism and heat generation by adjusting the norepinephrine-induced oxidative stress. A mutation in cytoplasmic dynein disturbs the metabolism of brown and white adipose in Huntington's disease-like 2[80]. Currently, this pathogenesis is thought to be non-specific. The relevance between junctophilin-3 gene mutation and acanthocytosis is unclear.
PANTOTHENATE KINASE-ASSOCIATED NEURODEGENERATION
Pantothenate kinase-associated neurodegeneration, a subtype of neurodegenerative disease due to brain iron deposition, is an autosomal recessive genetic disease. The main characteristic is progressive ataxia and significant globus pallidus iron deposition[81]. It was formerly termed as Hallervorden-Spatz syndrome or brain iron accumulation type 1. It was subsequently found that the disease is caused by PKAN2 gene[82] mutations, and thus renamed as pantothenate kinase-associated neurodegeneration. In 1992, it was confirmed that HARP (hypobetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration) syndrome is caused by PKAN2 gene mutations[83]. About 8% of the cases, hereditary or sporadic, showed acanthocytosis, both in childhood and adulthood[82]. Pantothenate kinase gene locates to autosomal chromosome site 20p13. The typical clinical manifestations appear in childhood, mainly as orofacial and limb dystonia, dance syndrome, and spasticity, and the majority of patients develop retinopathy pigmentosa. About one-third of typical cases have cognitive impairment[83]. The atypical cases are adult-onset, presenting as dystonia, rigidity, and gait stiffness in the early stages. Speech difficulties, particularly palilalia or dysarthria, are also prominent features of pantothenate kinase-associated neurodegeneration. Decreased cognition, impaired personality and pigment retinopathy are rare[83]. Kinase activity is relevant to the disease manifestations[84,85]. For typical cases, there is no kinase activity. Atypical cases are due to PKAN2 gene missense mutation, and there may be some residual enzyme function. Prebetalipoprotein evaluation is immaterial for neuroacanthocytosis syndromes[86]. The abnormal electrophoretic pattern of so-called aprebetalipoproteinemia described in a later case proved to be chorea-acanthocytosis, which may be observed in many normal subjects.
Imaging and pathological features
The typical MRI finding of pantothenate kinase-associated neurodegeneration is the “eye of the tiger” sign[85] (Figure 6), due to cysteine iron complex deposition in the globus pallidus and substantia nigra. Magnetic resonance spectroscopy showed that the nicotinic acid amide/creatine of the caudate nucleus decreased significantly, and gliosis increased in the substantia nigra[87]. Because of the diversity of clinical manifestations of this disease, autopsy of dementia cases found that Alzheimer's disease and pantothenate kinase-associated neurodegeneration were superimposed[88]. Atypical cases showed cortical Lewy bodies and rare tau protein-positive neurites. Positron emission tomography revealed that late-onset cases had decreased asymmetric striatum dopamine transporter[89].
Figure 6.
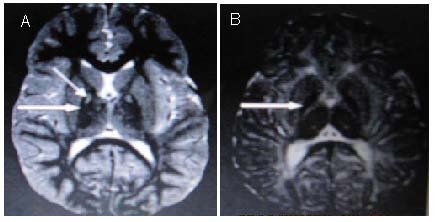
T2-weighted (A) and T1-weighted (B) brain MRI scans with PKAN demonstrating the “eye of the tiger” sign (arrows) in a pantothenate kinase-associated neurodegeneration patient.
DISCUSSION
Chorea-acanthocytosis and McLeod syndrome are multi-focal nervous system disorders with significant multi-system performances. Pantothenate kinase-associated neurodegeneration and Huntington's disease-like 2 are rare, and limited to the pathological changes of the central nervous system. There are many similarities between these syndromes, and because of gene mutations, basal ganglia neuron vulnerability is common. There are some initial studies on the etiology, clinical manifestations, and treatment for these diseases[4]. The symptoms are related to the degeneration of neurons caused by abnormal red cell morphology. Thereby, morphological change of erythrocytes is a feasible study target. Gene mutation is the basic cause of the pathogenesis. However, the relationship between mutation sites and the mutant form, and the clinical manifestations, are still unknown; therefore further research is needed. The etiology-related proteins play an important role in the recognition of membrane structure. Unfortunately, the physiological role of normal and abnormal protein expressed by these genes is unknown. The membrane instability caused by gene mutations may be the etiology of acanthocytes and neurodegeneration at the transcription and translation levels, which still needs further investigation. Treatment is mainly with drugs to mitigate symptoms, and there is little study on this aspect. Although these diseases show similar clinical manifestations with other nervous system diseases, drug treatment is not the same[68], indicating that they result from special pathological mechanisms. Further genomic research will provide a basis for screening of these diseases and may bring breakthroughs for gene therapy.
Footnotes
Conflicts of interest: None declared.
(Edited by Li XG, Cheng XD/Yang Y/Song LP)
REFERENCES
- [1].Rampoldi L, Danek A, Monaco AP. Clinical features and molecular bases of neuroacanthocytosis. J Mol Med (Berl) 2002;80(8):475–491. doi: 10.1007/s00109-002-0349-z. [DOI] [PubMed] [Google Scholar]
- [2].Ichiba M, Nakamura M, Sano A. Neuroacanthocytosis update. Brain Nerve. 2008;60(6):635–641. [PubMed] [Google Scholar]
- [3].Danek A, Jung HH, Melone MA, et al. Neuroacanthocytosis: new developments in a neglected group of dementing disorders. J Neurol Sci. 2005;229-230:171–186. doi: 10.1016/j.jns.2004.11.024. [DOI] [PubMed] [Google Scholar]
- [4].Danek A, Walker RH. Neuroacanthocytosis. Curr Opin Neurol. 2005;18(4):386–392. doi: 10.1097/01.wco.0000173464.01888.e9. [DOI] [PubMed] [Google Scholar]
- [5].Estes JW, Morley TJ, Levine IM, et al. A new hereditary acanthocytosis syndrome. Am J Med. 1967;42(6):868–881. doi: 10.1016/0002-9343(67)90068-x. [DOI] [PubMed] [Google Scholar]
- [6].Sibon I, Ghorayeb I, Arné P, et al. Distressing belching and neuroacanthocytosis. Mov Disord. 2004;19(7):856–869. doi: 10.1002/mds.20091. [DOI] [PubMed] [Google Scholar]
- [7].Gradstein L, Danek A, Grafman J, et al. Eye movements in chorea-acanthocytosis. Invest Ophthalmol Vis Sci. 2005;46(6):1979–1987. doi: 10.1167/iovs.04-0539. [DOI] [PubMed] [Google Scholar]
- [8].Lossos A, Dobson-Stone C, Monaco AP, et al. Early clinical heterogeneity in choreoacanthocytosis. Arch Neurol. 2005;62(4):611–614. doi: 10.1001/archneur.62.4.611. [DOI] [PubMed] [Google Scholar]
- [9].Huppertz HJ, Kröll-Seger J, Danek A, et al. Automatic striatal volumetry allows for identification of patients with chorea-acanthocytosis at single subject level. J Neural Transm. 2008;115(10):1393–1400. doi: 10.1007/s00702-008-0094-8. [DOI] [PubMed] [Google Scholar]
- [10].Marson AM, Bucciantini E, Gentile E, et al. Neuroacanthocytosis: clinical, radiological, and neurophysiological findings in an Italian family. Neurol Sci. 2003;24(3):188–189. doi: 10.1007/s10072-003-0123-1. [DOI] [PubMed] [Google Scholar]
- [11].Ishida C, Makifuchi T, Saiki S, et al. A neuropathological study of autosomal-dominant chorea-acanthocytosis with a mutation of VPS13A. Acta Neuropathol. 2009;117(1):85–94. doi: 10.1007/s00401-008-0403-1. [DOI] [PubMed] [Google Scholar]
- [12].Hardie RJ, Pullon HW, Harding AE, et al. Neuroacanthocytosis. A clinical, haematological and pathological study of 19 cases. Brain. 1991;114(Pt 1A):13–49. [PubMed] [Google Scholar]
- [13].Delecluse F, Deleval J, Gérard JM, et al. Frontal impairment and hypoperfusion in neuroacanthocytosis. Arch Neurol. 1991;48(2):232–234. doi: 10.1001/archneur.1991.00530140130031. [DOI] [PubMed] [Google Scholar]
- [14].Müller-Vahl KR, Berding G, Emrich HM, et al. Chorea-acanthocytosis in monozygotic twins: clinical findings and neuropathological changes as detected by diffusion tensor imaging, FDG-PET and (123)I-beta-CIT-SPECT. J Neurol. 2007;254(8):1081–1088. doi: 10.1007/s00415-006-0492-5. [DOI] [PubMed] [Google Scholar]
- [15].Kontoleon PE, Ilias I, Matsouka A, et al. Impaired hypothalamic endocrine function in neuroacanthocytosis. J Clin Neurosci. 2003;10(6):701–703. doi: 10.1016/s0967-5868(03)00166-8. [DOI] [PubMed] [Google Scholar]
- [16].Kito S, Itoga E, Hiroshige Y, et al. A pedigree of amyotrophic chorea with acanthocytosis. Arch Neurol. 1980;37(8):514–517. doi: 10.1001/archneur.1980.00500570062010. [DOI] [PubMed] [Google Scholar]
- [17].Gross KB, Skrivanek JA, Carlson KC, et al. Familial amyotrophic chorea with acanthocytosis. New clinical and laboratory investigations. Arch Neurol. 1985;42(8):753–756. doi: 10.1001/archneur.1985.04210090017005. [DOI] [PubMed] [Google Scholar]
- [18].Kageyama Y, Matsumoto K, Ichikawa K, et al. A new phenotype of chorea-acanthocytosis with dilated cardiomyopathy and myopathy. Mov Disord. 2007;22(11):1669–1670. doi: 10.1002/mds.21556. [DOI] [PubMed] [Google Scholar]
- [19].Machida M, Asai K, Sano M. Genome sequencing and analysis of Aspergillus oryzae. Nature. 2005;438(7071):1157–1161. doi: 10.1038/nature04300. [DOI] [PubMed] [Google Scholar]
- [20].Dobson-Stone C, Danek A, Rampoldi L. Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. Eur J Hum Genet. 2002;10(11):773–781. doi: 10.1038/sj.ejhg.5200866. [DOI] [PubMed] [Google Scholar]
- [21].Rampoldi L, Dobson-Stone C, Rubio JP, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001;28(2):119–120. doi: 10.1038/88821. [DOI] [PubMed] [Google Scholar]
- [22].Ueno S, Maruki Y, Nakamura M, et al. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet. 2001;28(2):121–122. doi: 10.1038/88825. [DOI] [PubMed] [Google Scholar]
- [23].Kurano Y, Nakamura M, Ichiba M, et al. In vivo distribution and localization of chorein. Biochem Biophys Res Commun. 2007;353(2):431–435. doi: 10.1016/j.bbrc.2006.12.059. [DOI] [PubMed] [Google Scholar]
- [24].Stege JT, Laub MT, Loomis WF. Tip genes act in parallel pathways of early Dictyostelium development. Dev Genet. 1999;25(1):64–77. doi: 10.1002/(SICI)1520-6408(1999)25:1<64::AID-DVG7>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- [25].Raymond CK, Howald-Stevenson I, Vater CA, et al. Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vps mutants. Mol Biol Cell. 1992;3(12):1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Brickner JH, Fuller RS. SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J Cell Biol. 1997;139(1):23–36. doi: 10.1083/jcb.139.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327(Pt 3):625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kurano Y, Nakamura M, Ichiba M, et al. Chorein deficiency leads to upregulation of gephyrin and GABA(A) receptor. Biochem Biophys Res Commun. 2006;351(2):438–442. doi: 10.1016/j.bbrc.2006.10.070. [DOI] [PubMed] [Google Scholar]
- [29].Bayreuther C, Borg M, Ferrero-Vacher C, et al. Chorea-acanthocytosis without acanthocytes. Rev Neurol. 2010;166(1):100–103. doi: 10.1016/j.neurol.2009.03.005. [DOI] [PubMed] [Google Scholar]
- [30].Sakai T, Antoku Y, Iwashita H, et al. Chorea-acanthocytosis: abnormal composition of covalently bound fatty acids of erythrocyte membrane proteins. Ann Neurol. 1991;29(6):664–669. doi: 10.1002/ana.410290615. [DOI] [PubMed] [Google Scholar]
- [31].Kay MM. Band 3 in aging and neurological disease. Ann N Y Acad Sci. 1991;621:179–204. doi: 10.1111/j.1749-6632.1991.tb16979.x. [DOI] [PubMed] [Google Scholar]
- [32].Bosman GJ, Kay MM. Alterations of band 3 transport protein by cellular aging and disease: erythrocyte band 3 and glucose transporter share a functional relationship. Biochem Cell Biol. 1990;68(12):1419–1427. doi: 10.1139/o90-205. [DOI] [PubMed] [Google Scholar]
- [33].Olivieri O, De Franceschi L, Bordin L, et al. Increased membrane protein phosphorylation and anion transport activity in chorea-acanthocytosis. Haematologica. 1997;82(6):648–653. [PubMed] [Google Scholar]
- [34].Kay MM, Goodman J, Goodman S, et al. Membrane protein band 3 alteration associated with neurologic disease and tissue-reactive antibodies. Exp Clin Immunogenet. 1990;7(3):181–199. [PubMed] [Google Scholar]
- [35].Danek A, Rubio JP, Rampoldi L, et al. McLeod neuroacanthocytosis; genotype and phenotype. Ann Neurol. 2001;50(6):755–764. doi: 10.1002/ana.10035. [DOI] [PubMed] [Google Scholar]
- [36].Jung HH, Danek A, Walker RH, et al. McLeod Neuroacanthocytosis Syndrome. In: Pagon RA, Bird TD, Dolan CR, editors. Seattle (WA): University of Washington, Seattle; 1993. [Google Scholar]
- [37].Jung HH, Hergersberg M, Kneifel S, et al. McLeod syndrome: a novel mutation, predominant psychiatric manifestations, and distinct striatal imaging findings. Ann Neurol. 2001;49(3):384–392. [PubMed] [Google Scholar]
- [38].Malandrini A, Fabrizi GM, Truschi F, et al. Atypical McLeod syndrome manifested as X-linked chorea-acanthocytosis, neuromyopathy and dilated cardiomyopathy: report of a family. J Neurol Sci. 1994;124:89–94. doi: 10.1016/0022-510x(94)90016-7. [DOI] [PubMed] [Google Scholar]
- [39].Nicholl DJ, Sutton I, Dotti MT, et al. White matter abnormalities on MRI in neuroacanthocytosis. J Neurol Neurosurg Psychiatry. 2004;75(8):1200–1201. doi: 10.1136/jnnp.2003.026781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Valko PO, Hänggi J, Meyer M, et al. Evolution of striatal degeneration in McLeod syndrome. Eur J Neurol. 2010;17(4):612–618. doi: 10.1111/j.1468-1331.2009.02872.x. [DOI] [PubMed] [Google Scholar]
- [41].Dotti MT, Battisti C, Malandrini A, et al. McLeod syndrome and neuroacanthocytosis with a novel mutation in the XK gene. Mov Disord. 2000;15:1282–1284. doi: 10.1002/1531-8257(200011)15:6<1282::aid-mds1042>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- [42].Dydak U, Mueller S, Sándor PS, et al. Cerebral metabolic alterationsin McLeod syndrome. Eur Neurol. 2006;56(1):17–23. doi: 10.1159/000095136. [DOI] [PubMed] [Google Scholar]
- [43].Oechsner M, Buchert R, Beyer W, et al. Reduction of striatal glucose metabolism in McLeod choreoacanthocytosis. J Neurol Neurosurg Psychiatry. 2001;70(4):517–520. doi: 10.1136/jnnp.70.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jung HH, Danek A, Frey BM. McLeod syndrome: a neurohaematological disorder. Vox Sang. 2007;93(2):112–121. doi: 10.1111/j.1423-0410.2007.00949.x. [DOI] [PubMed] [Google Scholar]
- [45].Jung HH, Hergersberg M, Kneifel S, et al. McLeod syndrome: a novel mutation, predominant psychiatric manifestations, and distinct striatal imaging findings. Ann Neurol. 2001;49:384–392. [PubMed] [Google Scholar]
- [46].Kawakami T, Takijama Y, Sakoe K, et al. A case of McLeod syndrome with unusually severe myopathy. J Neurol Sci. 1999;166:36–39. doi: 10.1016/s0022-510x(99)00108-2. [DOI] [PubMed] [Google Scholar]
- [47].Danek A, Witt TN, Stockmann HB, et al. Normal dystrophin in McLeod myopathy. Ann Neurol. 1990;28(5):720–722. doi: 10.1002/ana.410280521. [DOI] [PubMed] [Google Scholar]
- [48].Carter ND, Morgan JE, Monaco AP, et al. Dystrophin expression and genotypic analysis of two cases of benign X linked myopathy (McLeod's syndrome) J Med Genet. 1990;27(6):345–347. doi: 10.1136/jmg.27.6.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jung HH, Russo D, Redman C, et al. Kell and XK immunohistochemistry in McLeod myopathy. Muscle Nerve. 2001;24(10):1346–1351. doi: 10.1002/mus.1154. [DOI] [PubMed] [Google Scholar]
- [50].shikawa S, Tachibana N, Tabata KI, et al. Muscle CT scan findings in McLeod syndrome and chorea-acanthocytosis. Muscle Nerve. 2000;23(7):1113–1116. doi: 10.1002/1097-4598(200007)23:7<1113::aid-mus15>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- [51].Oechslin E, Kaup D, Jenni R, et al. Cardiac abnormalities in McLeod syndrome. Int J Cardiol. 2009;132(1):130–132. doi: 10.1016/j.ijcard.2007.07.167. [DOI] [PubMed] [Google Scholar]
- [52].Hewer E, Danek A, Schoser BG, et al. McLeod myopathy revisited: more neurogenic and less benign. Brain. 2007;130(Pt 12):3285–3296. doi: 10.1093/brain/awm269. [DOI] [PubMed] [Google Scholar]
- [53].Stanfield GM, Horvitz HR. The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol Cell. 2000;5(3):423–433. doi: 10.1016/s1097-2765(00)80437-2. [DOI] [PubMed] [Google Scholar]
- [54].Cooper RA Kell blood group. The human gene mutation database at the Institute of Medical Genetics in Cardiff (BIOBASE) 2007 [Google Scholar]
- [55].Lee S, Lin M, Mele A, et al. Proteolytic processing of big endothelin-3 by the kell blood group protein. Blood. 1999;94(4):1440–1450. [PubMed] [Google Scholar]
- [56].van den Buuse M, Webber KM. Endothelin and dopamine release. Prog Neurobiol. 2000;60(4):385–405. doi: 10.1016/s0301-0082(99)00034-9. [DOI] [PubMed] [Google Scholar]
- [57].Clapéron A, Hattab C, Armand V, et al. The Kell and XK proteins of the Kell blood group are not co-expressed in the central nervous system. Brain Res. 2007;1147:12–24. doi: 10.1016/j.brainres.2007.01.106. [DOI] [PubMed] [Google Scholar]
- [58].Lee S, Sha Q, Wu X, et al. Expression profiles of mouse Kell, XK, and XPLAC mRNA. J Histochem Cytochem. 2007;55(4):365–374. doi: 10.1369/jhc.6A7126.2006. [DOI] [PubMed] [Google Scholar]
- [59].Terada N, Fujii Y, Ueda H, et al. Ultrastructural changes of erythrocyte membrane skeletons in chorea-acanthocytosis and McLeod syndrome revealed by the quick-freezing and deep-etching method. Acta Haematol. 1999;101(1):25–31. doi: 10.1159/000040917. [DOI] [PubMed] [Google Scholar]
- [60].Ballas SK, Bator SM, Aubuchon JP, et al. Abnormal membrane physical properties of red cells in McLeod syndrome. Transfusion. 1990;30(8):722–727. doi: 10.1046/j.1537-2995.1990.30891020333.x. [DOI] [PubMed] [Google Scholar]
- [61].Redman CM, Avellino G, Pfeffer SR, et al. Kell blood group antigens are part of a 93,000-dalton red cell membrane protein. J Biol Chem. 1986;261(20):9521–9525. [PubMed] [Google Scholar]
- [62].Anstee DJ. Blood group-active surface molecules of the human red blood cell. Vox Sang. 1990;58(1):1–20. doi: 10.1111/j.1423-0410.1990.tb02049.x. [DOI] [PubMed] [Google Scholar]
- [63].Margolis RL, O’Hearn E, Rosenblatt A, et al. A disorder similar to Huntington's disease is associated with a novel CAG repeat expansion. Ann Neurol. 2001;50(6):373–380. doi: 10.1002/ana.1312. [DOI] [PubMed] [Google Scholar]
- [64].Walker RH, Rasmussen A, Rudnicki D, et al. Huntington's disease&like 2 can present as chorea-acanthocytosis. Neurology. 2003;61(7):1002–1004. doi: 10.1212/01.wnl.0000085866.68470.6d. [DOI] [PubMed] [Google Scholar]
- [65].Stevanin G, Camuzat A, Holmes SE. CAG/CTG repeat expansions at the Huntington's disease-like 2 locus are rare in Huntington's disease patients. Neurology. 2002;58(6):965–967. doi: 10.1212/wnl.58.6.965. [DOI] [PubMed] [Google Scholar]
- [66].Magazi DS, Krause A, Bonev V. Huntington's disease: genetic heterogeneity in black African patients. S Afr Med J. 2008;98(3):200–203. [PubMed] [Google Scholar]
- [67].Walker RH, Jankovic J, O’Hearn E, et al. Phenotypic features of Huntington's disease-like 2. Mov Disord. 2003;18(12):1527–1530. doi: 10.1002/mds.10587. [DOI] [PubMed] [Google Scholar]
- [68].Jung HH, Danek A, Walker RH. Neuroacanthocytosis syndromes. Orphanet J Rare Dis. 2011;6:68. doi: 10.1186/1750-1172-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Walker RH, Jung HH, Danek A. Neuroacanthocytosis. Handb Clin Neurol. 2011;100:141–151. doi: 10.1016/B978-0-444-52014-2.00007-0. [DOI] [PubMed] [Google Scholar]
- [70].Wilburn B, Rudnicki DD, Zhao J, et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70(3):427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Margolis RL, Holmes SE, Rosenblatt A, et al. Huntington's disease-like 2 (HDL2) in North America and Japan. Ann Neurol. 2004;56(5):670–674. doi: 10.1002/ana.20248. [DOI] [PubMed] [Google Scholar]
- [72].Holmes SE, O’Hearn E, Rosenblatt A, et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- [73].Wilburn B, Rudnicki DD, Zhao J, et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70(3):427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ranum LP, Day JW. Myotonic dystrophy: clinical and molecular parallels between myotonic dystrophy type 1 and type 2. Curr Neurol Neurosci Rep. 2002;2(5):465–470. doi: 10.1007/s11910-002-0074-6. [DOI] [PubMed] [Google Scholar]
- [75].Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15(8):925–932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- [76].Rodrigues GG, Walker RH, Brice A, et al. Huntington's disease-like 2 in Brazil--report of 4 patients. Mov Disord. 2008;23(15):2244–2247. doi: 10.1002/mds.22223. [DOI] [PubMed] [Google Scholar]
- [77].Takeshima H, Komazaki S, Nishi M, et al. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6(1):11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- [78].Nishi M, Mizushima A, Nakagawara K, et al. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 2000;273(3):920–927. doi: 10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- [79].Ito K, Komazaki S, Sasamoto K, et al. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol. 2001;154(5):1059–1067. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Eschbach J, Fergani A, Oudart H, et al. Mutations in cytoplasmic dynein lead to a Huntington's disease-like defect in energy metabolism of brown and white adipose tissues. Biochim Biophys Acta. 2011;1812(1):59–69. doi: 10.1016/j.bbadis.2010.09.009. [DOI] [PubMed] [Google Scholar]
- [81].Hartig MB, Prokisch H, Meitinger T, et al. Pantothenate kinase-associated neurodegeneration. Curr Drug Targets. 2012;13(9):1182–1189. doi: 10.2174/138945012802002384. [DOI] [PubMed] [Google Scholar]
- [82].Houlden H, Lincoln S, Farrer M, et al. Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology. 2003;61(10):1423–1426. doi: 10.1212/01.wnl.0000094120.09977.92. [DOI] [PubMed] [Google Scholar]
- [83].Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003;348(1):33–40. doi: 10.1056/NEJMoa020817. [DOI] [PubMed] [Google Scholar]
- [84].Seo JH, Song SK, Lee PH. A novel PANK2 mutation in a patient with Atypical Pantothenate-Kinase-Associated neurodegeneration presenting with adult-onset Parkinsonism. Clin Neurol. 2009;5(4):192–194. doi: 10.3988/jcn.2009.5.4.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hartig MB, Hörtnagel K, Garavaglia B, et al. Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol. 2006;59(2):248–256. doi: 10.1002/ana.20771. [DOI] [PubMed] [Google Scholar]
- [86].McNeill A, Birchall D, Hayflick SJ, et al. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology. 2008;70(18):1614–1619. doi: 10.1212/01.wnl.0000310985.40011.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Parashari UC, Aga P, Parihar A. Case report: MR spectroscopy in pantothenate kinase-2 associated neurodegeneration. Indian J Radiol Imaging. 2010;20(3):188–191. doi: 10.4103/0971-3026.69353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Dziewulska D, Domitrz I, Domzał-Stryga A. Dementia means number of things-the overlap of neurodegeneration with brain iron accumulation (NBIA) and Alzheimer changes: an autopsy case. Folia Neuropathol. 2010;48(2):129–133. [PubMed] [Google Scholar]
- [89].Antonini A, Goldwurm S, Benti R, et al. Genetic, clinical, and imaging characterization of one patient with late-onset, slowly progressive, pantothenate kinase-associated neurodegeneration. Mov Disord. 2006;21(3):417–418. doi: 10.1002/mds.20774. [DOI] [PubMed] [Google Scholar]