

Abstract
The extracellular signal-regulated kinase/cAMP response element-binding protein/brain-derived neurotrophic factor signal transduction pathway plays an important role in the mechanism of action of antidepressant drugs and has dominated recent studies on the pathogenesis of depression. In the present review we summarize the known roles of extracellular signal-regulated kinase, cAMP response element-binding protein and brain-derived neurotrophic factor in the pathogenesis of depression and in the mechanism of action of antidepressant medicines. The extracellular signal-regulated kinase/cAMP response element-binding protein/brain-derived neurotrophic factor pathway has potential to be used as a biological index to help diagnose depression, and as such it is considered as an important new target in the treatment of depression.
Keywords: neural regeneration, reviews, depression, mitogen-activated protein kinase, extracellular signal-regulated kinase, cAMP response element-binding protein, brain-derived neurotrophic factor, 5-hydroxytryptamine, grants-supported paper, neuroregeneration
Research Highlights
-
(1)
The monoamine hypothesis has long stood as the major research focus in the pathogenesis of depression. The majority of antidepressant drugs exert their effects by increasing the concentration of synaptic monoamine transmitters through the regulation of signal transduction pathways and transcription factors.
-
(2)
The neural plasticity hypothesis postulates that depression occurs not only because of receptor levels, but also their activation of signaling pathways, the initiation of gene transcription, and a change in the level of downstream target gene expression.
-
(3)
We focus on the extracellular signal-regulated kinase/cAMP response element-binding protein/brain-derived neurotrophic factor signal transduction pathway and explore the function of this pathway by discussing evidence from molecular biology studies.
INTRODUCTION
Depressive disorder is considered one of the most serious clinical diseases worldwide, yet its pathogenesis remains unclear. Depression presents with various symptoms including depressed mood, loss of interest, and anhedonia. The recently proposed neural plasticity hypothesis postulates that depression arises not only because of the levels of receptors present at the cell surface, but also due to their activation of signaling pathways, the initiation of gene transcription, and a change in the level of downstream target gene expression, which is correlated with synaptic plasticity[1,2,3]. We focus on the extracellular signal-regulated kinase/cAMP response element-binding protein/brain-derived neurotrophic factor signal transduction pathway, and discuss recent studies and future possibilities for exploiting this pathway, to find a new drug target and produce appropriate treatments.
PATHOGENESIS OF DEPRESSION AND THE ANTIDEPRESSANT SIGNALING PATHWAY
Many pathways, such as those involving cAMP, mitogen-activated protein kinase, and Ca2+/calmodulin dependent protein kinase, contribute to the pathogenesis of depression and the mechanism of action of antidepressant drugs. The mitogen-activated protein kinase cascade plays an important role in controlling a variety of cellular physiological processes (Figure 1)[1].
Figure 1.
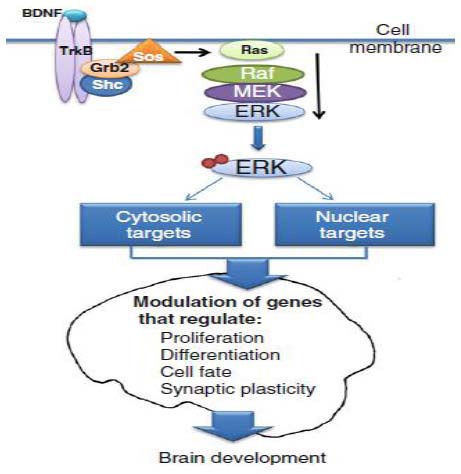
The extracellular signal-regulated kinase (ERK) signaling cascade in the regulation of brain development[1].
Typically, ERK1/2 activation occurs downstream from the binding of a ligand (e.g., brain-derived neurotrophic factor) to a tyrosine kinase receptor (e.g., TrkB), leading to receptor autophosphorylation. Subsequently, the Shc domain binds to phosphorylated TrkB and recruits the Grb2-Sos complex to activate the G-protein Ras. Ras activates Raf to phosphorylate MEK1/2. Activated MEK1/2 phosphorylates and activates ERK1/2, which subsequently activates multiple target proteins in the cell cytosol and nucleus by phosphorylation. Several of these proteins are involved in the regulation of cellular events central to brain development, including proliferation, differentiation, cell survival, and synaptic plasticity.
MEK: Mitogen-activated protein kinase/ERK kinase; TrkB: tropomyosin-receptor-kinase B.
Extracellular signal-regulated kinase is a core kinase of the mitogen-activated protein kinase family, and many studies support the hypothesis that inappropriate activation of extracellular signal-regulated kinase contributes to the pathogenesis of depression (Figure 2)[2,3,4,5].
Figure 2.
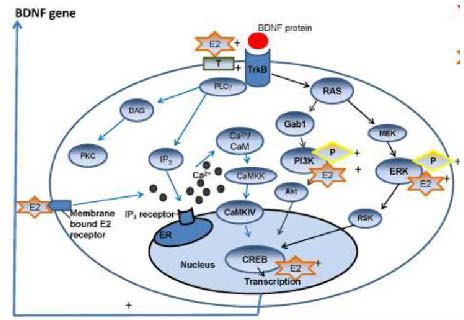
Interaction of drug and signaling pathway in relevance to depression[5].
Sex steroid hormones serve as an example. ER: Estrogen receptor; IP3: inositol-trisphosphate; DAG: diacyl glycerol; PKC: protein kinase C; MEK: mitogen-activated protein kinase/extracellular-signal-regulated kinase kinase; RSK: ribosomal S6 kinase; GAB1: Grb2-associated binder-1; CaMKK: Ca2+/calmodulin dependent protein kinase kinase; CaMKIV: Ca2+/calmodulin dependent protein kinase type IV; PI3-K: phosphatidylinositol 3-kinase; PLC: phospholipase C; BDNF: brain-derived neurotrophic factor.
EXTRACELLULAR SIGNAL-REGULATED KINASE IN DEPRESSION
There are five pathways in the mitogen-activated protein kinase cascade, comprising extracellular signal-regulated kinase 1/2, extracellular signal-regulated kinase 3/4, extracellular signal-regulated kinase 5, c-Jun N-terminal/stress activated protein kinase, and p38. Extracellular signal-regulated kinase 1/2 is involved in depressive disorder. The extracellular signal-regulated kinase family comprises five subtypes, extracellular signal-regulated kinase 1–5. Extracellular signal-regulated kinase 1 and 2 are the most widely expressed and have been studied extensively. Extracellular signal-regulated kinase 1/2 activation affects a number of physiological processes, including meiosis, karyokinesis, and the anaphase stage of mitosis. Activated extracellular signal-regulated kinase phosphorylates ribosome S6 kinase and other important transcription factors, resulting in downstream gene transcription[6]. Extracellular signal-regulated kinase 1/2 activation plays a key role in learning and memory, specifically in the process of memory consolidation[7,8]; the transformation from short- to long-term storage of memory.
cAMP response element-binding protein and brain-derived neurotrophic factor are located downstream in the extracellular signal-regulated kinase pathway[9]. An increased level of extracellular signal-regulated kinase1/2 phosphorylation may facilitate downstream gene transcription and induce the activation and expression of cAMP response element-binding protein[9,10]. Zhang et al[11] demonstrated that curcumin-induced increases in extracellular signal-regulated kinase phosphorylation in the amygdala were blocked by an extracellular signal-regulated kinase inhibitor, which diminished the antidepressant effects of curcumin. Di Benedetto et al[12] discovered that extracellular signal-regulated kinase 1 plays an important role in the treatment of depression. Norquetiapine (the principal metabolite of the atypical antipsychotic and antidepressant, quetiapine), like the antidepressant reboxetine, activated both extracellular signal-regulated kinase 1 and extracellular signal-regulated kinase 2 with consequent enhanced release of brain-derived neurotrophic factor, which was dependent on phosphorylated extracellular signal-regulated kinase, as demonstrated by its reversibility after pre-treatment with a pharmacological phosphorylated extracellular signal-regulated kinase inhibitor. Gourley et al[13] showed that antidepressant treatment significantly enhanced the level of phosphorylated extracellular signal-regulated kinase 1/2 in the dentate gyrus and corpus striatum. Nuttall et al[1] demonstrated that zinc could activate the extracellular signal-regulated kinase 1/2 signaling pathway at various levels (Figure 3).
Figure 3.
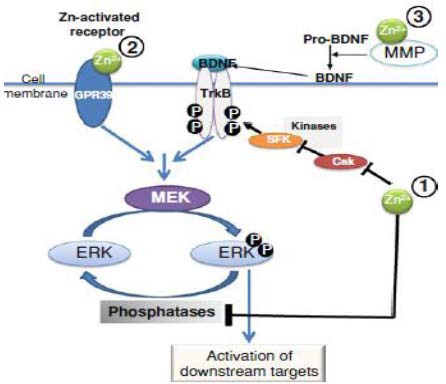
Regulation by zinc of the extracellular signal-regulated kinase (ERK) 1/2 signaling cascade.
Zinc can activate the ERK1/2 signaling pathway at various levels1: (1) by inhibiting phosphatases that are involved in ERK1/2 dephosphorylation, and inhibits kinases that indirectly regulate the phosphorylation levels of receptors that subsequently activate the ERK cascade; (2) by directly binding to and activating receptors (e.g., G protein-coupled receptor 39) which have ERK1/2 as a downstream signaling cascade; (3) by activating MMPs, which subsequently cleave and activate substrates (e.g., pro-BDNF) releasing ligands (e.g., BDNF) that activate receptors upstream from the ERK1/2 cascade.
MEK: Mitogen-activated protein kinase/ERK kinase; MMP: matrix metalloproteinases; BDNF: brain-derived neurotrophic factor.
cAMP RESPONSE ELEMENT-BINDING PROTEIN IN DEPRESSION
cAMP response element-binding protein is involved in the pathogenesis of depression and in the mechanism of action of antidepressant drugs[14]. Qi et al[15] found that rats with depressive-like behavior following chronic exposure to forced-swim stress had low levels of phosphorylated extracellular signal-regulated kinase 2 and phosphorylated cAMP response element-binding protein in the hippocampus and prefrontal cortex. Treatment with fluoxetine elevated the levels of both proteins. This suggests that extracellular signal-regulated kinase and cAMP response element-binding protein are involved in the neural mechanisms of depressive disorder.
cAMP response element-binding protein is a third messenger and intranuclear transcription factor. Phosphorylation of cAMP response element-binding protein at Ser133 promotes gene transcription, which can regulate neuron growth or regeneration, and synaptic plasticity (Figure 4)[16].
Figure 4.
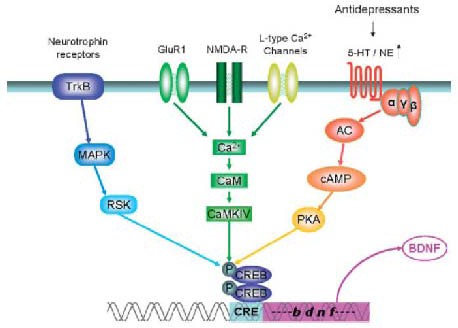
The convergence of several signal transduction pathways important in mood disorders on cAMP response element-binding (CREB) protein phosphorylation[16].
Since CREB acts as a transcription factor contributing to the control of cellular genetics, the term stimulus-transcription-coupling has been coined for this process. In these signaling cascades, postsynaptic cytoplasmic receptors, e.g., tropomyosin-receptor-kinase B (TrkB), glutamate receptors, monoaminergic receptors and others, are regarded as first messengers. They upregulate or downregulate a number of second messenger pathways, leading to the activation or inactivation of transcription factors such as CREB, a third messenger, which is activated by phosphorylation and acts via its cognate gene regulatory element (cAMP response element) in the promoter of target genes (the fourth messengers). As a representative target gene, brain-derived neurotrophic factor (BDNF) is shown, although there are several others known.
CaM: Calmodulin; MAPK: mitogen-activated protein kinase; RSK: ribosomal S6 kinase; AC: adenylate cyclase; PKA: cAMP-dependent protein kinase; NE: noradrenaline; 5-HT: 5-hydroxytryptamine; NMDA-R: N-methyl-D- aspartate receptor; CAMKIV: Ca2+/calmodulin-dependent protein kinase.
The mechanisms involved in cAMP response element-binding protein modulation of the effect of antidepressant drugs are highly complex. Animal studies have demonstrated that cAMP response element-binding protein expression varies over time in the central nervous systems[16,17]. Iga et al[18] found that the amount of cAMP response element-binding protein mRNA in peripheral leukocytes from untreated patients with depression was higher than in those treated with paroxetine. When the extracellular signal-regulated kinase pathway was inhibited in the hippocampus, subjects developed anhedonia as a result of the reduced level of cAMP response element-binding protein phosphorylation[19]. Despite differences between drugs, experimental protocols, and treatments across studies, cAMP response element-binding protein is consistently found to play a key role in the pathophysiology of depressive disorder[20,21]. Muschamp et al[22] used viral vectors to elevate or disrupt cAMP response element-binding protein in the nucleus accumbens shell in rats. Elevated levels of cAMP response element-binding protein produced increases in intracranial self-stimulation thresholds, reflecting anhedonia, while disruption of cAMP response element-binding protein function by the expression of a dominant negative cAMP response element-binding protein had the opposite effect. The authors also mimicked the downstream effects of cAMP response element-binding protein activation on the expression of the opioid peptide dynorphin, by injecting the kappa-opioid receptor agonist U50,488 into the nucleus accumbens shell. They concluded that activation of cAMP response element-binding protein in the nucleus accumbens shell produces multiple behavioral signs characteristic of experience-dependent psychiatric conditions such as posttraumatic stress disorder.
Zheng et al[23] demonstrated that while acute paroxetine did not improve the reduced activity of extracellular signal-regulated kinase 1/2 in a rat model of depression, its activity recovered after 4 weeks of treatment. The authors suggested that the antidepressant may target the extracellular signal-regulated kinase/cAMP response element-binding protein circuit. Paroxetine was found to improve memory at the same time as exerting an antidepressant effect. The effect on cAMP response element-binding protein after administration of paroxetine occurred before that on extracellular signal-regulated kinase, indicating that other signaling pathways were involved in the modulation of cAMP response element-binding protein.
Antidepressant drugs can modulate intracellular signal transduction pathways and produce a behavioral effect that corresponds to that seen in the clinic[24]. Gur et al[25] investigated the role of hippocampal neurogenesis in acute and chronic antidepressant treatment of wild-type and cAMP response element-binding protein-deficient rats. They concluded that increased hippocampal neurogenesis in the cAMP response element-binding protein-deficient rats allowed them to more efficiently respond to acute treatment than wild-type rats. Serretti et al[26] genotyped depressed patients for single nucleotide polymorphisms in the cAMP response element-binding protein 1 gene and monitored their response to antidepressants. They found that rs2253206-rs7569963 A-A and rs7569963-rs4675690 A-C haplotypes were associated with antidepressant treatment resistance, while the rs7569963GG genotype was associated with remission of depression. Some evidence suggests that genetic variants within cAMP response element-binding protein, cAMP response element-binding protein binding protein, and cAMP response element modulator are linked to several psychiatric disorders. Crisafulli et al[27] investigated whether some single nucleotide polymorphisms within these genes could be associated with major depressive disorder and bipolar disorder and whether they could predict clinical outcomes. Of the 14 single nucleotide polymorphisms studied, none were found to influence diagnosis or treatment response in patients with depression or bipolar disorder.
Many studies have suggested that cAMP response element-binding protein is involved in the pathogenesis of depression; however, the results are inconclusive. cAMP response element-binding protein may be one of the regulators of neurogenesis and of the mechanism of action of antidepressant drugs. It is necessary to identify the protein kinase that activates cAMP response element-binding protein, and to study the relationship between the controlling gene and neuronal plasticity downstream of cAMP response element-binding protein. In this way, we could come closer to understanding both the pathogenesis of depression and the mechanism of action of antidepressant drugs.
BRAIN-DERIVED NEUROTROPHIC FACTOR IN DEPRESSION
Antidepressant drugs act on physiological pathways such as those involving cAMP, mitogen-activated protein kinase, and Ca2+/calmodulin dependent protein kinase. cAMP response element-binding protein is activated last and controls brain-derived neurotrophic factor, which regulates neuronal plasticity. Thus, brain-derived neurotrophic factor may be responsible for the effects of antidepressants.
Brain-derived neurotrophic factor is a 12.3 kDa alkaline protein, which was separated from pig brain extract by German neurobiologist Barde in 1982. It is one of the most abundant neurotrophic factors, with widespread distribution in the cerebral cortex, hippocampus, basal forebrain, striatum and septal area, and, in particular, the cerebral cortex and hippocampus. The brain-derived neurotrophic factor gene, located at 11p13, is made up of 11 exons. It is widely expressed in the brain of mature mammals and contributes to neural regeneration, synaptic signal transduction and plasticity, and neurogenesis[28].
Brain-derived neurotrophic factor binds to tropomyosin-receptor-kinase B, causing its activation and subsequent physiological functions. Upregulation of brain-derived neurotrophic factor expression increases the level of postsynaptic tropomyosin-receptor-kinase B and activates signal transduction in the excited cells. The mitogen-activated protein kinase/extracellular signal-regulated kinase cascade plays an important role in this mechanism and activates cAMP response element-binding protein for transport and regulatory processes[29].
Many polymorphic markers have been found in the brain-derived neurotrophic factor gene, for example the single nucleotide polymorphism in the functional region at Val66Met (G196A) affects the transport and excretion of the protein. An association has been demonstrated between the Val66Met single nucleotide polymorphism and hippocampal volumes in drug-free depressed patients[30] using region of interest analysis on images acquired by magnetic resonance imaging. The study showed that the Val homozygote genotype may serve as a vulnerability factor in major depressive disorder in terms of hippocampal volume loss. Val66Met heterogenesis induces the precursor protein amino acid of brain-derived neurotrophic factor, which does not appear in the mature protein. The polymorphism of Val66Met may affect the secretion of brain-derived neurotrophic factor, and intracellular signal transduction, which plays a vital role in the pathogenesis of depression and cognition function deficits[31,32]. Brain-derived neurotrophic factor polymorphisms such as rs7124442 and rs6265 are associated with depressive symptoms in the clinic[33,34]. Domschke et al[35] identified that the brain-derived neurotrophic factor-rs7124442 allele C may be the anticipation factor for the antidepressant drug citalopram in German patients. Juhasz et al[36] found that minor allele carriers of brain-derived neurotrophic factor-rs6265 and cAMP response element-binding protein 1-rs2253206 were correlated closely to depression-related responses, and that childhood adversity increased the risk of lifetime depression in carriers of these alleles. Sarchiapone et al[37] showed that the Val66Met brain-derived neurotrophic factor polymorphism was associated with suicide attempts in depressed patients in a case control study. The suicide tendency of depressive patients with the Met/Met genotype was higher than that of those with the Met/Val and Val/Val genotype. Schenkel et al[38] thought the Val66Met polymorphism of may be an independent risk factor for high lethality in patients who had attempted suicide. Joffe et al[39] provided further evidence in support of this hypothesis, demonstrating that the Val66Met polymorphism affected hippocampal gray matter volume in a cross-sectional cohort of healthy subjects. Lower hippocampal gray matter volumes in Met carriers were associated with trait depression. Lee et al[40,41] found a reduction in brain-derived neurotrophic factor mRNA expression in peripheral blood mononuclear cells of depressed patients, using real-time quantitative PCR. However, no correlation was observed between the Val66Met polymorphism and the therapeutic effect of mirtazapine or a number of other antidepressants taken for up to 8 weeks[42,43]. The Val66Met polymorphism is related to other genes or environmental factors in the treatment of depression. These findings indicate that brain-derived neurotrophic factor single nucleotide polymorphisms and depression are associated with resistance to treatment with selective serotonin reuptake inhibitors[42,43].
Brain-derived neurotrophic factor expression was reduced in the hippocampal dentate gyrus of a mouse model of depression[44], and when stress-exposed mice were given antidepressants, the drop in brain-derived neurotrophic factor expression was reversed; thus, antidepressants also exert a neuroprotective effect[45]. Buchmann et al[46] showed that brain-derived neurotrophic factor levels were significantly reduced following exposure to high adversity situations in people homozygous for both the brain-derived neurotrophic factor Val allele and the 5-hydroxytryptamine transporter-linked polymorphic region allele. However, brain-derived neurotrophic factor levels were unaltered by early psychosocial adversity in carriers of these alleles. Whereas the former group showed to be most susceptible to depressive symptoms, the impact of early adversity was less pronounced in the latter group. Thus early-life stress could have lasting effects on plasma brain-derived neurotrophic factor levels in humans homozygous for Val. The vulnerability to this effect is diminished in Val66Met carriers. It is well established that depression and stress can be alleviated through physical exercise and/or the use of antidepressant drugs. Yang et al[47] assessed whether different antidepressants exerted neuroprotective effects in a model of nutrient deprivation stress, and evaluated whether key pro-survival pathways (mitogen-activated protein kinase, protein kinase A) were essential for such neuroprotection. They used quantitative western blot analysis to measure levels of brain-derived neurotrophic factor, phosphorylated mitogen-activated protein kinase, and phosphorylated cAMP response element-binding protein, and a live/dead cytotoxicity assay to evaluate cell survival. Their studies showed that norepinephrine, serotonin and three antidepressants were neuroprotective, and increased levels were seen in all the proteins measured. Intact pro-survival pathways were found to be more important for cell survival than the presence of external nutrients. Depressive-like symptoms in the mouse model were alleviated, and protein expression increased in the hippocampus and the cerebral cortex[48]. Taliaz et al[49] selectively knocked down brain-derived neurotrophic factor in the hippocampus; the hippocampal brain-derived neurotrophic factor knock-down mice displayed depressive-like behaviors, which were alleviated after administration of desipramine and citalopram. Adachi et al[50] proposed that brain-derived neurotrophic factor in the dentate gyrus might be critical in the treatment of depression. Furthermore, serum brain-derived neurotrophic factor levels in patients with major depression increased significantly after antidepressant treatment. Thus, peripheral brain-derived neurotrophic factor level could be taken as a biomarker for clinical efficacy[51,52].
Yasui-Furukori et al[53] assessed plasma brain-derived neurotrophic factor levels and paroxetine concentrations in 45 patients with major depression who were treated with paroxetine, using enzyme immunoassay and high-performance liquid chromatography. Changes in plasma brain-derived neurotrophic factor levels were correlated with plasma drug concentration after 2 and 6 weeks, but not after the first week of treatment. Freitas et al[54] found that fluoxetine regulates the hippocampal cell signaling pathways implicated in neuroplasticity in olfactory bulbectomized mice, by altering the activation of extracellular signal-regulated kinase 1/cAMP response element-binding protein/brain-derived neurotrophic factor in the hippocampus. Lee et al[41] demonstrated that serum or plasma brain-derived neurotrophic factor levels were decreased in untreated major depressive disorder patients. Antidepressant treatment lasting at least 4 weeks can restore the decreased brain-derived neurotrophic factor function to normal values. Brain-derived neurotrophic factor may also play an important role in the modulation of neuronal networks. Such a neuronal plastic change can positively influence mood or recover depressed mood. Brain-derived neurotrophic factor levels can therefore be useful markers for clinical response or improvement of depressive symptoms, but they are not diagnostic markers of major depression. Depressive patients frequently have altered cortisol secretion, which is related to impaired activity of the glucocorticoid receptor and the main regulator of the hypothalamic-pituitary adrenal axis (Figure 5)[55]. Glucocorticoids decrease the levels of brain-derived neurotrophic factor, and antidepressants increase the transcriptional activity of cAMP response element-binding protein, a regulator of brain-derived neurotrophic factor expression. Rojas et al[56] tried to find a biomarker to assess antidepressant action in depressed patients. Treated with venlafaxine, 34 depressed outpatients were assessed in this experiment. Levels of serum brain-derived neurotrophic factor and lymphocyte glucocorticoid receptor were found to be possible biomarkers that could be used to predict responses to venlafaxine treatment. Katsuki et al[57] studied 84 patients with major depressive disorder who were treated with mirtazapine for 4 weeks. The brain-derived neurotrophic factor Val66Met polymorphism was assessed by direct sequencing in the region, and levels of serum brain-derived neurotrophic factor and plasma catecholamine metabolites were measured by enzyme-linked immunosorbent assay and high-performance liquid chromatography with electrochemical detection, respectively. Serum levels of brain-derived neurotrophic factor, but not plasma levels of homovanillic acid or 3-methoxy 4-hydroxy phenyl glycol, and the brain-derived neurotrophic factor Val66Met polymorphism were associated with the mirtazapine response in major depressive disorder.
Figure 5.
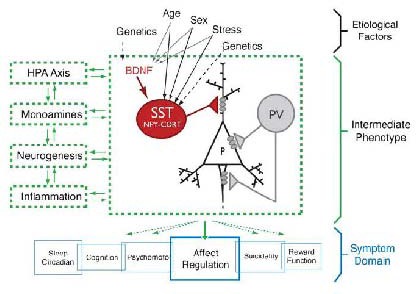
Proposed model of a brain-derived neurotrophic factor (BDNF)/somatostatin (SST)/gamma- aminobutyric acid (GABA) molecular intermediate phenotype within the complex multi-module pathophysiology of major depressive disorder (MDD)[55].
HPA: Hypothalamic-pituitary-adrenal; PV: parvalbumin.
Castrén et al[58] showed that stress and antidepressant drugs altered not only the level of brain-derived neurotrophic factor but also its activity in specific neural networks. The activity of brain-derived neurotrophic factor varied across encephalic regions and circuit loops. This could in turn affect the synaptic plasticity in these regions, and brain-derived neurotrophic factor injected directly into these regions could cause various behavioral changes.
Preclinical studies have shown that brain-derived neurotrophic factor can be measured through the blood-brain barrier, and serum brain-derived neurotrophic factor levels correlate positively with those in the cerebral cortex. Some studies have shown that serum brain-derived neurotrophic factor levels vary with the severity of depression. Tadić et al[59] performed a pilot study with 41 inpatients with major depressive disorder. Depression severity and serum brain-derived neurotrophic factor were measured weekly from baseline to week 6, using the 21-item Hamilton Depression Rating Scale and enzyme-linked immunosorbent assay. This study provided the first evidence that the absence of an early increase of serum brain-derived neurotrophic factor in conjunction with early non-improvement might be a highly specific peripheral marker predictive for treatment failure in patients with major depressive disorder. Okamoto et al[60] found that the serum level of brain-derived neurotrophic factor in patients who had received effective treatment increased significantly, but the patients for whom the treatment was ineffective had no change in serum brain-derived neurotrophic factor. Bay and Donders[61] hypothesized that the organism was in the broken compensation when people were in the long-term stress or in chronic nerve psychologic stress. The expression of brain-derived neurotrophic factor was downregulated after a series of middle factor accommodating leading to neuronal apoptosis. Vincze et al[62] found that serum brain-derived neurotrophic factor levels in depressed patients who had not taken antidepressants was low and correlated positively with the degree of depression. Calabrese et al[63] stated that brain-derived neurotrophic factor is a valuable marker of neuronal plasticity. Its expression is rapidly upregulated following acute stress, to preserve neuronal homeostasis. This kind of rapid reaction is the cell's acute defense mechanism. However, the expression of brain-derived neurotrophic factor becomes downregulated after chronic stress, and may lead to an impairment in neuronal plasticity. Other studies have indicated that a similar downregulation of brain-derived neurotrophic factor is observed in other psychiatric disorders, such as schizophrenia and Alzheimer's disease, meaning that brain-derived neurotrophic factor cannot be considered exclusively a biological marker of depression. Further research is required to identify a biological marker for major depression.
However, Martinowich et al[64] thought that brain-derived neurotrophic factor could reduce the reuptake of 5-hydroxytryptamine, thus increasing the extracellular concentration of 5-hydroxytryptamine. It is important to maintain a balance between brain-derived neurotrophic factor and 5-hydroxytryptamine. Aznar et al[65] found that antidepressant drugs can enhance the expression of brain-derived neurotrophic factor, which plays a role in synaptic plasticity in all kinds of neurons, including dopaminergic, serotonergic, cholinergic and adrenergic, and may influence the delivery of neurotransmitter to the synapse and its transmission to the postsynaptic cell. This may be the mechanism by which antidepressant action occurs. The brain-derived neurotrophic factor polymorphism Val66Met was not associated with serum brain-derived neurotrophic factor level, or with the response to selective serotonin reuptake inhibitors in depressed Japanese patients. A total of 132 patients were enrolled in Yoshimura's study[66]. These findings suggest that the brain-derived neurotrophic factor Val66Met polymorphism is independent of the response to treatment with selective serotonin reuptake inhibitors and serum brain-derived neurotrophic factor levels.
CONCLUSION
In summary, the extracellular signal-regulated kinase/cAMP response element-binding protein/brain-derived neurotrophic factor signal transduction pathway is very important in the pathogenesis of depression, and is activated by antidepressant drugs. The extracellular signal-regulated kinase/cAMP response element-binding protein/brain-derived neurotrophic factor pathway has potential as a biological marker in the diagnosis of depression and in the prevention of suicide, and might represent a new avenue of research in the production of antidepressants.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, No. 81202620, and the Domestic Visiting Scholar Program for Young Talent Teachers in University of Shandong Province.
Conflicts of interest: None declared.
(Edited by Yang Y/Song LP)
REFERENCES
- [1].Nuttall JR, Oteiza PI. Zinc and the ERK kinases in the developing brain. Neurotox Res. 2012;21(1):128–141. doi: 10.1007/s12640-011-9291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tronson NC, Schrick C, Fischer A, et al. Regulatory mechanisms of fear extinction and depression-like behavior. Neuropsychopharmacology. 2008;33(7):1570–1583. doi: 10.1038/sj.npp.1301550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rao JS, Ertley RN, Lee HT, et al. N-3 polyunsaturated fatty acid deprivation in rats decreases frontal cortex BDNF-via a p38-dependant mechamism. Mol Psychiatry. 2007;12(1):36–46. doi: 10.1038/sj.mp.4001888. [DOI] [PubMed] [Google Scholar]
- [4].Huang TY, Lin CH. Role of amygdale MAPK activation on immobility behavior of forced swim rats. Behav Brain Res. 2006;173(1):104–111. doi: 10.1016/j.bbr.2006.06.009. [DOI] [PubMed] [Google Scholar]
- [5].Hill RA. Interaction of sex steroid hormones and brain-derived neurotrophic factor-tyrosine kinase b signalling: relevance to schizophrenia and depression. J Neuroendocrinology. 2012;24(12):1553–1561. doi: 10.1111/j.1365-2826.2012.02365.x. [DOI] [PubMed] [Google Scholar]
- [6].Chambard JC, Lefloch R, Pouysségur J, et al. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773(8):1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- [7].Igaz LM, Winograd M, Cammarota M, et al. Early activation of extracellular signal-regulated kinase signaling pathway in the hippocampus is required for short-term memory formation of a fear-motivated learning. Cell Mol Neurobiol. 2006;26(4-6):989–1002. doi: 10.1007/s10571-006-9116-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5(3):173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- [9].Yuan P, Zhou R, Wang Y, et al. Altered levels of extracellular signal-regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J Affect Disord. 2010;124(1-2):164–169. doi: 10.1016/j.jad.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang JQ, Fibuch EE, Mao L. Regulation of mitogen-activated protein kinases by glutamate receptors. J Neurochem. 2007;100(1):1–11. doi: 10.1111/j.1471-4159.2006.04208.x. [DOI] [PubMed] [Google Scholar]
- [11].Zhang L, Xu T, Wang S, et al. Curcumin produces antidepressant effects via activating MAPK/ERK-dependent brain-derived neurotrophic factor expression in the amygdala of mice. Behav Brain Res. 2012;235(1):67–72. doi: 10.1016/j.bbr.2012.07.019. [DOI] [PubMed] [Google Scholar]
- [12].Di Benedetto B, Kühn R, Nothdurfter C, et al. N-desalkylquetiapine activates ERK1/2 to induce BDNF release in C6 glioma cells: A putative cellular mechanism for quetiapine as antidepressant. Neuropharmacology. 2012;62(1):209–216. doi: 10.1016/j.neuropharm.2011.07.001. [DOI] [PubMed] [Google Scholar]
- [13].Gourley SL, Wu FJ, Kiraly DD, et al. Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol Psychiatry. 2008;63(4):353–359. doi: 10.1016/j.biopsych.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006;59(12):1144–1150. doi: 10.1016/j.biopsych.2005.11.003. [DOI] [PubMed] [Google Scholar]
- [15].Qi X, Lin W, Li J, et al. Fluoxetine increases the activity of the ERK-CREB signal system and alleviates the depressive-like behavior in rats exposed to chronic forced swim stress. Neurobiol Dis. 2008;31(2):278–285. doi: 10.1016/j.nbd.2008.05.003. [DOI] [PubMed] [Google Scholar]
- [16].Gass P, Riva MA. CREB, neurogenesis and depression. Bioessays. 2007;29(10):957–961. doi: 10.1002/bies.20658. [DOI] [PubMed] [Google Scholar]
- [17].McCauslin CS, Heath V, Colangelo AM, et al. CAAT/Enhancer-binding protein delta and cAMP-response element-binding protein mediate inducible expression of the nerve growth factor gene in the central nervous system. J Biol Chem. 2006;281(26):17681–17688. doi: 10.1074/jbc.M600207200. [DOI] [PubMed] [Google Scholar]
- [18].Iga J, Ueno S, Yamauchi K, et al. Altered HDAC5 and CREB mRNA expressions in the peripheral leukocytes of major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):628–632. doi: 10.1016/j.pnpbp.2006.12.014. [DOI] [PubMed] [Google Scholar]
- [19].Lu J, Yang XY, Hua X, et al. cAMP response element binding protein: convergence of antidepressant signaling cascades. Shengli Kexue Jinzhan. 2008;39(4):371–374. [PubMed] [Google Scholar]
- [20].Dinieri JA, Nemeth CL, Parsegian A, et al. Altered sensitivity to rewarding and aversive drugs in mice with inducible disruption of cAMP response element-binding protein function within the nucleus accumbens. J Neurosci. 2009;29(6):1855–1859. doi: 10.1523/JNEUROSCI.5104-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wallace DL, Han MH, Graham DL, et al. CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nat Neurosci. 2009;12(2):200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Muschamp JW, Van’t Veer A, Parsegian A, et al. Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci. 2011;31(8):3095–3103. doi: 10.1523/JNEUROSCI.5973-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zheng H, Ma GY, Fu XC. Effect of Paroxetine on space learning memory and hippocampus ERK-CREB pathway in the rat model of depression. Zhongguo Yaoxue Zazhi. 2008;43(16):1234–1238. [Google Scholar]
- [24].Tardito D, Perez J, Tiraboschi E, et al. Signaling pathways regulating gene expression neuroplasticity and neurotrophic mechanisms in the action of antidepressants a critical overview. Pharmacol Rev. 2006;58(1):115–134. doi: 10.1124/pr.58.1.7. [DOI] [PubMed] [Google Scholar]
- [25].Gur TL, Conti AC, Holden J, et al. cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J Neurosci. 2007;27(29):7860–7868. doi: 10.1523/JNEUROSCI.2051-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Serretti A, Chiesa A, Calati R, et al. A preliminary investigation of the influence of CREB1 gene on treatment resistance in major depression. J Affect Disord. 2011;128(1-2):56–63. doi: 10.1016/j.jad.2010.06.025. [DOI] [PubMed] [Google Scholar]
- [27].Crisafulli C, Shim DS, Andrisano C, et al. Case-control association study of 14 variants of CREB1, CREBBP and CREM on diagnosis and treatment outcome in major depressive disorder and bipolar disorder. Psychiatry Res. 2012;198(1):39–46. doi: 10.1016/j.psychres.2011.08.022. [DOI] [PubMed] [Google Scholar]
- [28].Yu H, Chen ZY. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol Sin. 2011;32(1):3–11. doi: 10.1038/aps.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Peng CH, Chiou SH, Chen SJ, et al. Neuroprotection by imipramine against lipo- polysaccharide-induced apoptosis in hippocampus derived neural stem cells mediated by activation of BDNF and the MAPK pathway. Eur Neuropsychopharmacol. 2008;18(2):128–140. doi: 10.1016/j.euroneuro.2007.05.002. [DOI] [PubMed] [Google Scholar]
- [30].Gonul AS, Kitis O, Eker MC, et al. Association of the brain-derived neurotrophic factor Val66Met polymorphism with hippocampus volumes in drug-free depressed patients. World J Biol Psychiatry. 2011;12(2):110–118. doi: 10.3109/15622975.2010.507786. [DOI] [PubMed] [Google Scholar]
- [31].Duncan LE, Hutchison KE, Carey G, et al. Variation in brain-derived neurotrophic factor (BDNF) gene is associated with symptoms of depression. J Affect Disord. 2009;115(1-2):215–219. doi: 10.1016/j.jad.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Suchanek R, Owczarek A, Kowalczyk M, et al. Association between C-281A and val66met functional polymorphisms of BDNF gene and risk of recurrent major depressive disorder in Polish population. J Mol Neurosci. 2011;43(3):524–530. doi: 10.1007/s12031-010-9478-y. [DOI] [PubMed] [Google Scholar]
- [33].Czira ME, Wersching H, Baune BT, et al. Brain-derived neurotrophic factor gene polymorphisms, neurotransmitter levels, and depressive symptoms in an elderly population. Age (Dordr) 2012;34(6):1529–1541. doi: 10.1007/s11357-011-9313-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang K, Yang C, Xu Y, et al. Genetic association of the interaction between he BDNF and GSK genes and major depressive disorder in a Chinese population. J Neural Transm. 2010;117(3):393–401. doi: 10.1007/s00702-009-0360-4. [DOI] [PubMed] [Google Scholar]
- [35].Domschke K, Lawford B, Laje G, et al. Brain-derived neurotrophic factor (BDNF) gene: no major impact on antidepressant treatment response. Int J Neuropsychopharmacol. 2010;13(1):93–101. doi: 10.1017/S1461145709000030. [DOI] [PubMed] [Google Scholar]
- [36].Juhasz G, Dunham JS, McKie S, et al. The CREB1-BDNF-NTRK2 pathway in depression: multiple genecognition-environment interactions. Biol Psychiatry. 2011;69(8):762–771. doi: 10.1016/j.biopsych.2010.11.019. [DOI] [PubMed] [Google Scholar]
- [37].Sarchiapone M, Carli V, Roy A, et al. Association of polymorphism (Val66Met) of brain-derived neurotrophic factor with suicide attempts in depressed patients. Neuropsychobiology. 2008;57(3):139–145. doi: 10.1159/000142361. [DOI] [PubMed] [Google Scholar]
- [38].Schenkel LC, Segal J, Becker JA, et al. The BDNF Val66Met polymorphism is an independent risk factor for high lethality in suicide attempts of depressed patients. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(6):940–944. doi: 10.1016/j.pnpbp.2010.04.023. [DOI] [PubMed] [Google Scholar]
- [39].Joffe RT, Gatt JM, Kemp AH, et al. Brain derived neurotrophic factor Val66Met polymorphism, the five factor model of personality and hippocampal volume: Implications for depressive illness. Hum Brain Mapp. 2009;30(4):1246–1256. doi: 10.1002/hbm.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee BH, Kim YK. BDNF mRNA expression of peripheral blood mononuclear cells was decreased in depressive patients who had or had not recently attempted suicide. J Affect Disord. 2010;125(1-3):369–373. doi: 10.1016/j.jad.2010.01.074. [DOI] [PubMed] [Google Scholar]
- [41].Lee BH, Kim YK. The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Investig. 2010;7(4):231–235. doi: 10.4306/pi.2010.7.4.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rajewska-Rager A, Skibińska M, Szczepankiewicz A, et al. Association between polymorphisms of Val66Net in the BDNF gene and the response to escitalpram and nortriptyline treatment in the light the neurodevelopmental hypothesis of depression. Psychiatr Pol. 2008;42(6):915–923. [PubMed] [Google Scholar]
- [43].Domschke K, Lawford B, Laje G, et al. Brain-derived neurotrophic-factor (BDNF) gene no major in pact on antidepressant treatment response. Int J Neuropsychopharmacol. 2010;13(1):93–101. doi: 10.1017/S1461145709000030. [DOI] [PubMed] [Google Scholar]
- [44].Molteni R, Calabrese F, Chourbaji S, et al. Depression-prone mice with reduced glucocorticoid receptor expression display an altered stress-dependent regulation of brain-derived neurotrophic factor and activity-regulated cytoskeleton-associated protein. J Psychopharmacol. 2010;24(4):595–603. doi: 10.1177/0269881108099815. [DOI] [PubMed] [Google Scholar]
- [45].Castrén E, Rantamaki T. Role of brain-derived neurotrophic factor in the aetiology of depression: implications for pharmacological treatment. CNS Drugs. 2010;24(1):1–7. doi: 10.2165/11530010-000000000-00000. [DOI] [PubMed] [Google Scholar]
- [46].Buchmann AF, Hellweg R, Rietschel M, et al. BDNF Val 66 Met and 5-HTTLPR genotype moderate the impact of early psychosocial adversity on plasma brain-derived neurotrophic factor and depressive symptoms: A prospective study. Eur Neuropsychopharmacol. doi: 10.1016/j.euroneuro.2012.09.003. in press. [DOI] [PubMed] [Google Scholar]
- [47].Yang D, Chen M, Russo-Neustadt A. Antidepressants are neuroprotective against nutrient deprivation stress in rat hippocampal neurons. Eur J Neurosci. 2012;36(5):2573–2587. doi: 10.1111/j.1460-9568.2012.08187.x. [DOI] [PubMed] [Google Scholar]
- [48].Antunes PB, Rosa MA, Belmonte-de-Abreu PS, et al. Electroconvulsive therapy in major depression current aspects. Rev Bras Psiquiatr. 2009;31(Suppl 1):S26–33. doi: 10.1590/s1516-44462009000500005. [DOI] [PubMed] [Google Scholar]
- [49].Taliaz D, Stall N, Dar DE, et al. Knockdown of brain-derived neurotrophic factor in specific brain sites precipitates behaviors associated with depression and reduces neurogenesis. Mol Psychiatry. 2010;15(1):80–92. doi: 10.1038/mp.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Adachi M, Barrot M, Autry AE, et al. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry. 2008;63(7):642–649. doi: 10.1016/j.biopsych.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Matrisciano F, Bonaccorso S, Ricciardi A, et al. Changes in BDNF serum levels in patients with major depression disorder (MDD) after 6 months treatment with sertraline, escitalopram, or venlafaxine. J Psychiatr Res. 2009;43(3):247–254. doi: 10.1016/j.jpsychires.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169–1180. doi: 10.1017/S1461145708009309. [DOI] [PubMed] [Google Scholar]
- [53].Yasui-Furukori N, Tsuchimine S, Nakagami T, et al. Association between plasma paroxetine concentration and changes in plasma brain-derived neurotrophic factor levels in patients with major depressive disorder. Hum Psychopharmacol. 2011;26(3):194–200. doi: 10.1002/hup.1192. [DOI] [PubMed] [Google Scholar]
- [54].Freitas AE, Machado DG, Budni J, et al. Fluoxetine modulates hippocampal cell signaling pathways implicated in neuroplasticity in olfactory bulbectomized mice. Behav Brain Res. 2013;237:176–184. doi: 10.1016/j.bbr.2012.09.035. [DOI] [PubMed] [Google Scholar]
- [55].Guilloux JP, Douillard-Guilloux G, Kota R, et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2012;17(11):1130–1142. doi: 10.1038/mp.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rojas PS, Fritsch R, Rojas RA, et al. Serum brain-derived neurotrophic factor and glucocorticoid receptor levels in lymphocytes as markers of antidepressant response in major depressive patients: A pilot study. Psychiatry Res. 2011;189(2):239–245. doi: 10.1016/j.psychres.2011.04.032. [DOI] [PubMed] [Google Scholar]
- [57].Katsuki A, Yoshimura R, Kishi T, et al. Serum levels of brain-derived neurotrophic factor (BDNF), BDNF gene Val66Met polymorphism, or plasma catecholamine metabolites, and response to mirtazapine in Japanese patients with major depressive disorder (MDD) CNS Spectr. 2012;17(3):155–163. doi: 10.1017/S109285291200051X. [DOI] [PubMed] [Google Scholar]
- [58].Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev Neurobiol. 2010;70(5):289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- [59].Tadić A, Wagner S, Schlicht KF, et al. The early non-increase of serum BDNF predicts failure of antidepressant treatment in patients with major depression: A pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(2):415–420. doi: 10.1016/j.pnpbp.2010.08.011. [DOI] [PubMed] [Google Scholar]
- [60].Okamoto T, Yoshimura R, Ikenouchi-Sugita A, et al. Efficacy of electroconvulsive therapy is associated with changing blood levels of homovanillic acid and brain-derived neurotrophic factor (BDNF) in refractory depressed patients: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(5):1185–1190. doi: 10.1016/j.pnpbp.2008.02.009. [DOI] [PubMed] [Google Scholar]
- [61].Bay E, Donders J. Risk factors for depressive symptoms after-mild-to-moderate traumatic brain injury. Brain Inj. 2008;22(3):233–241. doi: 10.1080/02699050801953073. [DOI] [PubMed] [Google Scholar]
- [62].Vincze I, Perroud N, Buresi C, et al. Association between brain-derived neurotrophic factor gene and a severe form of bipolar disorder, but no interaction with the seroton transporter gene. Bipolar Disord. 2008;10(5):580–587. doi: 10.1111/j.1399-5618.2008.00603.x. [DOI] [PubMed] [Google Scholar]
- [63].Calabrese F, Molteni R, Racagni G, et al. Neuronal plasticity: A link between stress and mood disorders. Psychoneuroendocrinology. 2009;34(Suppl 1):S208–216. doi: 10.1016/j.psyneuen.2009.05.014. [DOI] [PubMed] [Google Scholar]
- [64].Martinowich K, Lu B. Interaction between BDNF and serotonin role in mood disorders. Neuropsychopharmacology. 2008;33(1):73–83. doi: 10.1038/sj.npp.1301571. [DOI] [PubMed] [Google Scholar]
- [65].Aznar S, Klein AB, Santini MA, et al. Aging and depression vulnerability interaction results in decreased serotonin innervation associated with reduced BDNF levels in hippocampus of rats bred for learned helplessness. Synapse. 2010;64(7):561–565. doi: 10.1002/syn.20773. [DOI] [PubMed] [Google Scholar]
- [66].Yoshimura R, Kishi T, Suzuki A, et al. The brain-derived neurotrophic factor (BDNF) polymorphism Val66Met is associated with neither serum BDNF level nor response to selective serotonin reuptake inhibitors in depressed Japanese patients. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(4):1022–1025. doi: 10.1016/j.pnpbp.2011.02.009. [DOI] [PubMed] [Google Scholar]