

Abstract
Mitochondria play an important role in neuronal apoptosis caused by cerebral ischemia, and the role is mediated by the expression of mitochondrial proteins. This study investigated the involvement of mitochondrial proteins in hippocampal cell apoptosis after transient cerebral ischemia-reperfusion injury in aged rats using a comparative proteomics strategy. Our experimental results show that the aged rat brain is sensitive to ischemia-reperfusion injury and that transient ischemia led to cell apoptosis in the hippocampus and changes in memory and cognition of aged rats. Differential proteomics analysis suggested that this phenomenon may be mediated by mitochondrial proteins associated with energy metabolism and apoptosis in aged rats. This study provides potential drug targets for the treatment of transient cerebral ischemia-reperfusion injury.
Keywords: nerve regeneration, cerebral ischemia, reperfusion injury, hippocampus, cognitive function, apoptosis, mitochondria, differential proteomics, rats, aged, neural regeneration
Introduction
Ischemia may cause neuronal necrosis and apoptosis; long-term cerebral ischemia triggers necrosis, while short-term cerebral ischemia triggers apoptosis, which plays a crucial role in delayed neuronal death (Muller et al., 2004; Ahlemeyer et al., 2012). Previous studies have mainly investigated ischemia-reperfusion injury in young animals; however, cerebral vascular trauma is more prevalent in aged people owing to the high vulnerability to ischemic injury and the decline in protection against cerebral ischemia. Whether transient cerebral ischemia conclusively induces neurological impairment in the brain of aged people remains unclear.
Mitochondria play an important role in neuronal apoptosis caused by cerebral ischemia, and the role is mediated by the expression of mitochondrial proteins. These proteins may also play a crucial role in the prevention and treatment of ischemic neuronal injury, and are considered ideal drug targets (Zhang et al.,1995; Cwerman et al., 2012). The rise of proteomics provides a new strategy to investigate ischemia-reperfusion injury mechanisms, and differential analysis of the mitochondrial proteome under different pathological conditions may help our understanding of this complex process (Datta, 2010; Scornavacca et al., 2012).
This study investigated the effect of short-term cerebral ischemia-reperfusion injury on cognitive function and cell apoptosis in the hippocampus of aged rats. In addition, to further study the mechanism of transient cerebral ischemic injury and to identify potential drug targets, a comparative proteomics analysis was performed to explore mitochondrial protein expression in the hippocampus of aged rats after transient ischemic injury.
Materials and Methods
Experimental animals
Forty healthy, male, Wistar rats, aged 19–21 months, weighing 520–750 g, were provided by the Experimental Animal Institute of Changsha, China (license No. SCXK (Xiang) 2009-0012). Animals were housed at 20–25°C in 40–70% humidity, on a 12-hour light/dark cycle, with ad libitum access to food and water. All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23).
Establishment of transient cerebral ischemia model
The animals were equally and randomly divided into control and ischemia groups. The transient global cerebral ischemia model was established in the ischemia group using the four-vessel occlusion method, as previously described (Pulsinelli and Brierley, 1979). In brief, rats were anesthetized with 10% chloral hydrate (0.4 mL/100 g) via intraperitoneal injection and fixed in the prone position. The neck hair was shaved and the skin disinfected. An incision was made between the first and second cervical vertebrae, and the first cervical vertebra bilateral foramen alares were exposed. The downward vertebral artery was cauterized two or three times using a self-made cauterizer, then the incision was sutured. The rats were then fixed in the supine position and a median neck incision was cut along the ventral midline between the two forelimbs, exposing the bilateral carotid sheath. The bilateral common carotid arteries were then exposed and threads were bilaterally placed and then crossed to form a loop. A needle electrode was inserted subcutaneously into the head and connected to an electroencephalograph (EEG) for EEG monitoring. The thread loop was then lifted to expose the bilateral common carotid arteries, which were then occluded using a noninvasive vascular clamp to produce cerebral ischemia. According to a preliminary experimental study (Ji et al., 2006), the ischemia time was predetermined at 5 minutes. After 5 minutes, vascular clamps were removed to restore blood supply in the common carotid artery, initiating reperfusion. The incision was then sutured. During surgery, rats were illuminated under an incandescent lamp, and rectal temperature was maintained at 36.5–37.5°C. The model establishment procedure is shown in Figure 1.
Figure 1.
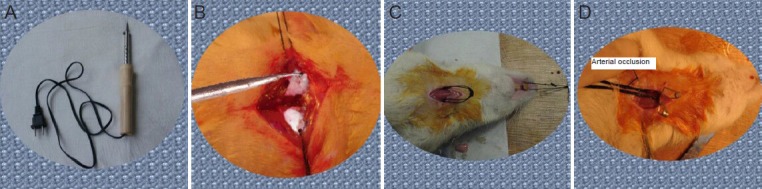
Establishing the transient global cerebral ischemia model in rats.
(A) Self-made cauterizer. (B) Vertebral artery was cauterized using the self-made cauterizer. (C) Threads were placed under the common carotid arter-ies and crossed to form thread loop. (D) Bilateral carotid arteries were occluded using a noninvasive vascular clamp to produce cerebral ischemia.
Criteria for establishment of the model: The success of model was defined by the appearance of white color of the eyeball, mydriasis, loss of righting reflex, a linear wave in the electroencephalogram, and loss of consciousness immediately after bilateral carotid artery occlusion (within 30–60 seconds). Animals in which the model failed were excluded from the experiment. In the control group, the bilateral foramen alares were exposed but the vertebral artery was not cauterized, and the bilateral carotid arteries were exposed and the threads were inserted, but the arteries were not clamped. The remaining steps were the same as those in the ischemia group.
Behavioral assessment
On days 2–7 after model establishment, rats were behaviorally assessed in a Morris water maze (Mulder and Pritchett, 2003). The Morris water maze was composed of a water tank (diameter 130 cm, height 50 cm), an automatic recording device, and a computer analysis device. The water depth was 30 cm (1 cm above the platform). The experiment was performed in a dark, soundproof room at 26 ± 1°C. The water maze was divided into four quadrants, and the entry point was in the quadrant opposite that containing the platform. The escape latency was recorded as the time from entering the water to finding the platform. The rats were tested twice per day, once in the morning and again in the afternoon, for 5 days. The platform was withdrawn after the final test. The time for rats to find the platform was recorded as the learning latency. Those that failed to find the platform were recorded as 120 seconds.
Pathological analysis
Harvesting specimens
After the behavioral tests were completed, ten rats in each group were randomly selected and anesthetized by intraperitoneal injection of 10% chloral hydrate (0.5 mL/100 g), and a thoracoabdominal incision was made to expose the heart. Then an infusion needle was inserted into the apex of the right atrium for perfusion of 500 mL of saline and 4% paraformaldehyde phosphate buffer at 4°C at a slow speed for 2 hours. Subsequently, the brain was harvested and fixed in paraformaldehyde for 24 hours. After fixation, a coronal slice was cut 1–4 mm posterior to the optic chiasm and rinsed in phosphate buffer for 4 hours. The slice was then dehydrated in an ethanol gradient, cleared in xylene, and embedded in paraffin. The slice was then cut into serial 5 μm coronal sections and stored at room temperature until use.
Hematoxylin-eosin staining
Three paraffin sections from each rat were conventionally dewaxed, stained with Harris hematoxylin solution for 5 minutes, differentiated with 75% hydrochloric acid and ethanol for 30 seconds, and acidified using eosin ethanol for 1–2 minutes. Afterwards, conventional dehydration, clearing, and mounting were performed. The morphology of hippocampal neurons was observed under light microscopy (BH-2 type, Olympus, Tokyo, Japan).
Apoptosis in the hippocampus detected by TUNEL staining
The detection of apoptosis was performed using a terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) kit according to the manufacturer's instructions (Wuhan Boster Biotechnology, Wuhan, China). In brief, paraffin sections were conventionally hydrated through a series of ethanol washes, and treated with freshly prepared 3% H2O2 at room temperature for 10 minutes. The sections were then digested with proteinase K (1:200) at 37°C for 10 minutes and then incubated with 18 μL labeling buffer containing 1 μL TdT and 1 μL DIG-d-UTP in a humidified box at 37°C for 2 hours. Thereafter, the samples were treated with blocking solution, 50 μL/slice, at room temperature for 30 minutes, then with biotinylated anti-digoxin antibody (1:200), 50 μL/slice, for 30 minutes, followed by SABC (1:100), 50 μL/slice, for 30 minutes at 37°C. Finally, the sections were developed with 3,3′-diaminobenzidine. Three successive sections from each rat, 10 rats per group, were selected. Two non-overlapping fields of view in the hippocampal CA1 region were randomly selected from each section under light microscopy (Olympus, Shinjuku-ku, Tokyo, Japan) at 200× magnification. TUNEL-positive cells were identified by brown-yellow granules in the nucleus and were counted. The results are expressed as the number of TUNEL-positive cells in a 200 × magnification field of view.
Proteomic analysis of differentially expressed mitochondrial proteins
Sample preparation
After behavioral testing was complete, the remaining ten rats in each group were decapitated and the hippocampus was harvested on ice.
Purification of hippocampal mitochondria
Mitochondria were purified from the hippocampus as previously described (Jiang et al., 2005). Briefly, the hippocampus was harvested and immediately placed in separation medium (10 mmol/L Tris-HCl, 5 mmol/L K+-EDTA, 0.25 mol/L sucrose, pH 7.4, 4°C). The tissues were homogenized in a homogenizer and subjected to Ficoll density gradient centrifugation. The mitochondria-containing precipitates were then resuspended in 2.4 mL 25% Nycodenz and centrifuged in an ultracentrifugation tube in the following discontinuous Nycodenz density gradient: 1 mL 34%, 1.6 mL 25%, 2.4 mL 25%, 1.6 mL 23%, 0.6 mL 25%, at 4°C for 90 minutes, at 52,000 × g. The mitochondria between 20 and 30% were recovered and dissolved in Nycodenz separation medium (5 mmol/L Tris-HCl, 1 mmol/L Na+-EDTA, 0.5 mmol/L EGTA, 0.25 mol/L sucrose, pH 7.4, 4°C), then centrifuged for 10 minutes at 10,000 × g. The resulting precipitates contained the purified mitochondria.
Observation of purified mitochondria by transmission electron microscopy
The mitochondrial precipitates were placed overnight in PBS containing 2.5% glutaraldehyde (pH 7.4) at 4°C, then rinsed with PBS three times, fixed in 1% osmium tetroxide for 1.5 hours, rinsed once with PBS, dehydrated in a graded series of ethanol and embedded in epoxy resin. The mitochondria were then cut into ultrathin slices at a thickness of 70 nm. The slices were observed under transmission electron microscopy (JEX-1200EM type, Olympus) after lead citrate and uranyl acetate staining (Phillips, 1998).
Preparation of protein samples for two-dimensional electrophoresis
Mitochondrial precipitates were incubated with protein lysate cocktail (2 mol/L thiourea, 7 mol/L urea, 40 mmol/L Tris-base, 4% CHAPS, 1% DTT, 1 mmol/L EDTA,) in an ice bath for 30 minutes and then subjected to ultrasound treatment on ice for 1 minute. Samples were then centrifuged for 1 hour at 4°C, 25,000 × g and the supernatant containing total mitochondrial protein harvested and stored at –80°C.
Two-dimensional electrophoresis and gel image acquisition and analysis
150 μg of mitochondrial protein sample was made up to a volume to 350 μL using hydration solution. To prepare the first isoelectric focusing system, the protein samples were obtained using the swelling method (Görg et al., 2004). The strips were balanced immediately after one-dimensional electrophoresis, and then transferred to a 13% SDS gel for two-dimensional electrophoresis. The analysis gel was stained with silver nitrate and the preparation gel was stained with Coomassie blue (Görg et al., 2004). The stained stripes were scanned with an image scanner and the difference between the groups was compared using ImageMaster 2D Platinum Software (General Electric Company, Fairfield, CT, USA). A differentially expressed protein was defined to exhibit a greater than three times change in protein level (upregulation or downregulation) and to exhibit clear boundaries, without any interference.
Identification of differentially expressed proteins by mass spectrometry
The differentially expressed protein spots were successively subjected to particle bleaching, enzyme digestion, peptide extraction, resin filtration, desalination and filtration, and finally matrix-assisted laser desorption ionization-time of flight mass spectrometry analysis to produce peptide mass finger print maps. The significance of the results was defined as an expectation < 0.5 with determination of an apparent molecular weight and isoelectric point.
Statistical analysis
Data are expressed as mean ± SD and were analyzed using SPSS 11.1 software (SPSS, Chicago, IL, USA). The difference between groups was compared using one-way analysis of variance and the least significant difference test. A level of P < 0.05 was considered statistically significant.
Results
Cognitive function of aged rats with transient cerebral ischemia
Morris water maze test results showed that the cognitive function of aged rats with transient cerebral ischemia was declined compared with that of control rats (P < 0.05, P < 0.01; Table 1).
Table 1.
Morris water maze test results in aged rats with transient cerebral ischemia
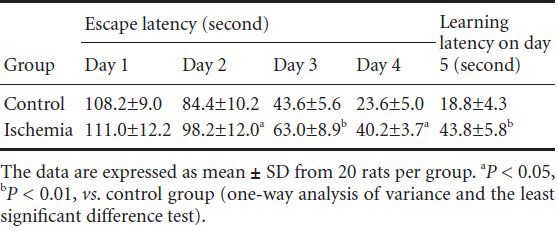
Morphology of hippocampal neurons
In the control group, hippocampal neurons exhibited normal and slightly cone-shaped morphology; the nuclear membrane was clearly visible, the chromatin was uniform and the space surrounding cells and capillaries was normal with dense stroma. A small number of pyramidal cells were slightly shrunken, with condensed and deeply stained nuclear chromatin. In the ischemia group, the number of normal neurons in the hippocampal CA1 region was reduced, neuronal degeneration was seen and some neurons and glial cells were significantly swollen and had round cell bodies. The space surrounding cells and capillaries was widened, and interstitial edema and pyknosis were seen (Figure 2).
Figure 2.
Hematoxylin-eosin staining of rat hippocampus in the two groups (× 200).
(A) In the control group, only a small number of hippocampal pyra-midal cells were shrunken with condensed and deeply stained nuclear chromatin (arrows). (B) In the ischemia group, the majority of hippo-campal neurons exhibited pyknosis and deep staining (arrows). Some neurons and glial cells were significantly swollen and were round, and the space surrounding cells and capillaries was widened.
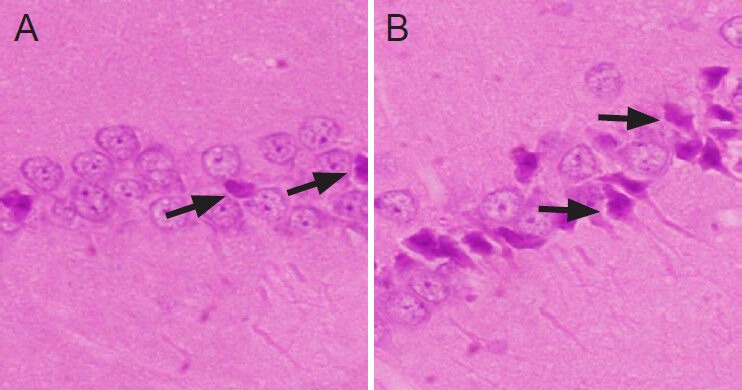
Apoptotic cells in rat hippocampus
TUNEL staining showed that in the control group, a small number of hippocampal cells were apoptotic, and that the number of apoptotic cells was significantly increased in the ischemia group (Figure 3). The number of apoptotic cells counted in high-power fields was 3.21 ± 3.76 and 20.50 ± 5.83 in the control and ischemia groups, respectively, and these counts were significantly different (P < 0.01).
Figure 3.
Cell apoptosis in the hippocampus of rats after cerebral ischemia-reperfusion injury (× 200).
The TUNEL-positive apoptotic cells contained brown granules. In the control group (A), a small number of hippocampal cells were apoptot-ic, while the number of apoptotic cells was significantly increased in the ischemia group (B).
Mitochondrial purification
Transmission electron microscopy confirmed the extracted precipitates to be mainly mitochondria, which were oval or round shaped. The majority of mitochondria maintained an intact ridge structure and an intact membrane structure, while a few exhibited swelling and sparse ridges. The mitochondrial purity was over 97%.
Two-dimensional gel electrophoresis of mitochondrial proteins
The electrophoresis chromatogram revealed similar protein distribution patterns between ischemia and control groups. Most proteins were located in the PI 4-8 region, and 300–380 protein spots were detected (Figure 4). The matching rate between the two groups was 73 ± 6.3% (n = 15). Ten spots showed significant difference in protein abundance between ischemia and control groups. Among these 10 spots, the expression was increased in the ischemia group in seven and decreased in three.
Figure 4.
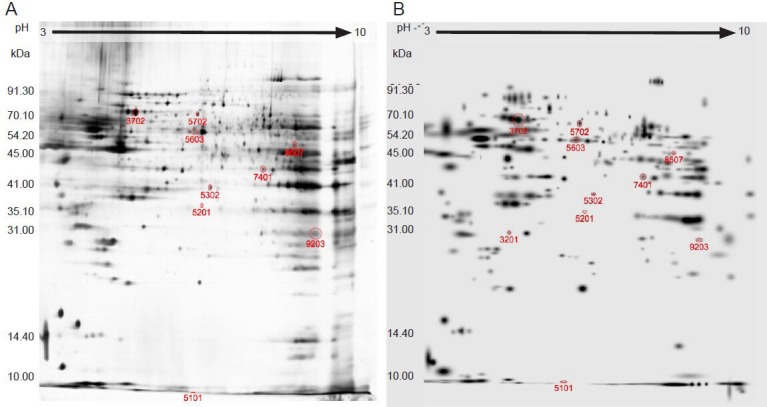
Electrophoresis chromatogram of mitochondrial proteins in the two groups (silver staining).
(A) Control group; (B) ischemia group.
Mass spectrogram of mitochondrial proteins
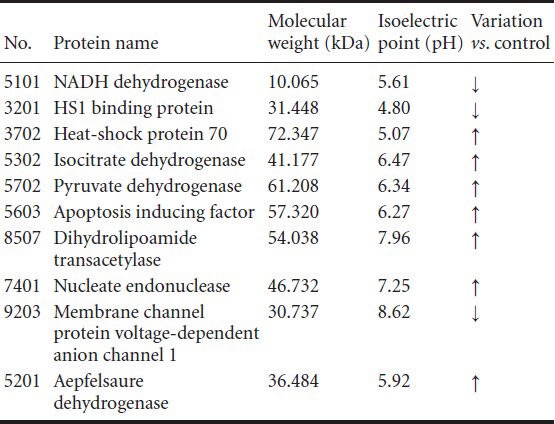
The 10 differential protein spots were analyzed with matrix-assisted laser desorption ionization-time of flight mass spectrometry, and the obtained PMF spectrum was retrieved using a Mascot Distiller search of the NCBI nr database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Ten proteins were successfully identified (Table 2).
Table 2.
Ten differential protein spots identified successfully
Discussion
To investigate the pathophysiology, pathogenesis, and treatment methods of ischemic cerebrovascular disease, selection and application of a reliable animal model of the disease is very important (Krafft et al., 2012). The global-brain ischemia model (four-vessel occlusion) is a recognized model for global cerebral ischemia, especially for delayed neuronal injury. Many scholars have adopted an electrocoagulation method to block vertebral arteries; however, this operation is likely to cause spinal cord and brain stem injury (McBean et al., 1998). To avoid these shortcomings, we modified the cauterization method, using a 30 W self-made electric cauterizer to block the vertebral arteries. Preliminary experiments and this study both found that the modified cauterization method achieved a success rate of over 80%, which compares favorably with the 30–45% success rate of the traditional coagulation method (Pulsinelli and Buchan, 1988; Zhao and Su, 1996; Jiang et al., 2005). Therefore, this method is simple and repeatable.
In this study, we used aged rats, which exhibit comparable cerebrovascular disease symptoms to those seen in the clinic. Apoptosis is one of the characteristics of aging (Salminen et al., 2011) and the present study showed that the number of apoptotic cells in the hippocampus was significantly increased in rats with cerebral ischemia compared with that in the control group. The behavioral analysis showed both escape latency and memory latency were prolonged in ischemia rats, indicating that cognition and memory abilities were affected. The present study confirmed the aged rat brain is sensitive to ischemia-reperfusion injury, and that the safe time limit for cerebral ischemia is less than traditionally thought (5 minutes) (Allen and Buckberg, 2012); A 5 minute bout of ischemia increased neuronal apoptosis in the hippocampus of aged rats and led to changes in memory and cognition. This evidence suggests that the safe time limit of cerebral ischemia should be carefully considered after accounting for apoptosis. However, the mechanism of increased sensitivity and targets for the prevention and treatment of transient cerebral ischemia remain unclear.
In recent years, advances in proteomics technology have allowed the comprehensive understanding of mitochondrial proteome changes during physiological and pathological processes. This has enabled the study of differential expression of mitochondrial proteins after ischemia (Datta, 2010; Kumari et al., 2012; Scornavacca et al., 2012). In this study, we identified 12 protein spots that were differentially expressed by at least three-fold. These proteins were identified and are highly involved in energy metabolism and apoptosis (Villa et al., 2009); NADH dehydrogenase, isocitrate dehydrogenase, pyruvate dehydrogenase, malate dehydrogenase, mitochondrial creatine kinase isoenzyme and dihydrolipoamide transacetylase are closely related to the respiratory chain or the tricarboxylic acid cycle in energy metabolism, while cytochrome c oxidase, heat shock protein 70, membrane channel protein voltage-dependent anion channel 1, apoptosis-inducing factor, nucleate endonuclease G, and HS1 binding protein are mainly involved in apoptosis.
The present study showed that brain energy metabolism was altered in aged rats after cerebral ischemic injury, with the following manifestations: (1) The expression of proteases involved in oxidative phosphorylation complexes, such as NADH dehydrogenase, was downregulated, while that of mitochondrial creatine kinase isoenzyme was upregulated. The oxidative phosphorylation process is mainly carried out on the inner membrane of mitochondria, so its functional activity directly affects mitochondrial ATP synthesis. This change will lead to a reduction of mitochondrial ATP synthesis and an increased production of reactive oxygen species (James et al., 2012). This may lead to a lack of compensatory energy after cerebral ischemia, and the mechanism needs further elucidation. (2) In this study, we found increased expression in the ischemia group of malate dehydrogenase, isocitrate dehydrogenase, pyruvate dehydrogenase and dihydrolipoamide transacetylase, which are involved in the tricarboxylic acid cycle. These findings confirm the presence of energy metabolism disruption at the acute cerebral ischemic stage and the need for more energy for damaged neurons and key proteins in the compensatory energy metabolic pathways.
There are three signal transduction pathways involved in apoptosis, namely the mitochondrial pathway, the death receptor pathway and the endoplasmic reticulum pathway. Among them, the mitochondrial pathway plays a dominant role (James et al., 2012; Kumari et al., 2012). As the site for cellular energy synthesis and storage, substance metabolism and energy conversion, mitochondria play a crucial role in the regulation and execution of apoptosis (Villa, 2009). In this study, compared with the control group, the expression of apoptosis-related proteins was increased in the ischemia group. (1) Cytochrome c is located in the outer membrane, and is closely linked with the apoptosis. After mitochondrial damage occurs, cytochrome c serves as a sensor and is released into the cytoplasm and ATP synthesis in the impaired mitochondrial respiratory chain is inhibited, thus triggering apoptosis (Cali, 2012). (2) HAX-1 binding protein is expressed at low levels. Accumulating evidence suggests that HAX-1 in several cells and viruses interacts with anti-apoptotic proteins, such as Bcl-2, to promote cell survival and regulate apoptosis (Diaz-Moreno et al., 2011; Simmen, 2011). (3) Voltage-dependent anion channels are located on the outer mitochondrial membrane, and can inhibit mitochondrial function, resulting in changes of mitochondrial membrane permeability (Vafiadaki et al., 2009). Our findings revealed that the voltage-dependent anion channel content was decreased in aged rats after transient cerebral ischemia, which was consistent with decrease ATP/ADP exchange, thus affecting mitochondrial energy supply and mitochondria-mediated apoptosis. (4) Apoptosis-inducing factor and nucleate endonuclease expression was increased. Apoptosis-inducing factor is a recently discovered cytokine that plays an important role in neuronal apoptosis after cerebral ischemia (De Stefani et al., 2012). Apoptosis-inducing factor-dependent apoptosis is an important non-caspase-dependent, apoptotic pathway widespread in eukaryotic cells. (5) Expression of heat shock protein 70 is increased, which can reduce apoptosis (Oxler et al., 2012). Expression of heat shock protein 70 determines the degree of nerve cell damage after hypoxia and ischemia, while its expression also benefits the repair of nerve cells after injury (Lee et al., 2004; Chaitanya and Babu, 2008). The present study indicated that the oxidative stress reaction was enhanced after cerebral ischemia, and that a protective mechanism to mitigate cerebral ischemia began to function. The upregulated expression of heat shock protein 70 may repair the damage.
In summary, we observed the effect of cerebral ischemia-reperfusion injury on cognitive function and neuronal apoptosis in the hippocampus of aged rats. The results confirmed that aged rats are sensitivity to cerebral ischemia-reperfusion injury, and that cerebral ischemia for a shorter time than the traditional safety limit could cause neuronal apoptosis in the hippocampus and lead to changes in memory and cognition. The mitochondrial changes in the hippocampus after transient cerebral ischemia were investigated using a comparative proteomics strategy. Preliminary analysis of the biological function of differentially expressed proteins reveals that they may be associated with neuronal apoptosis after transient cerebral ischemia. However, many factors affect cerebral ischemia and the functional meaning of the correlation between differential protein expression and cerebral ischemia remains unclear. How differentially expressed proteins participate in the regulation of neuronal apoptosis after cerebral ischemia deserves further exploration.
Acknowledgments:
We would like to express our thanks to Prof. Wang SD, Affiliated Hospital of Qingdao University, China and Prof. Wang MS, Qingdao Municipal Hospital, China. We are particularly grateful to Prof. Guo YL, Institute of Cerebrovascular Disease, Medical College, Qingdao University, China, for guidance in animal experiments.
Footnotes
Conflicts of interest: None declared.
Funding: This study was supported by a grant from Ministry of Science and Technology of Qingdao City, No. 10-3-4-7-8-JCH.
Copyedited by Li YT, Allen J, Raye W, Li CH, Song LP, Zhao M
References
- 1.Ahlemeyer B, Gottwald M, Baumgart-Vogt E. Deletion of a single allele of the Pex11β gene is sufficient to cause oxidative stress, delayed differentiation and neuronal death in mouse brain. Dis Model Mech. 2012;5:125–140. doi: 10.1242/dmm.007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen BS, Buckberg GD. Studies of isolated global brain ischaemia: I Overview of irreversible brain injury and evolution of a new concept-redefining the time of brain death. Eur J Cardiothorac Surg. 2012;41:1132–1137. doi: 10.1093/ejcts/ezr315. [DOI] [PubMed] [Google Scholar]
- 3.Calì T, Ottolini D, Brini M. Mitochondrial Ca(2+) and neurodegeneration. Cell Calcium. 2012;52:73–85. doi: 10.1016/j.ceca.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaitanya GV, Babu PP. Multiple apoptogenic proteins are involved in the nuclear translocation of apoptosis inducing factor during transient focal cerebral ischemia in rat. Brain Res. 2008;30:178–190. doi: 10.1016/j.brainres.2008.09.075. [DOI] [PubMed] [Google Scholar]
- 5.Cwerman-Thibault H, Sahel JA, Corral-Debrinski M. Mitochondrial medicine: to a new era of gene therapy for mitochondrial DNA mutations. Inherit Metab Dis. 2011;34:327–344. doi: 10.1007/s10545-010-9131-5. [DOI] [PubMed] [Google Scholar]
- 6.Datta A, Park JE, Li X, Zhang H, Ho ZS, Heese K, Lim SK, Tam JP, Sze SK. Phenotyping of an in vitro model of ischemic penumbra by iTRAQ-based shotgun quantitative proteomics. J Proteome Res. 2010;9:472–484. doi: 10.1021/pr900829h. [DOI] [PubMed] [Google Scholar]
- 7.De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012;19:267–273. doi: 10.1038/cdd.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Díaz-Moreno I, García-Heredia JM, Díaz-Quintana A. Cytochrome c signalosome in mitochondria. Eur Biophys J. 2011;40:1301–1315. doi: 10.1007/s00249-011-0774-4. [DOI] [PubMed] [Google Scholar]
- 9.Görg A, Weiss W, Dunn MJ. Current two-dimensional electrophoresis technology for proteomics. Proteomics. 2004;4:3665–3685. doi: 10.1002/pmic.200401031. [DOI] [PubMed] [Google Scholar]
- 10.James R, Searcy JL, Le Bihan T, Martin SF, Gliddon CM, Povey J, Deighton RF, Kerr LE, McCulloch J, Horsburgh K. Proteomic analysis of mitochondria in APOE transgenic mice and in response to an ischemic challenge. J Cereb Blood Flow Metab. 2012;32:164–176. doi: 10.1038/jcbfm.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji XY, Wang SR, Wang MS. Qingdao, China: Qingdao University; 2006. Effects of transient cerebral ischemia and reperfusion injury on apoptosis and expression of Caspase-3 protein to aged rat's hippocampal neurons. [Google Scholar]
- 12.Jiang XS, Dai J, Sheng QH, Zhang L, Xia QC, Wu JR, Zeng R. A comparative proteomic strategy for subcellular proteome research: ICAT approach coupled with bioinfomatics prediction to ascertain rat liver mitochondrial proteins and indication of mitochondrial localization for catalase. Mol Cell Proteomies. 2005;4:12–34. doi: 10.1074/mcp.M400079-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Krafft PR, Bailey EL, Lekic T, Rolland WB, Altay O, Tang J, Wardlaw JM, Zhang JH, Sudlow CL. Etiology of stroke and choice of models. Int J Stroke. 2012;7:398–406. doi: 10.1111/j.1747-4949.2012.00838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumari S, Anderson L, Farmer S, Mehta SL, Li PA. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res. 2012;3:296–304. doi: 10.1007/s12975-012-0158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SH, Kwon HM, Kim YJ, Lee KM, Kim M, Yoon BW. Effects of hsp70.1 gene knockout on the mitochondrial apoptotic pathway after focal cerebral ischemia. Stroke. 2004;35:2195–2199. doi: 10.1161/01.STR.0000136150.73891.14. [DOI] [PubMed] [Google Scholar]
- 16.McBean DE, Kelly PA. Rodent models of global cerebral ischemia: a comparison of two-vessel occlusion and four-vessel occlusion. Gen Pharmacol. 1998;30:431–434. doi: 10.1016/s0306-3623(97)00284-x. [DOI] [PubMed] [Google Scholar]
- 17.Mulder GB, Pritchett K. The Morris water maze. Contemp Top Lab Anim Sci. 2003;42:49–50. [PubMed] [Google Scholar]
- 18.Muller GJ, Stadeimann C, Basthoim L. Ischemia leads to apoptosis and necrosis-like neuron death in the ischemic rat hippocampus. Brain Pathol. 2004;14:415–424. doi: 10.1111/j.1750-3639.2004.tb00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oxler EM, Dolga A, Culmsee C. AIF depletion provides neuroprotection through a preconditioning effect. Apoptosis. 2012;17:1027–1038. doi: 10.1007/s10495-012-0748-8. [DOI] [PubMed] [Google Scholar]
- 20.Phillips DM. Electron microscopy: use of transmission and scanning electron microscopy to study cells in culture. Methods Cell Biol. 1998;57:297–311. doi: 10.1016/s0091-679x(08)61587-3. [DOI] [PubMed] [Google Scholar]
- 21.Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- 22.Pulsinelli WA, Buchan AM. The four-vessel occlusion rat model: method for complete occlusion of vertebral arteries and control of collateral circulation. Stroke. 1988;19:913–914. doi: 10.1161/01.str.19.7.913. [DOI] [PubMed] [Google Scholar]
- 23.Salminen A, Ojala J, Kaarniranta K. Apoptosis and aging: increased resistance to apoptosis enhances the aging process. Cell Mol Life Sci. 2011;68:1021–1031. doi: 10.1007/s00018-010-0597-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scornavacca G, Gesuete R, Orsini F, Pastorelli R, Fanelli R, de Simoni MG, Airoldi L. Proteomic analysis of mouse brain cortex identifies metabolic down-regulation as a general feature of ischemic pre-conditioning. J Neurochem. 2012;122:1219–1229. doi: 10.1111/j.1471-4159.2012.07874.x. [DOI] [PubMed] [Google Scholar]
- 25.Simmen T. Hax-1: a regulator of calcium signaling and apoptosis progression with multiple roles in human disease. Expert Opin Ther Targets. 2011;15:741–751. doi: 10.1517/14728222.2011.561787. [DOI] [PubMed] [Google Scholar]
- 26.Vafiadaki E, Arvanitis DA, Pagakis SN. The anti-apoptotic protein HAX-1 interacts with SERCA2 and regulates its protein levels to promote cell survival. Mol Biol Cell. 2009;20:306–318. doi: 10.1091/mbc.E08-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villa RF, Gorini A, Hoyer S. Effect of ageing and ischemia on enzymatic activities linked to Krebs’ cycle, electron transfer chain, glutamate and aminoacids metabolism of free and intrasynaptic mitochondriaof cerebral cortex. Neurochem Res. 2009;34:2102–2116. doi: 10.1007/s11064-009-0004-y. [DOI] [PubMed] [Google Scholar]
- 28.Zhang CY, Miao H, Tian HC. Anatomic observation on rat vertebrobasilar artery for the model of cerebral ischemia. Zhongguo Linchuang Jiepouxue Zazhi. 1995;139:53–55. [Google Scholar]
- 29.Zhao G, Su XC. Animal models of brain ischemia in stroke. Zuzhong yu Shenjing Jibing. 1996;3:99–100. [Google Scholar]