

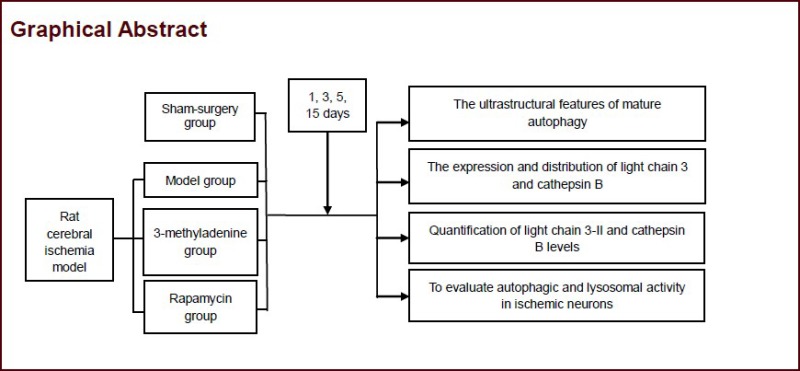
Keywords: neural regeneration, brain injury, autophagy, lysosome, light chain 3, cathepsin B, cerebral ischemia, neuron, neuroprotection, grants-supported paper, neuroregeneration
Abstract
Autophagy is involved in neural cell death after cerebral ischemia. Our previous studies showed that rapamycin-induced autophagy decreased the rate of apoptosis, but the rate of apoptosis was creased after the autophagy inhibitor, 3-methyladenine, was used. In this study, a suture-occluded method was performed to generate a rat model of brain ischemia. Under a transmission electron microscope, autophagic bodies and autophagy lysosomes were markedly accumulated in neurons at 4 hours post brain ischemic injury, with their numbers gradually reducing over time. Western blotting demonstrated that protein levels of light chain 3-II and cathepsin B were significantly increased within 4 hours of ischemic injury, but these levels were not persistently upregulated over time. Confocal microscopy showed that autophagy was mainly found in neurons with positive light chain 3 signal. Injection of rapamycin via tail vein promoted the occurrence of autophagy in rat brain tissue after cerebral ischemia and elevated light chain 3 and cathepsin B expression. However, injection of 3-methyladenine significantly diminished light chain 3-II and cathepsin B expression. Results verified that autophagic and lysosomal activity is increased in ischemic neurons. Abnormal components in cells can be eliminated through upregulating cell autophagy or inhibiting autophagy after ischemic brain injury, resulting in a dynamic balance of substances in cells. Moreover, drugs that interfere with autophagy may be potential therapies for the treatment of brain injury.
INTRODUCTION
Cell death due to cerebral ischemia has been attributed to necrosis and apoptosis[1,2]; however, there is increasing evidence that autophagy may be involved in mediating neuronal death in cerebral ischemia[3,4,5]. Autophagy is a regulatory process that is activated for bulk removal of cellular proteins and organelles[6]. For autophagic degradation, proteins are targeted and transported to membrane-enclosed vesicles. The vesicles are fused with lysosomes and their contents are degraded by lysosomal enzymes, predominantly cathepsin B[7,8]. Microtubule-associated protein light chain 3, a mammalian homolog of yeast ATG8, is synthesized and split into light chain 3-I, which activates transcription factor 4 protease, thereby activating autophagy process. Light chain 3-I and lipidic phosphatidyl ethanolamines are then connected and form light chain 3-II[9]. The protein levels of the membrane components of light chain 3-II are often used to measure autophagic activity[10]. Autophagic cell death is characterized by the existence of a large amount of autophagy or lysosomal autophagy. In neurons, autophagy is enhanced in several conditions, including nutrient starvation, development, and neurodegeneration[11]. Autophagic cell death occurs in neurons during normal development[12]. Theoretically, autophagy may help promote cell survival, either by purging the cell of damaged organelles, toxic metabolites, and intracellular pathogens or by generating the intracellular building blocks required to maintain vital functions during nutrient-limiting conditions. By contrast, autophagy may also promote cell death through excessive self-digestion and degradation of essential cellular constituents[13,14]. In fact, there is increasing evidence that autophagy may be involved in mediating neuronal death in cerebral ischemia[6,9,10,15].
When cerebral infarction occurs, up-regulation or inhibition of cell autophagy can allow the elimination of abnormal cell components, as well as maintain the dynamic balance of energy substrates. This phenomenon may provide a further theoretical basis for the treatment of cerebral infarction and may present a possible novel drug target. In the present study, a rat model of permanent middle cerebral artery occlusion was used as a brain ischemic model to examine the role of autophagy at different time points in ischemic brain.
RESULTS
Quantitative analysis of experimental animals
Sixty adult rats were equally and randomly assigned to four groups: sham-surgery, model, rapamycin, or 3-methyladenine. Animals in the model, rapamycin, and 3-methyladenine groups were injected with normal saline, rapamycin and 3-methyladenine respectively, via the tail vein at 1, 3, 5, 7, and 15 days after model induction. All 60 rats were included in the final analysis.
Ultrastructural features of mature autophagy after brain ischemia
The gold standard for identifying mature autophagy under a transmission electron microscope is the formation of foamy cytoplasmic vacuoles[16]. In the sham-surgery group, pre-existing autophagic bodies were occasionally observed in cortical neurons. These normal cortical neurons contain a number of ribosomes, nuclei, rough endoplasmic reticulum, and mitochondrion. In the model group, significant changes were observed at 4 hours after brain ischemia at the ultrastructural level. These changes included a baseline level of autophagy and lysosomal bodies, as well as a small amount of mitochondria. An accumulation of autophagic and lysosomal bodies occurred in neurons, while the polyribosomes, nuclei, Golgi complex, and mitochondrion appeared to be normal.
In the rapamycin group, double-membrane vesicles formed in the cytoplasm, indicating the presence of autophagic bodies. In the 3-methyladenine group, no double-membrane structures of autophagic bodies or lysosomes were observed. At 24 hours after brain ischemia, the model group showed a large number of newly produced autophagic and lysosomal bodies. In the rapamycin group, the formation of cytoplasmic autophagic bodies with double membrane structure and dense stained particles was observed. In the 3-methyladenine group, there were no double-membrane structures of autophagic or lysosomal bodies observed, but vacuole formation at the outer edge of the nucleus was seen (Figure 1).
Figure 1.

Ultrastructural features of brain tissue after cerebral ischemia (transmission electron microscopy).
Brain sections were obtained from the neocortical area near the injury site and stained using the osmium-uranyl-lead method. Arrows show polyribosomes. Scale bars: 500 nm. AL: Autolysosome; G: Golgi apparatus; M: mitochondrion; N: nucleus; MCAO: middle cerebral artery occlusion. 4 hours: accumulation of autophagic bodies and autophagic lysosomes; 1 day: increase in the number of autophagic bodies; 3, 5, 15 days: the number of autophagic bodies did not increase.
At 5 days following brain ischemia, the amount of autophagic and lysosomal bodies decreased compared with the 3-day animals in the model group. In the rapamycin group, besides the formation of cytoplasmic double membrane autophagic bodies and densely stained particles, autophagic bodies with multiple membrane structures and mitochondrion with normal morphology were also visible. No further changes in the 3-methyladenine group were visible at the 5-day time point (Figure 1). At 15 days after brain ischemia, only a small number of autophagic and lysosomal bodies were observed in the model group. In the rapamycin group, some double membrane autophagic bodies were seen in the cytoplasm, and could sometimes be observed in high density stained particles. The 3-methyladenine group remained the same as at previous time points (Figure 1).
Expression and distribution of light chain 3 and cathepsin B after brain ischemia
Positive signal for light chain 3 was mainly located in the membrane of neocortical neurons. Cathepsin B was mainly located in the cytoplasm, which is consistent with previous studies[9,17] (Figure 2). Light chain 3 expression quickly reached its peak at 3 days after an ischemic brain insult and then began to decrease. Cathepsin B did not sustain its expression after brain ischemia either, showing a similar pattern of changes to light chain 3. In the sham-surgery, model, and rapamycin groups, light chain 3 and cathepsin B expression significantly increased, but significantly decreased in the 3-methyladenine group (Figure 3).
Figure 2.
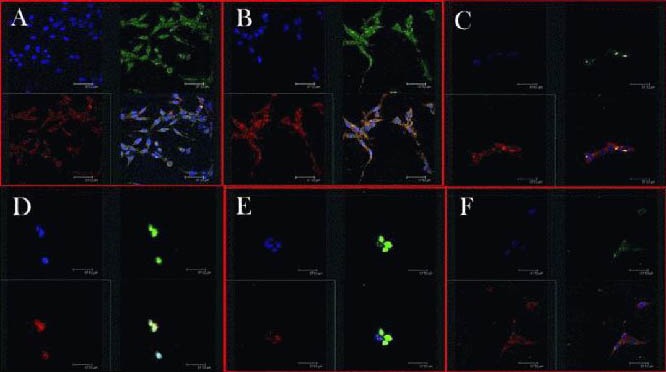
Light chain 3 (red) and Cathepsin B (green) immunohistochemistry in the brain of rats with middle cerebral artery occlusion (laser scanning confocal microscope).
Three kinds of staining were used in the brain sections. Nuclei of neurons were labeled with blue, light chain 3 red and Cathepsin B was labeled green. Drugs were injected via the tail vein at 0, 4 hours, 1, 3, 5, 15 days after middle cerebral artery occlusion (A–F, respectively). Light chain 3-II signal significantly increased at 4 hours until 3 days after brain ischemia, and then decreased after 5 and 15 days. Scale bars: 32.7 nm.
Figure 3.
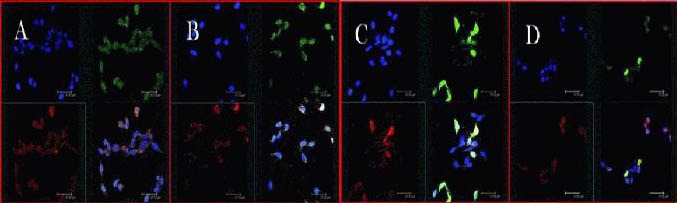
Confocal microscopic images of light chain 3 (red) and cathepsin B (green) immunohistochemistry at 1 day after middle cerebral artery occlusion.
Three kinds of staining were used in brain sections. Nuclei of neurons were labeled with blue, light chain 3 red and cathepsin B was labeled green. (A) Sham-surgery group; (B) model group; (C) rapamycin group; (D) 3-methyladenine (an autophagy inhibitor) group. Drugs were injected via the tail vein at 1 day after middle cerebral artery occlusion. The groups treated with 3-methyladenine showed an increased number of cells and enhanced cellular activities. Scale bars: 32.7 nm.
Quantitative changes of light chain 3-II and cathepsin B after brain ischemia
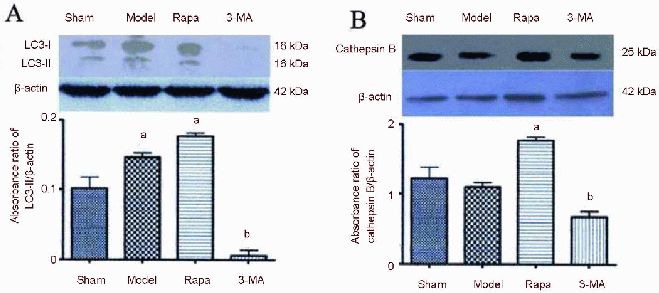
N terminal light chain 3 antibody could recognize a light chain 3-II band[17]. In the present study, western blot showed changes in light chain 3-II (Figure 4A) and Cathepsin B (Figure 4B) expression in ischemic rat brain at different time points. Consistent with previous immunohistochemical results, the expression of light chain 3-II peaked at 3 days after the injury and decreased afterwards; while Cathepsin B peaked at 1 day post injury and then decreased until the 3-day time point (P < 0.05). Additionally, compared with the sham-surgery group and model group, light chain 3-II and Cathepsin B showed an increase in expression in the rapamycin group, and a decrease in expression in the 3-methyladenine group (P < 0.05; Figure 5).
Figure 4.

Middle cerebral artery occlusion-induced changes of light chain 3 (LC3; A) and cathepsin B (B) protein expression in ischemic rat brain over time.
Extracts from the ischemic and sham-operated (Sham) cortex were separated using gel electrophoresis and protein levels of LC3 and cathepsin B were detected. β-actin was used as the loading control. Drugs were injected via the tail vein at 4 hours, 1, 3, 5, 15 days after middle cerebral artery occlusion. Results are represented as mean ± SD. Groups with heterogeneity of variance were analyzed with paired t-test. aP < 0.05, vs. sham-surgery group.
Figure 5.
Expression levels of light chain 3 (LC3) and cathepsin B at 1 day after middle cerebral artery occlusion.
Extracts from the ischemic and sham-operated (Sham) cortex were separated via gel electrophoresis and protein levels of LC3 and cathepsin B were detected using immunoblotting.
β-actin was used as the loading control. Drugs were injected via the tail vein at 1 day after middle cerebral artery occlusion. Results are represented as mean ± SD. Groups with heterogeneity of variance were analyzed with paired t-test. aP < 0.05, vs. sham-surgery group (sham); bP < 0.05, vs. model group. (A) Sham-surgery group; (B) model group; (C) rapamycin (Rapa) group; (D) 3-methyladenine (3-MA; an autophay inhibitor) group.
DISCUSSION
Transmission electron microscopy revealed signs of autophagy following brain ischemia, including double membraned structures and cytoplasmic material or abnormal autophagic organelles.
Autophagic lysosomes containing some heterogeneous or homogeneous dense material were also observed. All of these changes provide strong morphological evidence of autophagy pathway activation. In the ultra-early phase (before 4 hours) of ischemic brain insult, brain tissues showed few changes, with swelling of some vascular cells, neurons and astrocytes, as well as mitochondria. In the early phase (4 hours), electron microscopy revealed accumulation of autophagic bodies and autophagic lysosomes. In the acute phase of brain ischemia (1 day), the ischemic area appeared to be pale in color as well as displaying mild tissue swelling. Neurons, glial cells, and endothelial cells showed characteristics of ischemic changes. Under a transmission electron microscope, the double membrane structure of the autophagic bodies and autophagic lysosomes could be observed, showing a significant increase in the presence of this structure. At 3 days after cerebral ischemia, many neurons disappeared, infiltration of neutral granulocytes, lymphocytes, and macrophages into the tissue was observed, as well as brain edema. The double membrane structure of the autophagic bodies and autophagic lysosomes could be observed, but the number did not further increase compared with that at 1 day following ischemia. At 5 and 15 days after ischemia, the infarcted tissues liquefied and showed atrophy. The bilayer membrane structure, and autophagic bodies, could no longer be seen at this stage, and few autophagic lysosomes were present.
Microtubule associated protein light chain 3 is a mammal ATG8-PE conjugate, which remains on the autophagosomal membrane even after the fusion of autophagic bodies and autophagic lysosomes.
The light chain 3-II protein level was therefore used to determine the number of autophagic bodies in the cytoplasm. Light chain 3-II was mainly located in autophagic bodies, and lower levels were present in the autophagic lysosomal membrane. The present study showed that light chain 3-II increased significantly at 4 hours to 3 days after brain ischemia, and then decreased at 5 days and 15 days post-insult. This observation in the present study is not consistent with a previous study showing the sustained expression of light chain 3-II[18]. However, these findings still support the theory that autophagic bodies and autophagic lysosomal membrane light chain 3-II is mainly distributed in the living cell membrane of neurons. Light chain 3-II protein expression increased 1 day after ischemia, but quickly decreased after this peak, and Cathepsin B showed a similar change in pattern of expression. Light chain 3 immunopositive signal was mainly located in surviving neurons, and an increased level of immunofluorescence in brain sections was observed from 1 day post-ischemia. Taken together, the cell ultrastructure and biochemical results showed that upon ischemic brain injury, a lysosomal autophagy pathway in neurons was activated, but not with sustained activity over time.
Additionally, in some studies into light chain 3 expression following brain hypoxia-ischemia, increased light chain 3 was reported in adults rather than in newborns[19,20]. In the adult mouse model of brain hypoxia-ischemia, light chain 3-I decreased, but there was no increase in light chain 3-II[3,4]. In another study that used the mouse brain ischemia model, light chain 3-II increased within 1 day, with activation of the autophagy pathway leading to loss of neurons[18].
With the fact that neuronal loss happened after the activation of the autophagy pathway, we pharmacologically inhibited the autophagy to see if autophagy could be neuroprotective at this time period. We also showed that the groups with autophagy inhibitor showed an increased number of cells and enhanced cellular activity, consistent with the theory that the activation of autophagy may be a mechanism for the neuroprotective response[21]. In the study with ATG gene knockout, it was found that autophagy is neuroprotective in damaged protein aggregation-induced cellular injury[22]. However, the over-activation of the autophagy pathway could result in excessive cell death, as shown previously[23] and in the present study, named autophagic cell death. Characteristic autophagy is considered as autophagic vesicle accumulation in the cytoplasm[24]. However, whether autophagy is a failure to rescue dying cells or is a programmed cell death reaction is yet to be understood[12,25]. Increasing evidence suggests that autophagy is critical in maintaining the cellular stability of the neuron. For example, the genetic loss of this pathway led to neurodegeneration and other diseases[26,27]; the conditional knockout of key genes in the autophagy pathway, such as ATG5 or ATG7, resulted in intracellular accumulation of proteins and neuronal death[28,29]. Considering all the evidence, autophagy could function as the induced cellular reaction to stressful stimuli, such as ischemia and hypoxia, and could partly ameliorate ischemic damage. This process may be limited in its overall function and incomplete in its regulation so excessive autophagy cannot be prevented[11,30].
In summary, balancing the autophagy pathway to maximize neuroprotection without the effects of excessive autophagy is an important consideration for post-infraction therapies. It would be ideal to pharmacologically target the natural autophagic pathway at different time points after brain ischemia with different compounds that diversely regulate this pathway to precisely enable neuroprotection. Moreover, the activity of the autophagic pathway could be used in the diagnosis of the progression of brain infarction and assist in the prediction of recovery in a clinical setting.
MATERIALS AND METHODS
Design
A randomized, controlled, in vivo animal study.
Time and setting
All experiments were performed at the Sixth People's Hospital of Shanghai, Shanghai Jiao Tong University, China from April 2009 to September 2012.
Materials
Animals
A total of 60 adult male Sprague-Dawley rats of specific pathogen-free grade, aged 6–8 weeks and weighing 270–320 g were purchased from Poole BK Zhongyinghezi Animals Hercynian Limited (certificate No. 20020008, grade II). NIH Guidelines for Care and Use of the Laboratory Animals were followed in all animal procedures.
Drugs
Rapamycin powder was purchased from Sigma, St. Louis, MO, USA, [assay ≥ 95% (high performance liquid chromatography). The chemical structure of rapamycin is shown in Figure 6.
Figure 6.
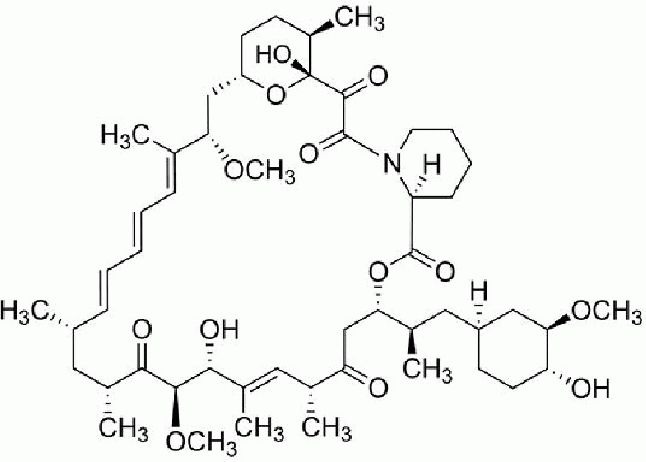
The chemical structure of rapamycin.
3-Methyladenine powder was purchased from Sigma, [assay ≥ 95% (high performance liquid chromatography]. 3-Methyladenine The chemical structure of 3-methyladenine is shown in Figure 7.
Figure 7.

The chemical structure of 3-methyladenine.
Methods
Animal model of rat cerebral ischemia
The animals were anesthetized with 3% halothane gas (30% oxygen and 70% nitrogen) prior to surgery. During surgery, the thigh artery was catheterized and pancuronium bromide (0.5 mg/kg per hour) was infused. Endotracheal intubation was performed and anesthesia was maintained with 1% halothane administered using a ventilator. A 4.8 mm craniotomy was then performed (3.8 mm caudal to bregma, 2.5 mm lateral to midline). Blood gas, blood glucose, and hemoglobin volume were monitored before the surgery, 15 minutes after surgery and again at 4 hours post-procedure. During the entire surgery, the brain temperature was controlled at 37°C and the animal was maintained in the following normal physiological range: mean arterial pressure: 120– 140 mmHg; oxygen partial pressure: 105–170 mmHg; CO2 and gas pressure 35–45 mmHg; blood pH: 7.38–7.41. The suture model of permanent middle cerebral artery occlusion was adopted and modified from Longa et al[31] using a suture with a diameter of 0.26 mm, and insertion depth at 18.5 ± 0.5 mm. After recovery, the animals received an evaluation for neurological deficits using the Longa scale as follows[31]: 0, normal; 1, cannot fully extend the left forelimb; 2, left limb paralysis and walking to the left; 3, falls to the left when walking, cannot stand or roll; 4, no spontaneous locomotor activity with a depressed level of consciousness. Animals that received 1 to 3 points were divided into successful models, while animals that received 0 or 4 points were removed. In the sham-surgery group, only the craniotomy was performed.
Drug intervention
After surgery, animals in the sham-surgery, model, rapamycin, and 3-methyladenine groups were intravenously injected via the tail vein with normal saline (0.1 mL), normal saline (0.1 mL) + rapamycin [0.1 mL (20 g/mL)], and 3-methyladenine [0.1 mL (600 nmol/L)], respectively, at 1, 3, 5, and 15 days after surgery.
Tissue sample preparation
Animals were sacrificed at 4 hours after the last injection, and two animals in each group were perfused with 4% paraformaldehyde. Frozen brain sections (50 μm) were then processed for immunohistochemistry. The striatum[32] from the two animals was isolated for western blot studies. For transmission electron microscopy, rats were perfused with 2% glutaraldehyde containing 0.1 mmol/L cacodylate.
Electron microscopy of neocortical neuron ultrastructural features
Brain tissues at 4 hours, 1, 3, 5, and 15 days after surgery were processed with conventional osmium-uranyl lead staining for electron microscopy analysis. Briefly, coronal brain slices (100 μm) were fixed in 2% glutaraldehyde in 0.1 mmol/L of dimethyl arsenate buffer (pH 7.4) for 1 hour, and then placed in 0.1 mmol/L dimethyl arsenate buffer containing 1% osmium tetroxide for 2 hours. Slices were then rinsed in distilled water and placed in 1% aqueous uranyl acetate overnight, followed by graded alcohol submersion (100%), dehydration, acetone processing, and finally embedded in Durcupan ACM. Slices (0.1 μm) were then prepared and stained with lead citrate before electron microscopy (JEM-1230, JEOL, Tokyo, Japan).
Immunohistochemistry
Protocols were adopted from Hu et al[16]. Antigen retrieval was performed with 0.01 mol/L sodium citrate buffer and blocked with 3% bovine serum albumin in 0.1% Triton X-100 containing PBS before incubation with the primary antibody. The primary antibodies included light chain 3 antigen (1:1 000; Abgent, San Diego, CA, USA) and cathepsin B (1:1 000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Before secondary antibody incubation, the sections were washed again and mounted onto slides for confocal microscopy (Nicolet Almega XR, Thermo, Marietta, OH, USA).
Western blot assay
Harvested brain tissues were preserved in a cryochamber at –15°C and then homogenized at 4°C before sodium dodecyl sulfate polyacrylamide gel electrophoresis. Proteins were then transferred to an immobilon-P membrane and incubated with the following primary antibodies: light chain 3 (1:300; Sigma) and cathepsin B (1:300; Sigma). The levels of β-actin (Sigma) were used as an internal control. The final results were visualized with enhanced chemical fluorescence Kodak X-omat LS film (Eastman Kodak, Rochester, NY, USA) and absorbance was assessed using Kodak ID image analysis software (Eastman Kodak).
Statistical analysis
SAS 6.10 software (SAS Institute Inc., Cary, NC, USA) was used for data analysis. Results were represented as mean ± SD. Groups with heterogeneity of variance were analyzed with paired t-test. P < 0.05 was considered to be statistically significant.
Research background: Current research has shown that cerebral ischemia may induce neuronal autophagic death. Autophagy is a constantly ongoing process, and is considered to be the response to further restore normal tissue following injury.
Research frontiers: After cerebral ischemia, activation of the autophagy-lysosomal pathway maintains cellular homeostasis, which is a protective mechanism underlying ischemic neuronal survival and also provides nutrition and energy.
Clinical significance: Rapamycin promotes the intracellular degradation of damaged organelles by regulating the autophagy signaling pathway. This mechanism may become a future target for therapies aimed at treating cerebral infarction.
Academic terminology: “Autophagy-lysosomal pathway” - the main pathway for degradation of abnormal organelles, protein aggregates and foreign matter.
Peer review: This article describes an in-depth study of the autophagy-lysosomal pathway in cerebral ischemic neuronal injury in rats. Results showed that at 4 hours after cerebral ischemia, autophagy, autophagy-lysosome and microtubule-associated protein light chain 3 increased significantly in neurons. These studies show a degree of innovation and potential clinical value.
Footnotes
Funding: This work was supported by grants from the National Natural Science Foundation of China, No. 31171014 and No. 30970869; the Project of Science and Technology Commission of Shanghai City, No. 09DZ1950400; Board of Health of Shanghai, China, No. 2008086; the Youth Projects of National Natural Science Foundation of China, No. 31100783; Youth Key Project in Shanghai College of Medicine of Fudan University, No. 09-L37.
Conflicts of interest: None declared.
Ethical approval: The study was approved by the Local Institutional Ethical Committee of the Sixth People's Hospital of Shanghai, China.
(Reviewed by Apricò K, de Souza M, Sui RB, Di ZL)
(Edited by Yu J, Qiu Y, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- [2].Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- [3].Adhami F, Liao G, Morozov YM, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169(2):566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Adhami F, Schloemer A, Kuan CY. The roles of autophagy in cerebral ischemia. Autophagy. 2007;3(1):42–44. doi: 10.4161/auto.3412. [DOI] [PubMed] [Google Scholar]
- [5].Koike M, Shibata M, Tadakoshi M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172(2):454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Punnonen EL, Autio S, Kaija H, et al. Autophagic vacuoles fuse with the prelysosomal compartment in cultured rat fibroblasts. Eur J Cell Biol. 1993;61(1):54–66. [PubMed] [Google Scholar]
- [8].Glaumann H, Ericsson JL, Marzella L. Mechanisms of intralysosomal degradation with special reference to autophagocytosis and heterophagocytosis of cell organelles. Int Rev Cytol. 1981;73:149–182. doi: 10.1016/s0074-7696(08)61288-7. [DOI] [PubMed] [Google Scholar]
- [9].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117(Pt 13):2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- [11].Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29(9):528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- [12].Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181(3):195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- [13].Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1(1):11–21. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu K, Gui B, Sun Y, et al. Inhibition of L-type Ca(2+) channels by curcumin requires a novel protein kinasetheta isoform in rat hippocampal neurons. Cell Calcium. 2013;53(3):195–203. doi: 10.1016/j.ceca.2012.11.014. [DOI] [PubMed] [Google Scholar]
- [15].Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269(8):1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- [16].Hu BR, Martone ME, Jones YZ, et al. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20(9):3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Klionsky DJ. Neurodegeneration: good riddance to bad rubbish. Nature. 2006;441(7095):819–820. doi: 10.1038/441819a. [DOI] [PubMed] [Google Scholar]
- [18].Wen YD, Sheng R, Zhang LS, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4(6):762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- [19].Zhu C, Wang X, Xu F, et al. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005;12(2):162–176. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]
- [20].Zhu C, Xu F, Wang X, et al. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J Neurochem. 2006;96(4):1016–1027. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]
- [21].Moore MN, Allen JI, Somerfield PJ. Autophagy: role in surviving environmental stress. Mar Environ Res. 2006;62(Suppl):S420–425. doi: 10.1016/j.marenvres.2006.04.055. [DOI] [PubMed] [Google Scholar]
- [22].Swanson MS. Autophagy: eating for good health. J Immunol. 2006;177(8):4945–4951. doi: 10.4049/jimmunol.177.8.4945. [DOI] [PubMed] [Google Scholar]
- [23].Shi JJ, Liu KY, Yang YP. Autophagy in the pathogenesis of Parkinson's disease. Sheng Li Ke Xue Jin Zhan. 2009;40(1):67–71. [PubMed] [Google Scholar]
- [24].Bowen ID, Mullarkey K, Morgan SM. Programmed cell death during metamorphosis in the blow-fly Calliphora vomitoria. Microsc Res Tech. 1996;34(3):202–217. doi: 10.1002/(SICI)1097-0029(19960615)34:3<202::AID-JEMT3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- [25].Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- [26].Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27(5-6):495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- [27].Nishino I. Autophagic vacuolar myopathy. Semin Pediatr Neurol. 2006;13(2):90–95. doi: 10.1016/j.spen.2006.06.004. [DOI] [PubMed] [Google Scholar]
- [28].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- [29].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- [30].Liu K, Shi N, Sun Y, et al. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem Res. 2013;38(1):201–207. doi: 10.1007/s11064-012-0909-8. [DOI] [PubMed] [Google Scholar]
- [31].Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- [32].Paxinos G, Watson C. 5th ed. London: Academic Press, UK; 2005. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]