

Abstract
The retina of Wistar rats within 1–3 days of birth were dissociated into a retinal cell suspension using 0.05% trypsin digestion. The cell suspension was incubated in Dulbecco's modified Eagle's medium for 24 hours, followed by neurobasal medium for 5–7 days. Nissl staining showed that 79.86% of primary cultured retinal cells were positive and immunocytochemical staining showed that the purity of anti-neurofilament heavy chain antibody-positive cells was 71.53%, indicating that the primary culture system of rat retinal neurons was a reliable and stable cell system with neurons as the predominant cell type. The primary cultured retinal neurons were further treated with 0, 5.5, 15, 25, and 35 mM glucose for 24, 48, and 72 hours. The thiazolyl blue tetrazolium bromide test and flow cytometry showed that with increasing glucose concentration and treatment duration, the viability of retinal neurons was reduced, and apoptosis increased. In particular, 35 mM glucose exhibited the most significant effect at 72 hours. Thus, rat retinal neurons treated with 35 mM glucose for 72 hours can be used to simulate a neuronal model of diabetic retinopathy.
Keywords: neural regeneration, peripheral nerve injury, retina, neurons, apoptosis, hyperglycemia model, diabetic retinopathy, glucose, grants-supported paper, photographs-containing paper, neuroregeneration
Research Highlights
(1) Retinal neurons from neonatal Wistar rats (1–3 days) were cultured with plating medium and maintenance medium to establish a reliable and stable primary culture system using neurons as the predominant cells.
(2) The retinal neurons were treated with glucose at different concentrations for different time, and 35 mM for 72 hours was identified as the optimal concentration and intervention time.
(3) A retinal neuron model of high glucose was successfully established, which is significant for further studies of structure characteristics and biological functions of cell components in the retina, retinal pathological changes, and drug reactions.
INTRODUCTION
Retinal neurons are retinal photoreceptors and executors for nerve information transfer. Retinal functional damage caused by various causative factors is mainly manifested by loss of neuronal functions[1]. Thus, it is important to study physiologic and biochemical characteristics of retinal neurons in vitro. Isolation from the retina greatly damages neurons[2], so the isolation and culture of retinal neurons must be given careful attention. In addition, a large number of studies have focused on cell-cell and cell-extracellular matrix interactions in the retina, but complex in vivo environmental effects greatly limits in vivo studies[3]. Therefore a stable and reliable in vitro culture system for retinal nerve cells is important for further understanding the structural characteristics and biological functions of components of various cells in the retina. In addition, it is important in the retina to understand the mechanisms of drug reactions and pathological conditions.
Diabetic retinopathy is a major oculopathy which can eventually lead to blindness. It is therefore a major focus and challenge of clinical and basic studies regarding oculopathy. Thus, it is clinically significant to study the pathogenesis and prevention of this disorder. Recent evidence has indicated that changes in retinal neurons and glial cells occur earlier than retinal capillary alterations in the initial stages of diabetes[4]. The changes in retinal neurons directly influence progression of diabetic retinopathy and may be the leading cause of diabetes-induced retinal capillary lesions[5]. Thus, understanding of retinal neuron changes is useful for prevention of diabetic retinopathy. In addition, in vivo experiments have been conducted in animal models to investigate the changes of retinal neurons in diabetic retinopathy[6]. Establishment of an in vitro retinal neuron model of high glucose to simulate the in vivo microenvironment of diabetes can eliminate extraneous interference of other factors in vivo, and allow direct observation of cell changes in specific stages of diabetes. This is an advantage over animal experiments.
Numerous studies have demonstrated that changes in retinal vascular endothelial cells[7,8], retinal pigment epithelium[9], retinal Müller cells[6], and retinal neurons[10,11] are highly correlated with the incidence and progression of diabetic retinopathy. A single in vitro cell model of high glucose injury could help elucidate the precise mechanism by which high glucose damages retinal cells. It would also provide experimental and theoretical evidence for prevention and treatment of diabetic retinopathy. There have been no reports of an in vitro cultured retinal neuron model of high glucose-induced injury. Takano et al[12] cultured retinal tissue blocks with 57 mM glucose for 3–10 days to investigate the influence of high glucose on axonal regeneration of retinal neurons from normal and streptozocin-induced diabetic mice, and found that high glucose suppressed axonal regeneration of retinal neurons in both groups. Another study established an apoptosis model using primary cultures of retinal neurons with 15 mM glucose for 9 days, to investigate the influence of erythropoietin, glucose, and nitric oxide on neurons[10]. Results showed that erythropoietin reduced nitric oxide levels and neuronal death in neuron cultures containing low concentrations of glucose. Santiago et al[11] utilized 25 mM glucose to culture retinal neurons in vitro for 7 days to establish a high glucose model, and found that neuronal apoptosis was associated with the caspase pathway. All the above retinal neuronal models of high glucose utilized different concentrations and durations of glucose treatment, providing a reference for our study. However, they were simply based on previously described methods, and did not continuously observe apoptotic retinal neurons in response to glucose treatment at different concentrations for different durations. Moreover, some of them did not utilize culture media specific for neurons. Thus, it is important for ophthalmology studies to develop an optimal method for in vitro culture of retinal neurons, to establish a stable and reliable model to study the effects of high glucose.
Our objective in this study was to establish an optimal culture system of retinal neurons, and a neuron model of high glucose, to establish a foundation for future studies of mechanisms by which drugs could protect retinal neurons in the presence of high glucose.
RESULTS
Morphology and phenotype of primary cultures of retinal neurons
The retina was harvested from Wistar rats within 1–3 days of birth for primary cultures of retinal neurons. After incubation for 30 minutes, the body of retinal cells was small and round shaped (Figure 1A). At 24 hours, the majority of cells adhered to the wall, and short processes emanated from some cells and accumulated at the center (Figure 1B). At 2–3 days, the processes were extended, about 1/2 to 1–2 folds longer than cell body length. The neurons were polygon- and oval-shaped with plump bodies, with surrounding visible nuclei and nucleoli (Figure 1C). After culturing for 5–6 days, the processes further enlarged and increased, accompanied by surrounding non-neuron cells (glial cells; Figure 1D). At 7–10 days, neurons continued growing, and the length of processes extended over 10-fold longer than cell bodies, gradually forming a complex network, with a gradual reduction in the number of non-neuron cells (Figure 1E). Up to 15 days, the processes of most cells became shortened, and neurons disintegrated or died (Figure 1F).
Figure 1.
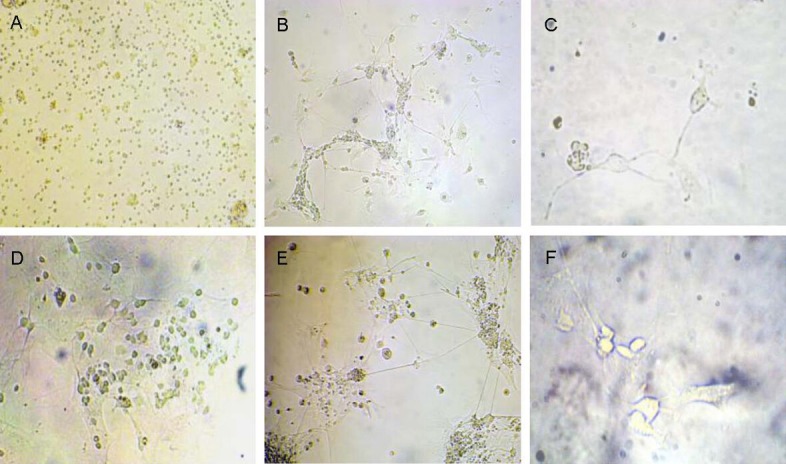
Morphology of primary cultured retinal cells from Wistar rats aged 1–3 days (inverted microscope).
(A) Retinal cells appear as small cell bodies with round shape after culture for 30 minutes (× 200).
(B) Short processes were observed, accumulating at the center, after retinal cells were adhered for 24 hours (× 200).
(C) Retinal cells were polygon- and oval-shaped, with plump cell bodies and surrounding halation. Nuclei and nucleoli were also observed, and the length of processes reached 1/2- to 1–2-folds longer than cell bodies at 2–3 days (× 400).
(D) The processes further increased and enlarged, and a few non-neuron cells (glial cells) were observed at 5–6 days (× 200).
(E) The retinal cells grew further, and the length of process reached 10-fold longer than cell bodies and gradually formed a complex network. The number of non-neuron cells decreased gradually at 7–10 days (× 200).
(F) The processes of most cells were shortened, and cells disintegrated or even died at 15 days (× 400).
Nissl staining of retinal neurons cultured for 5–7 days showed a blue-violet stained cytoplasm, and granular Nissl bodies with clear structures. The cytoplasm of non-neuron cells was not stained, with light violet, round nuclei and clear nucleoli (Figure 2). The percentage of neurons was approximately 79.86%.
Figure 2.
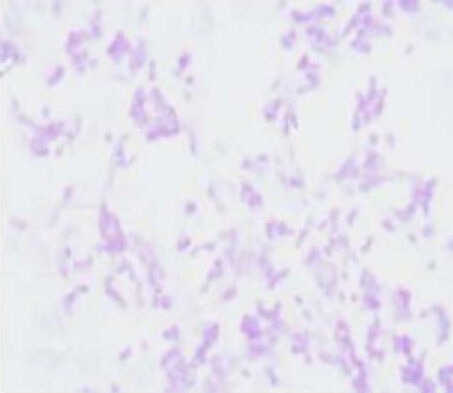
Nissl staining of primary cultured retinal neurons from Wistar rats (× 200).
Light microscopic observation showed blue-violet stained cytoplasm and granular Nissl bodies. The cytoplasm of non-neuron cells was lightly stained, and the nuclei were round and light violet, with clear nucleoli.
Immunocytochemical staining of retinal cells cultured for 5–7 days showed that 71.53% of cells were positive for anti-neurofilament antibody (Figure 3A), but only 21.13% were positive for anti-glial fibrillary acidic protein antibody (Figure 3B).
Figure 3.
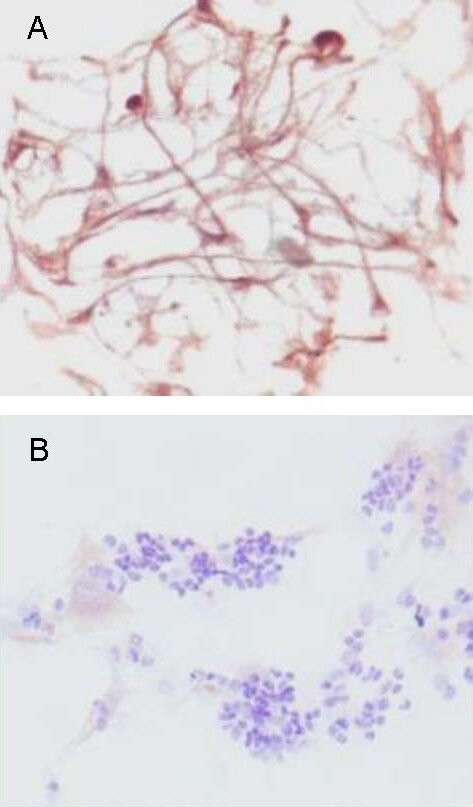
Neurofilament and glial fibrillary acidic protein expression in primary cultured retinal cells from Wistar rats 1–3 days old (immunocytochemical staining, × 400).
(A) Anti-neurofilament immunocytochemical staining showed brownish-yellow particles in cell bodies and processes. The cell bodies were darkly stained, but processes were lightly stained.
(B) Anti-glial fibrillary acidic protein immunocytochemical staining showed brownish-yellow or brown particles in cytoplasm and membrane, which were positively stained glial cells.
Influence of high glucose on retinal neuron viability
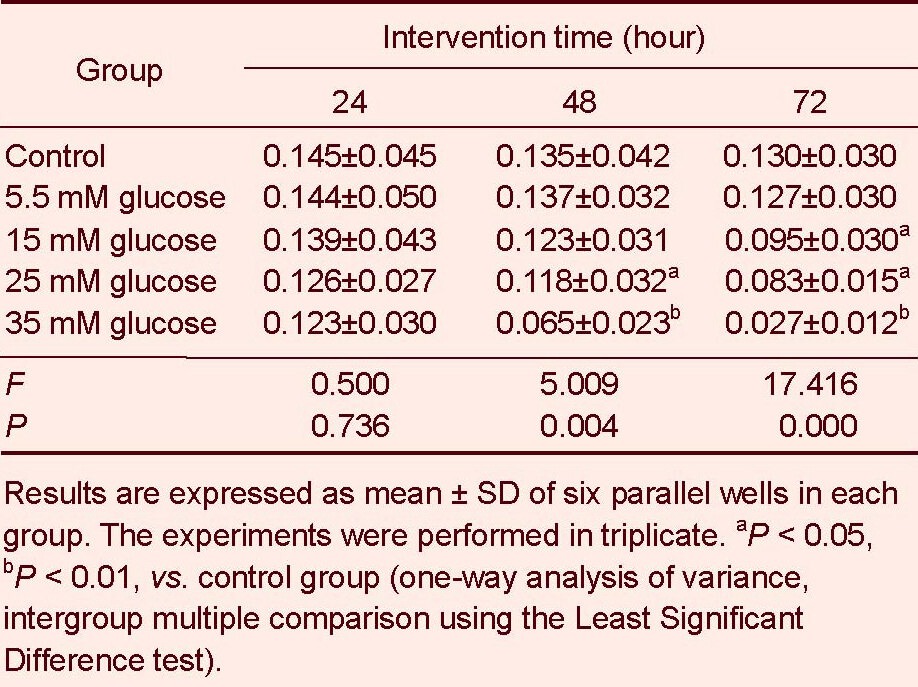
MTT assay showed that retinal neuron viability remained unchanged following 5.5 mM glucose treatment for 24–72 hours (P > 0.05). However, cell viability significantly decreased with increasing glucose concentration and intervention duration, compared with the control group (15 mM for 72 hours, P < 0.05; 25 mM for 48, 72 hours, P < 0.05; 35 mM for 48, 72 hours, P < 0.01). Most importantly, 35 mM glucose treatment for 72 hours significantly decreased cell viability (P < 0.01, Table 1).
Table 1.
Influence of glucose treatment at different concentrations for different time on retinal neuron viability (absorbance)
Influence of high glucose on apoptosis of retinal neurons
Annexin/propidium iodide double staining was used to detect apoptosis. Flow cytometry showed that 5.5 mM glucose treatment for 24–72 hours did not influence apoptosis of retinal neurons (P > 0.05). However, the apoptosis rate was significantly increased with increasing glucose concentration and intervention duration compared with the control group (15 mM for 72 hours; 25 mM for 48, 72 hours; 35 mM for 48, 72 hours, P < 0.05 or P < 0.01). In particular, the apoptosis rate was significantly higher after 35 mM glucose treatment for 72 hours compared with the other groups (P < 0.01; Figures 4–6, Table 2).
Figure 4.
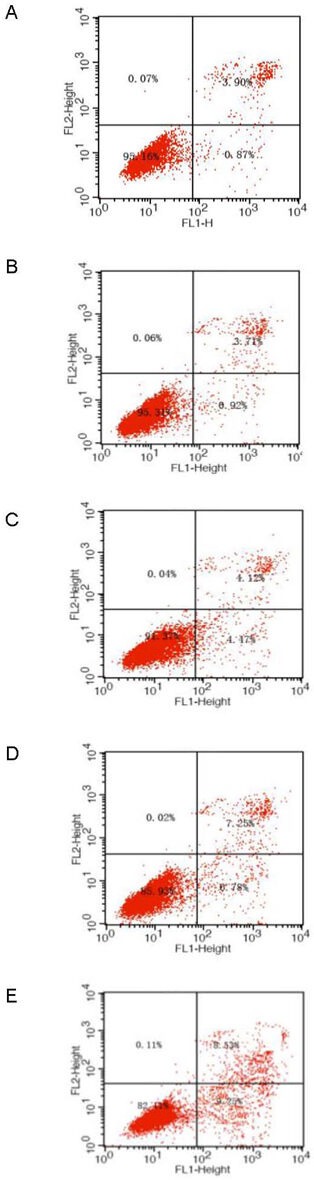
Apoptosis of primary cultured retinal neurons exposed to glucose at different concentrations for 24 hours.
(A) Normal control group; (B–E) 5.5, 15, 25, and 35 mM glucose groups. Flow cytometry detected and divided cells into four subgroups, including necrotic and non-viable apoptotic cells [annexin−/propidium iodide (PI)+] in the left upper quadrant, necrotic cells (annexin+/PI+) in the right upper quadrant, normal living cells (annexin−/PI−) in the left lower quadrant, and viable apoptotic cells (annexin+/PI−) in the right lower quadrant.
Figure 6.
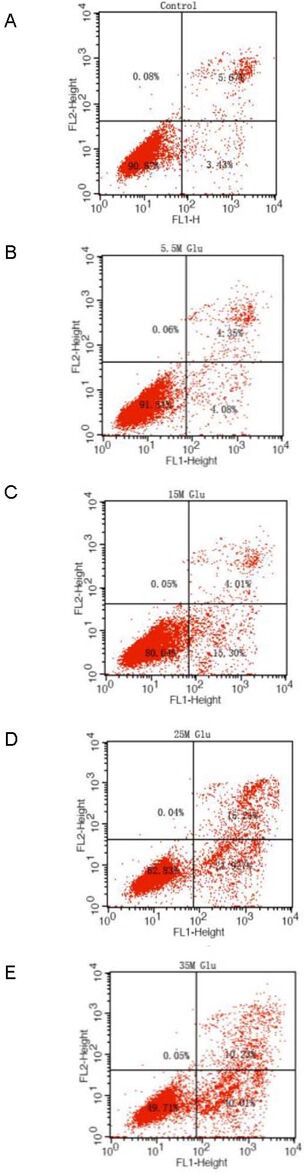
Apoptosis of primary cultured retinal neurons exposed to glucose at different concentrations for 72 hours.
(A) Normal control group; (B–E) 5.5, 15, 25, and 35 mM glucose groups.
Flow cytometry detected and divided cells into four subgroups, including necrotic and non-viable apoptotic cells [annexin−/propidium iodide (PI)+] in the left upper quadrant, necrotic cells (annexin+/PI+) in the right upper quadrant, normal living cells (annexin−/PI−) in the left lower quadrant, and viable apoptotic cells (annexin+/PI−) in the right lower quadrant.
Table 2.
Influence of glucose treatment at different concentrations for different times on apoptosis rate (%) of retinal neurons
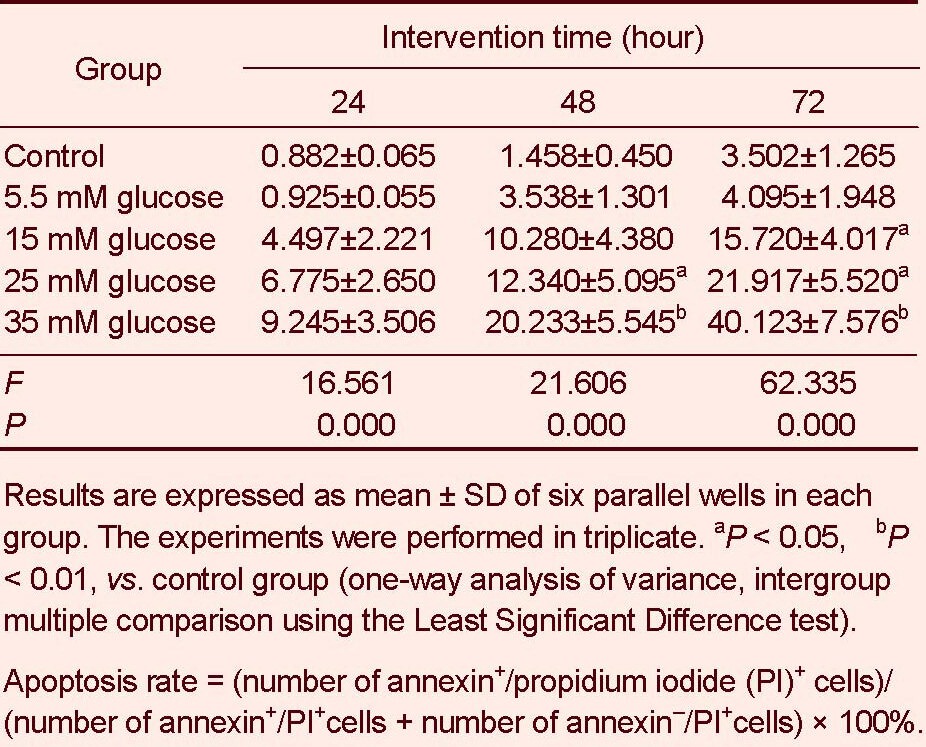
Figure 5.
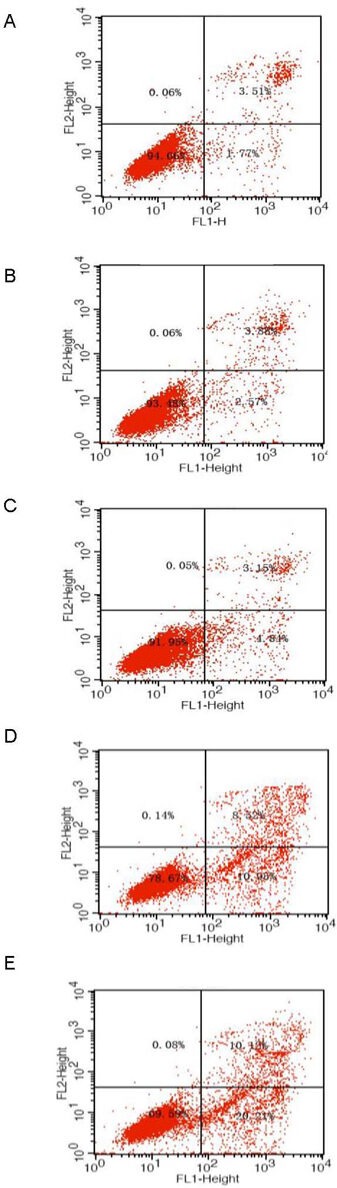
Apoptosis of primary cultured retinal neurons exposed to glucose at different concentrations for 48 hours.
(A) Normal control group; (B–E) 5.5, 15, 25, and 35 mM glucose groups.
Flow cytometry detected and divided cells into four subgroups, including necrotic and non-viable apoptotic cells [annexin−/propidium iodide (PI)+] in the left upper quadrant, necrotic cells (annexin+/PI+) in the right upper quadrant, normal living cells (annexin−/PI−) in the left lower quadrant, and viable apoptotic cells (annexin+/PI−) in the right lower quadrant.
DISCUSSION
Primary culture conditions of retinal neurons are controllable, allowing studies that cannot be conducted in vivo. Retinal neurons cannot divide, and can easily be contaminated by proliferating cells. Moreover, the conditions for retinal neuron survival are very specific. Thus, culture of retinal neurons is more difficult than other types of cells[13].
Several factors for successful primary culture of retinal neurons include: (1) Selection of experimental animals: rats have an advanced nervous system similar to humans, and the retina is not completely layered in Wistar rats within 1–3 days of birth, with an observable gap between neuroepithelium and pigment epithelium, allowing complete isolation of neuroepithelium from the pigment epithelium[14].
Thus, we used rats as experimental animals to obtain retinal neurons of high purity. (2) Time of sampling: generally, the survival rate of in vitro cultured neurons is associated with the age of the donor animals[15].
During isolation, neurons from embryos are in early development. The axons and dendrites have not extensively branched, nutritional dependence to target cells is low, and the epithelium of connective tissue is easily isolated[16]. However, it is difficult to confirm the age of the embryo and to obtain eyeball samples because of pigment epithelium contamination[17]. (3) Preparation of retinal cell suspension: the retinal cell suspension was prepared using a complete digestion method, so the quantity of obtained neurons depended on trypsin concentration and digestion duration. Following comparison of different methods, we utilized a method previously described by Santiago et al[11], which resulted in a large quantity of retinal neurons after retina tissues were digested with 0.05% trypsin at 37°C for 15 minutes. Low concentrations of trypsin or short durations of digestion resulted in nerve cell aggregation, while high concentrations of trypsin or long durations of digestion decreased cell viability. Our results demonstrated that enzyme digestion is a stable and reliable method for acquiring retinal neurons, providing a good experimental model for studying retinal disease. (4) Importance of adherence in cell growth and development: firm adherence is critical for neuron cultures, because the cell body can enlarge and the number of extended processes can increase. Takahashi et al[18] inoculated retinal ganglion cells on a glial cell monolayer, resulting in an increase in viability and an increase of neurite outgrowth. However, it is difficult to distinguish or isolate retinal ganglion cells and glial cells. Some studies also cultured cells on poly-L-Lysine- or poly-D-Lysine-pretreated glass slides to facilitate attachment[19,20]. In addition, a tectal extract of polylysine plus rat tail collagen increased adherence of cells compared with polylysine or rat tail collagen alone[21,22], and a tectal extract of cellular adhesion molecule L1, anti-Thy-1 monoclonal antibody, and vitronectin were also used[23]. The present study utilized polylysine (molecular weight 70 000)-coated glass slides. Polylysine can support neuronal growth and increase cell adherence and growth. In addition, Dulbecco's modified Eagle's medium containing newborn calf serum, F-12 nutrient mixtures, and glutamine were used, which provided sufficient nutrition and energy for neuronal adherence, to shorten the time of adherence to approximately 24 hours after inoculation. (5) Selection of maintenance medium: serum-containing Dulbecco's modified Eagle's medium has been frequently utilized for in vitro culture of neurons, generally supplemented with mitotic inhibitors (cytarabine) to prevent hyperplasia of glial cells and fibroblasts[24]. However, this medium is designed to supply the required nutrition for continuous proliferation, but is not suitable for neuronal growth. During isolation of cell suspensions of nerve tissues, glial cells, fibroblasts, and other non-neuron cells cannot be completely removed[25]. They are toxic to neurons, and could possibly lead to cell death[26]. Thus, the present study replaced Dulbecco's modified Eagle's medium with neurobasal medium/B27 serum-free supplements to maintain cultured cells that stably adhered. Serum-free neurobasal medium is utilized specifically for nerve cells. This medium can support purified neuronal growth, promote process differentiation and extension, and not allow continuous division or proliferation of glial cells and fibroblasts[27]. The medium also inhibits the effects of various components in the serum, resulting in neurons with active growth, high density and purity, but with a small number of non-neuron cells.
Neurofilament is a cytoskeletal protein and a marker for mature axons. It is specifically distributed in the body and processes of neurons[28]. Neurofilament-positive staining can display the cell body, dendrites, and thick axons of neurons. Moreover, anti-neurofilament immunocytochemical staining has high sensitivity that can help visualize neuronal morphology[29]. In this study, the majority of retinal cells cultured for 5–7 days were positive for anti-neurofilament antibody, manifested as brownish-yellow particles in the cell body and processes, with darkly stained cell body and lightly stained processes. This indicated that neurofilament mainly accumulated in the cell body and processes, demonstrating that these cells have properties of retinal neurons[30], consistent with Nissl staining. Glial fibrillary acidic protein is specific to glial cells. Anti-glial fibrillary acidic protein immunocytochemical studies showed that the percentage of glial fibrillary acidic protein antibody-positive cells was less than 30% of total retinal cells. This confirmed that the purity of the neurons was over 70%, supporting previous results[31]. Thus, this method can be used to culture retinal neurons with high purity, long survival time, and intact cell morphology, which provides conditions for studies of neuroprotection and drug action of retinal neurons.
High glucose is an important factor during onset of diabetic retinopathy. An earlier study showed that glucose at concentrations of 25–30 mM in an in vitro culture system is equivalent to a high glucose state of diabetes in vivo[5]. The MTT assay and flow cytometry showed that 5.5 mM glucose had minimal effect on neurons, but cell viability gradually decreased and apoptosis rate increased with prolonged glucose concentration and culture time. In particular, the effects of glucose treatment at 35 mM for 72 hours were the most significant. Preliminary studies showed that all cells almost died when cultured with glucose over 40 mM, consistent with the results of a previous study that retinal vascular endothelial cells were badly damaged in response to glucose over 80 mM[32]. This study utilized an annexin V/fluorescein isothiocyanate apoptosis detection kit, and flow cytometry demonstrated that apoptosis was the main injury to retinal neurons induced by high glucose. The early apoptosis rate was high in the 15–35 mM glucose treatment groups, possibly because either annexin V-propidium iodide detection was highly sensitive[33] and/or reduced viability and increased apoptosis resulted from long durations of in vitro culture[34]. In addition, the cell source may influence the apoptosis rate. For example, a large number of neurons in neonatal rats apoptosed within one week after birth[35,36]. MTT and flow cytometry confirmed that 35 mM glucose treatment for 72 hours was the optimal concentration and intervention time for simulating the high glucose state in vivo.
Recent studies have utilized glucose at 15–33 mM to establish a neuron model of high glucose[10,11]. There are some differences in high glucose models due to the objectives of the studies, the culture media used, the purity of retinal neurons, and the varied experimental animals and conditions. For example, Dulbecco's modified Eagle's medium/F-12 culture media containing cytarabine to inhibit glial cell proliferation was used, but the glucose concentration was very low. In the present study, Neurobasal medium was used to culture retinal neurons for a short period of time (72 hours). As we did not use cytarabine, there were some glial cells, which could promote neuronal growth, partly attenuating the high glucose-induced injury. Thus, the glucose concentration used here was slightly higher than previous studies.
This also demonstrates interaction between neurons and glial cells under high glucose conditions. Additional studies may further focus on these relationships. Regarding the natural death of neurons, we established a normal control group and treated cells cultured for 3–4 days with glucose at different concentrations not more than 3 days. This can maintain cells in the best condition during experimentation, so that the experimental data were more relevant.
In summary, a retinal neuron model of high glucose was successfully established. This model can prevent overdose of glucose-induced severe neuronal injury, and inhibit natural death after long periods of intervention. Moreover, MTT and flow cytometry demonstrated neuron apoptosis in response to high glucose. It should therefore be feasible to study the neuroprotective role of drugs in early diabetic retinopathy. Furthermore, the results of this study provide novel ideas for treating neuronal changes in early diabetic retinopathy.
MATERIALS AND METHODS
Design
Parallel study of cytology.
Time and setting
The experiments were performed at the Central Laboratory of Third Xiangya Hospital, Central South University, China from October 2009 to October 2010.
Materials
A total of 20 Wistar neonatal rats, within 1–3 days of birth, were provided by the Department of Experimental Animals, Xiangya Medical College, Central South University, China [license No. SCXK (Xiang) 2006-0002]. All protocols were conducted in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, formulated by the Ministry of Science and Technology of China[37].
Methods
Isolation and primary culture of retinal neurons
The neonatal rats were sacrificed by drowning in 75% alcohol. After disinfection for 5 minutes, the eyeballs were harvested in a sterile manner, and washed twice in D-Hanks solution. The eyeballs were cut open at 0.5 mm posterior to corneal limbus using a dissecting microscope (Olympus, Tokyo, Japan). The retina was dissociated in D-Hanks solution and cut into pieces, triturated into a single cell suspension, and digested with 0.05% trypsin (Gibco, Carlsbad, CA, USA) at 37°C for 15 minutes, which was terminated by Dulbecco's modified Eagle's medium/F-12 culture solution (Gibco) containing fetal bovine serum. The suspension was filtrated through a stainless steel mesh, centrifuged at 800–1 000 r/min for 5 minutes, resuspended in medium, and centrifuged. This process was repeated twice. The upper suspension was harvested, and the number of cells was quantified using a light microscope (Olympus). Cells at a density of 1 × 106/mL were added to 12 mm × 12 mm coverslips and incubated in polylysine-coated 6-well culture plates with 80% Dulbecco's modified Eagle's medium/F-12 in 5% CO2 at 37°C. Maintenance medium 1 [Neurobasal (Gibco), B27 supplement, 0.06 g/L glutamine, 100 U/mL penicillin, and 50 μg/mL streptomycin] was used after 24 hours, and maintenance medium 2 (Neurobasal, B27 supplement, 100 U/mL penicillin, and 50 μg/mL streptomycin) was used at 72 hours. One third of the medium was renewed every 1–2 days. Cell morphology was observed daily using an inverted microscope (Olympus) to identify quantity and length of processes and cell adherence.
Identification of retinal neurons
For Nissl staining, cell slices of 5–6 day primary cultures of retinal neurons were fixed in 4% paraformaldehyde for 25 minutes, washed with double distilled water three times, 5 minutes each, mixed with 10 g/L toluidine blue solution (Invitrogen, Carlsbad, CA, USA), incubated at 54°C for 30 minutes, cooled, and washed twice with double distilled water with a wash of 5 minutes each, rapidly washed with 95% alcohol, dehydrated with absolute alcohol, cleared with xylene, and mounted in neutral gum. Five high power fields (400 ×) from each coverslip were randomly observed, and the number of positive neurons in every 100 cells in every field of view was quantified using a light microscope. The positive neuron amount in each field of view was calculated using the formula: percentage of neurons (%) = number of neurons/total number of cells × 100%[1]. Measurements were conducted in triplicate, and the mean value was calculated.
For immunocytochemical staining, retinal neuronal and glial cells were observed using anti-neurofilament and anti-glial fibrillary acidic protein immunocytochemical staining methods[2]. Briefly, cells were harvested from the coverslips, fixed with 4% paraformaldehyde at room temperature for 30 minutes, washed with PBS three times, 3 minutes each, blocked with 0.3% methanol in H2O2 at room temperature for 20 minutes, incubated with PBS containing 1% fetal bovine serum and 0.5% Triton X-100 for 20 minutes, followed by PBS containing 5–10% normal goat serum for 20 minutes. The cells were incubated with rabbit anti-rat neurofilament (1:100; Maixin-Bio, Fujian, China) and glial fibrillary acidic protein (1:50; Sigma, St. Louis, MO, USA) monoclonal antibody at room temperature for 2 hours, then overnight at 4°C, then washed with PBS three times, for 3 minutes each. The negative control was treated with PBS rather than primary antibody. Cells were incubated with biotinylated goat anti-rabbit IgG (1:200; Sigma) and peroxide-labeled streptavidin (Gibco), followed by coloration with 0.01 M Tris-HCl (pH 7.4) containing 0.05% diaminobenzidine and 0.03% H2O2. Coloration time was controlled using a microscope to terminate the reaction time. Cells were dehydrated and photographed. Glial cells were identified as follows: after nuclei were hematoxylin counterstained for 25 seconds, cells were washed with tap water, dehydrated with gradient alcohol, cleared with xylene, and mounted with neutral gum. Cells stained brownish-yellow were neuron-positive staining, cells with brownish-yellow or brown stained particles in the cytoplasm and membrane were glial cell-positive staining. Five high power fields (400 ×) from each coverslip were randomly observed, and the number of neurofilament and glial fibrillary acidic protein positive neurons in every 100 cells in every field of view was quantified. The percentage of positive cells out of the total number of cells was calculated. Measurements were conducted in triplicate, and the mean value was calculated.
Establishment of a retinal neuron model of high glucose
According to a previously described method, cells were treated with 0 mM (normal control), 5.5, 15, 25, and 35 mM glucose (Sigma) for 24, 48, and 72 hours. Cell survival rate was determined using the MTT assay, and the apoptosis rate detected was determined using flow cytometry. The optimal glucose concentration and intervention duration for establishing the high glucose model were confirmed according to experimental results.
MTT assay for retinal neuron survival
Primary cultures of retinal neurons for 3–4 days were trypsinized (0.25%). A single cell suspension was prepared using serum-free culture medium, and cells at a concentration of 1–3 × 105 cells/mL were seeded into 96-well culture plates, with 100 μL in each well. The culture media containing different concentrations of glucose was replaced at a cell confluency of 80%. After culture for 24, 48, and 72 hours, 20 μL MTT solution (0.5 mg/mL, Sigma) was added to each well and cultured for 4 hours, treated with 100 μL dimethyl sulfoxide after the supernatant was discarded, and shaken at room temperature for 10 minutes to completely dissolve crystals. Absorbance at 570 nm was detected using an enzyme-linked immunosorbent assay reader (Perkin Elmer, Turku, Finland). The experiment was conducted in triplicate, and the mean value was calculated. Higher absorbance value represented more surviving cells.
Flow cytometry for apoptosis
Cells treated with different concentrations of glucose for 24, 48, and 72 hours were harvested, and digested with 0.25% trypsin (200 μL) in each well. The reaction was terminated by adding 10% fetal bovine serum after the cells shrunk. Cells were detached by trituration, centrifuged at 1 000 r/min for 5 minutes, washed with PBS after the supernatant was discarded, and centrifuged. The process was conducted in triplicate. A single cell suspension was prepared using PBS, diluted with deionized water at a ratio of 1:4, and washed with PBS twice. Cells were resuspended using 250 μL binding buffer, and cell concentration was adjusted to 1 × 106 cells/ mL. The cell suspension (100 μL) was placed in a 5 μL flow tube, incubated with 1 μL annexin/fluorescein isothiocyanate (Bender MedSystems, Vienna, Austria) and 5 μL propidium iodide (20 μg/mL) for 15 minutes in the dark, followed by 400 μL PBS. Cell apoptosis was analyzed using flow cytometry (Becton Dickinson, San Jose, CA, USA).
Statistical analysis
Data were expressed as mean ± SD and analyzed using SPSS 13.0 software (SPSS, Chicago, IL, USA). Comparison of the multiple group mean was conducted using one-way analysis of variance, and multiple comparisons among groups were analyzed using the Least Significant Difference test. A value of P < 0.05 was considered statistically significant.
Acknowledgments:
We thank Renhong Tang and his team for technical assistance and the staff from the Third Xiangya Hospital of Central South University in China for their help.
Footnotes
Yu Liu, M.D., Attending physician.
Funding: This study was supported by the Department of Health of Hunan Province, No. B2009-050, and the Science and Technology Foundation of Hunan Province, No. 2012FJ4077.
Conflicts of interest: None declared.
Ethical approval: This study received permission from the Animal Ethics Committee of Central South University, China.
(Edited by Wang YS, Xiu B/Su LL/Song LP)
REFERENCES
- [1].Wu XY, Zhang D, Liu SZ. Primary culture of retinal neurons of newborn rat. Guoji Yanke Zazhi. 2008;8(1):45–46. [Google Scholar]
- [2].Bai HQ, Wang JH, Wang DB, et al. Short period culture of retinal neurons and ganglion cells of neonatal rat in vitro. Yanke Xin Jinzhan. 2004;2(2):107–110. [Google Scholar]
- [3].Yao J, Li XX. In vitro culture of retinal neuron cells of rat. Yanke Yanjiu. 2004;22(4):340–342. [Google Scholar]
- [4].Zhang YJ, Xu HX, Zeng KH, et al. Influence of Taurine on the expression of GFAP and TAUT in the Müller cells from retina in high glucose culture. Xiandai Shengwu Yixue Jinzhan. 2007;7(5):649–652. [Google Scholar]
- [5].Fletcher EL, Phipps JA, Ward MM, et al. Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr Pharm Des. 2007;13(26):2699–2712. doi: 10.2174/138161207781662920. [DOI] [PubMed] [Google Scholar]
- [6].Zeng K, Xu H, Mi M, et al. Effects of taurine on glial cells apoptosis and taurine transporter expression in retina under diabetic conditions. Neurochem Res. 2010;35(10):1566–1574. doi: 10.1007/s11064-010-0216-1. [DOI] [PubMed] [Google Scholar]
- [7].McBain VA, Robertson M, Muckersie E, et al. High glucose concentration decreases insulin-like growth factor type 1-mediated mitogen-activated protein kinase activation in bovine retinal endothelial cells. Metabolism. 2003;52(5):547–551. doi: 10.1053/meta.2003.50046. [DOI] [PubMed] [Google Scholar]
- [8].Kane R, Stevenson L, Godson C, et al. Gremlin gene expression in bovine retinal pericytes exposed to elevated glucose. Br J Ophthalmol. 2005;89(12):1638–1642. doi: 10.1136/bjo.2005.069591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yuan Z, Feng W, Hong J, et al. p38MAPK and ERK promote nitric oxide production in cultured human retinal pigmented epithelial cells induced by high concentration glucose. Nitric Oxide. 2009;20(1):9–15. doi: 10.1016/j.niox.2008.09.001. [DOI] [PubMed] [Google Scholar]
- [10].Layton CJ, Wood JP, Chidlow G, et al. Neuronal death in primary retinal cultures is related to nitric oxide production, and is inhibited by erythropoietin in a glucose-sensitive manner. J Neurochem. 2005;92(3):487–493. doi: 10.1111/j.1471-4159.2004.02876.x. [DOI] [PubMed] [Google Scholar]
- [11].Santiago AR, Cristóvão AJ, Santos PF, et al. High glucose induces caspase-independent cell death in retinal neural cells. Neurobiol Dis. 2007;25(3):464–472. doi: 10.1016/j.nbd.2006.10.023. [DOI] [PubMed] [Google Scholar]
- [12].Takano M, Sango K, Horie H, et al. Diabetes alters neurite regeneration from mouse retinal explants in culture. Neurosci Lett. 1999;275(3):175–178. doi: 10.1016/s0304-3940(99)00768-5. [DOI] [PubMed] [Google Scholar]
- [13].Xu HX, Mi MT, Huang GR, et al. Purified retinal ganglion cells cultured in serum-free neurobasal medium. Zhonghua Yandibing Zazhi. 2006;22(3):200–202. [Google Scholar]
- [14].Hattar S, Kumar M, Park A, et al. Central projections of melanopsin-expressing retinal ganglion cells in the mouse. J Comp Neurol. 2006;497(3):326–349. doi: 10.1002/cne.20970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu FL, Niu YJ, Wang JB, et al. Primary culture of retinal neuron cells of neonatal rat. Shandong Daxue Xuebao: Yixueban. 2006;44(7):684–688. [Google Scholar]
- [16].Zheng ZH, Lin L. Beijing: Science Press; 2002. Theory and Practice of Neurons Culture. [Google Scholar]
- [17].Benowitz L, Yin Y. Rewiring the injured CNS: lessons from the optic nerve. Exp Neurol. 2008;209(2):389–398. doi: 10.1016/j.expneurol.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Takahashi N, Cummins D, Caprioli J. Rat retinal ganglion cells in cultures. Exp Eye Res. 1991;53(2):565–572. doi: 10.1016/0014-4835(91)90214-y. [DOI] [PubMed] [Google Scholar]
- [19].Carri NG, Rubin K, Gullberg D, et al. Neuritogenesis on collagen substrates. Involvement of integrin-like matrix receptors in retinal fibre outgrowth on collagen. Int J Dev Neurosci. 1992;10(5):393–405. doi: 10.1016/0736-5748(92)90029-y. [DOI] [PubMed] [Google Scholar]
- [20].Seil FJ. The extracellular matrix molecule, laminin, induces purkinje cell dendritic spine proliferation in granule cell depleted cerebellar cultures. Brain Res. 1998;795(1-2):112–120. doi: 10.1016/s0006-8993(98)00265-0. [DOI] [PubMed] [Google Scholar]
- [21].Hayashida Y, Partida GJ, Ishida AT. Dissociation of retinal ganglion cells without enzymes. J Neurosci Methods. 2004;137(1):25–35. doi: 10.1016/j.jneumeth.2004.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou MH, Zhao LP. The growth effect of tectal extract on neonatal rat retinal neurons cultured on various substrata. Jiepou Xuebao. 1991;22(3):195–198. [Google Scholar]
- [23].Brunetti V, Maiorano G, Rizzello L, et al. Neurons sense nanoscale roughness with nanometer sensitivity. Proc Natl Acad Sci U S A. 2010;107(14):6264–6269. doi: 10.1073/pnas.0914456107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wada T, Honda M, Minami I, et al. Highly efficient differentiation and enrichment of spinal motor neurons derived from human and monkey embryonic stem cells. PLoS One. 2009;4(8):e6722. doi: 10.1371/journal.pone.0006722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Graham JM. Isolation of rat and human hippocampal neuron fractions in a discontinuous density gradient. Sci World J. 2002;2:1634–1637. doi: 10.1100/tsw.2002.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lilley S, Robbins J. The rat retinal ganglion cell in culture: an accessible CNS neurone. J Pharmacol Toxicol Methods. 2005;51(3):209–220. doi: 10.1016/j.vascn.2004.08.009. [DOI] [PubMed] [Google Scholar]
- [27].Xu DF, Zhu CT, Liu DM, et al. A comparative study of three sorts of nutrient medium on primary hippocampal neurons culture. Anhui Yike Daxue Xuebao. 2006;41(5):512–514. [Google Scholar]
- [28].Kino T, Jaffe H, Amin ND, et al. Cyclin-dependent kinase 5 modulates the transcriptional activity of the mineralocorticoid receptor and regulates expression of brain-derived neurotrophic factor. Mol Endocrinol. 2010;24(5):941–952. doi: 10.1210/me.2009-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Julien JP, Mushynski WE. Neurofilaments in health and disease. Prog Nucleic Acid Res Mol Biol. 1998;61(1):1–23. doi: 10.1016/s0079-6603(08)60823-5. [DOI] [PubMed] [Google Scholar]
- [30].Kikuchi M, Kashii S, Honda Y, et al. Protective effects of methylcobalamin, a vitamin B12 analog, against glutamate-induced neurotoxicity in retinal cell culture. Invest Ophthalmol Vis Sci. 1997;38(5):848–854. [PubMed] [Google Scholar]
- [31].Weinberg JB, Chen Y, Jiang N, et al. Inhibition of nitric oxide synthase by cobalamins and cobinamides. Free Radic Biol Med. 2009;46(12):1626–1632. doi: 10.1016/j.freeradbiomed.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xia XG, Yin Z. Protective function of 7-Difluoromethylyl-5,4º-Di-methoxyl isoflavone on the damage of hyperglycemic simian retinal endothelial cells. Yanke Xin Jinzhan. 2008;28(2):113–115. [Google Scholar]
- [33].Gou L, Moss SE, Alexander RA, et al. Retinal ganglion cell apoptosis in glaucoma is related to intraocular pressure and IOP-induced effects on extracellular matrix. Invest Ophthalmol Vis Sci. 2005;46(1):175–182. doi: 10.1167/iovs.04-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Spalding KL, Rush RA, Harvey AR. Target-derived and locally derived neurotrophins support retinal ganglion cell survival in the neonatal rat retina. J Neurobiol. 2004;60(3):319–327. doi: 10.1002/neu.20028. [DOI] [PubMed] [Google Scholar]
- [35].Hu H, Lu W, Zhang M, et al. Stimulation of the P2X7 receptor kills rat retinal ganglion cells in vivo. Exp Eye Res. 2010;91(3):425–432. doi: 10.1016/j.exer.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Johnson EC, Guo Y, Cepurna WO, et al. Neurotrophin roles in retinal ganglion cell survival: lessons from rat glaucoma models. Exp Eye Res. 2009;88(4):808–815. doi: 10.1016/j.exer.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals 2006-09-30 [Google Scholar]