

Keywords: neural regeneration, brain injury, hypobaric hypoxia, cerebral edema, mitochondria, proteomics, differential expression, energy metabolism, electron transport chain, cognitive function, grants-supported paper, neuroregeneration
Abstract
Hypobaric hypoxia can cause severe brain damage and mitochondrial dysfunction, and is involved in hypoxic brain injury. However, little is currently known about the mechanisms responsible for mitochondrial dysfunction in hypobaric hypoxic brain damage. In this study, a rat model of hypobaric hypoxic brain injury was established to investigate the molecular mechanisms associated with mitochondrial dysfunction. As revealed by two-dimensional electrophoresis analysis, 16, 21, and 36 differential protein spots in cerebral mitochondria were observed at 6, 12, and 24 hours post-hypobaric hypoxia, respectively. Furthermore, ten protein spots selected from each hypobaric hypoxia subgroup were similarly regulated and were identified by mass spectrometry. These detected proteins included dihydropyrimidinase-related protein 2, creatine kinase B-type, isovaleryl-CoA dehydrogenase, elongation factor Ts, ATP synthase beta-subunit, 3-mercaptopyruvate sulfurtransferase, electron transfer flavoprotein alpha-subunit, Chain A of 2-enoyl-CoA hydratase, NADH dehydrogenase iron-sulfur protein 8 and tropomyosin beta chain. These ten proteins are all involved in the electron transport chain and the function of ATP synthase. Our findings indicate that hypobaric hypoxia can induce the differential expression of several cerebral mitochondrial proteins, which are involved in the regulation of mitochondrial energy production.
INTRODUCTION
High-altitude environments can cause altitude sickness, and with the increasing numbers of people working, traveling and living in high-altitude environments, the incidence of high-altitude illness is growing[1,2]. High-altitude illness encompasses an array of conditions that may occur in individuals who travel to high elevations, including acute mountain sickness, high-altitude cerebral edema and high-altitude pulmonary edema[3,4]. Hypoxia is generally considered the root cause of altitude spectrum illnesses, and high-altitude environments have been shown to cause hypobaric hypoxia because of diminished oxygen pressure. The partial pressure of oxygen falls with increasing altitude, resulting in an anoxic challenge to any individual ascending to high altitudes. Especially after rapid ascent to a high altitude, an individual can develop hypobaric hypoxia-related illnesses. As more people visit high altitudes, altitude related medical problems are gaining wider attention, and much greater awareness has been given to hypobaric hypoxia studies.
Cerebral oxygen consumption is known to occupy nearly 20% of total body oxygen consumption. Thus, the brain is greatly affected at high-altitudes. Although compensatory hyperventilation, tachycardia, and increased cerebral blood flow can partially maintain cerebral oxygen delivery at high altitudes, a spectrum of cerebral damage can occur when the hypoxic stress outweighs the subject's ability to acclimatize. Hypobaric hypoxia causes severe brain injury in humans and rats with a variety of alterations in cerebral blood flow, energy metabolism and cognitive functions such as learning and memory[5,6,7]. Recently, hypobaric hypoxia- induced cerebral damage has been recognized, but the exact molecular mechanisms remain speculative.
What happens to brain tissue during hypoxia? What organelles are the most important to maintain nerve cell survival? Mitochondria are unique organelles that supply cellular energy as ATP[8,9] and play an important role in brain energy metabolism[10,11,12]. In mitochondria, sufficient oxygen is required to generate ATP for cell survival, and hypoxic conditions may hamper the energy metabolic pathway leading to cell damage and death in the brain. High-latitude environments can cause decreased oxygen partial pressure, affect respiratory activity and energy synthesis in brain mitochondria, thus triggering brain energy metabolism disorders[13]. Therefore, mitochondrial dysfunction is critically involved in cerebral hypoxic damage.
Some studies have shown that hypoxic mitochondrial dysfunction is associated with the inhibition of the electron transport chain and a decrease in ATP synthesis[14]. However, what is the molecular mechanism responsible for mitochondrial dysfunction in hypobaric hypoxia-induced cerebral impairment? Therefore, the purpose of this study was to evaluate the degree of brain damage with different exposure times to hypoxia and further investigate the differential expression of cerebral mitochondrial proteins by comparative proteomic analysis, in a broader attempt to search for treatment targets of hypobaric hypoxia brain injury.
RESULTS
Quantitative analysis of experimental animals
Sixty female rats were randomly divided into four groups with 15 rats in each group: 6-hour, 12-hour, 24-hour hypobaric hypoxia groups and a control group. Hypobaric hypoxia groups were exposed to a barometric pressure (0.06 MPa) for 6, 12 and 24 hours in a specially designed decompression chamber. Control group rats were kept at a normal atmospheric pressure. All 60 rats were involved in the final analysis.
Effect of hypobaric hypoxia on the general behavior of rats
The survival rate of rats was 100% in both the control and hypobaric hypoxia groups. The activity and diet of rats decreased, and their respiration increased in frequency after exposure to hypobaric hypoxia.
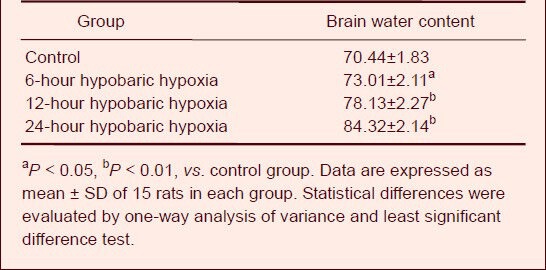
Effect of hypobaric hypoxia on cerebral cortical morphology and brain water content
Following hypobaric hypoxia treatment, the morphology of injured neurons appeared to change and included cell shrinkage, karyopyknosis and karyolysis. In addition, brain damage became more severe with increasing duration of hypobaric hypoxia (Figure 1). The brain water content of rats in the hypobaric hypoxia groups was markedly elevated in a time-dependent manner (P < 0.05 or P < 0.01) when compared with the control group (Table 1).
Figure 1.
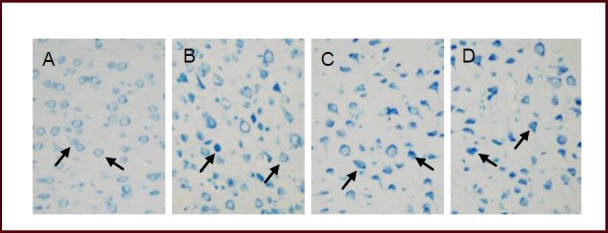
Effect of hypobaric hypoxia on the morphology of cerebral cortical nerve cells (arrows) using toluidine blue staining (× 400).
(A) Neurons in the control group exhibited a normal size, shape and an ordered arrangement.
(B–D) Following exposure to hypobaric hypoxia for 6, 12 and 24 hours, a large number of neurons exhibited cell shrinkage, karyopyknosis as well as karyolysis, and the degree of injury was enhanced with increasing duration.
Table 1.
Effect of hypobaric hypoxia on brain water content (%) in each group
Purification and identification of mitochondria
Cerebral mitochondria were extracted and purified with the Nycodenz density gradient centrifugation method, with over 95% purity. A transmission electron microscope was used to assess the ultrastructure of the isolated mitochondria, and the results showed that mitochondria in control group were isolated with an intact structure (Figure 2). This evidence suggests that complete mitochondria can be extracted using this method.
Figure 2.
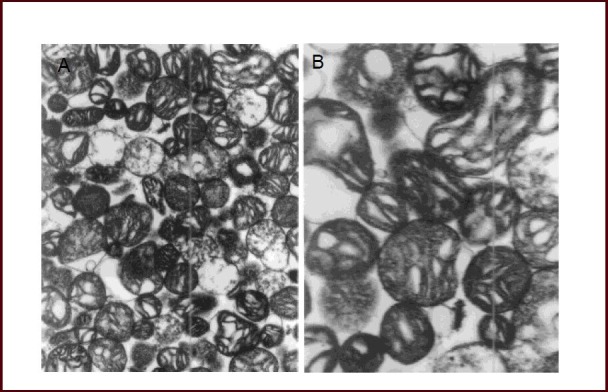
Intact structure of cerebral mitochondria in normal rats (transmission electron microscope).
The ultrastructure of mitochondria was observed in (A, × 10 000) and (B, × 20 000) using transmission electron microscopy (PHILIPS CM120). Intact cerebral mitochondria with integrated structure were isolated.
Effect of hypobaric hypoxia on differentially expressed mitochondrial proteins in the rat brain
After exposure to hypobaric hypoxia, the differential expression of mitochondrial proteins was determined using two-dimensional electrophoresis. Compared with spots from the control group, the quantities of several protein spots were found to be different in the hypobaric hypoxia groups, including the increase of 12 spots and decrease of 4 spots in the 6-hour hypobaric hypoxia group, the increase of 16 spots and decrease of 5 spots in the 12-hour hypobaric hypoxia group, and the increase of 30 spots and decrease of 6 spots in the 24-hour hypobaric hypoxia group (Figure 3). Finally, ten protein spots whose expression changed in a time-dependent manner were selected and further identified using mass spectrometry (Table 2). Proteins A–E and G were down- regulated, while proteins F, H, I and J were up-regulated after hypoxia (Figure 4).
Figure 3.
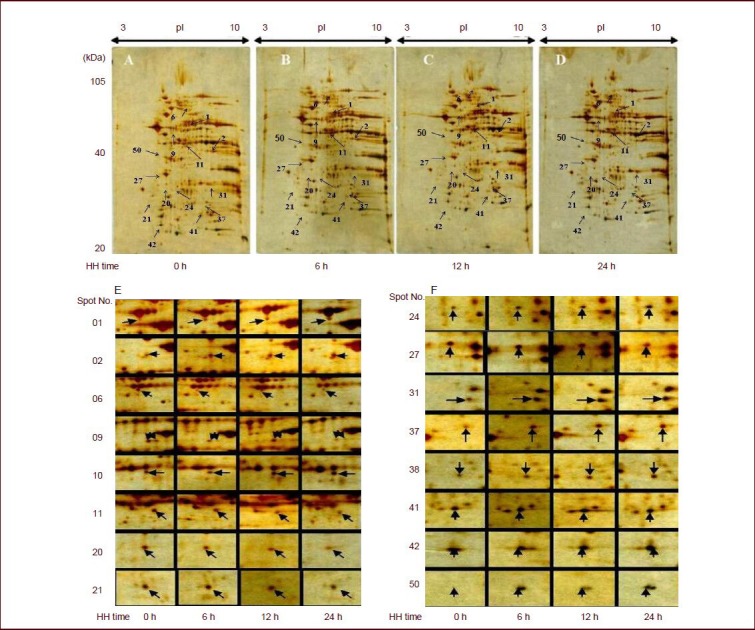
Effect of hypobaric hypoxia (HH) on the expression of mitochondrial proteins in rat brain tissue (two-dimensional electrophoresis analysis, silver staining, 17 cm immobilized pH gradient strip pH 3–10).
Samples of mitochondria in control (A), 6- (B), 12- (C) and 24-hour (D) hypobaric hypoxia groups are shown. Enlarged two-dimensional electrophoresis images (E, F) showed differential expression of mitochondrial proteins after hypobaric hypoxia in the rat cerebral cortex when compared with the control group. The expression of protein 06, 09, 11, 20, 21, and 27 was down-regulated and the expression of protein 01, 02, 10, 11, 24, 31, 37, 38, 41, 42, and 50 was up-regulated after hypoxia.
Table 2.
Mitochondrial proteins identified by matrix-assisted laser desorption/ionization-time of flight mass spectrometry after exposure to hypobaric hypoxia
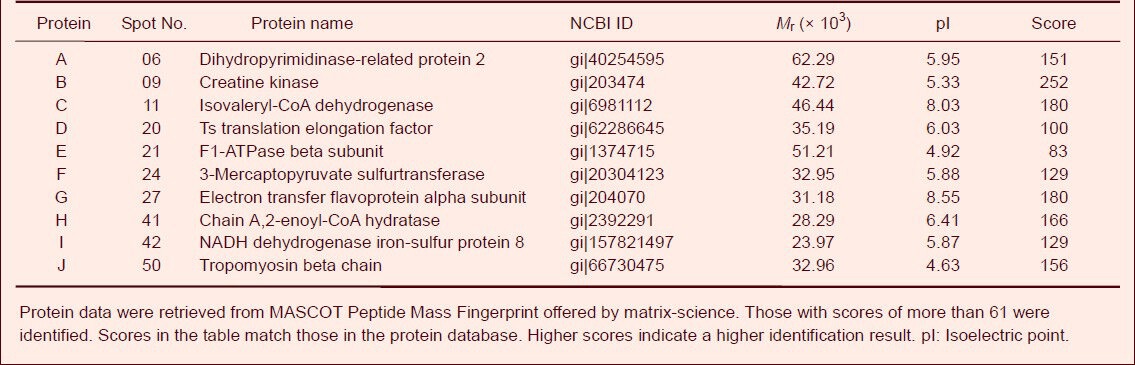
Figure 4.
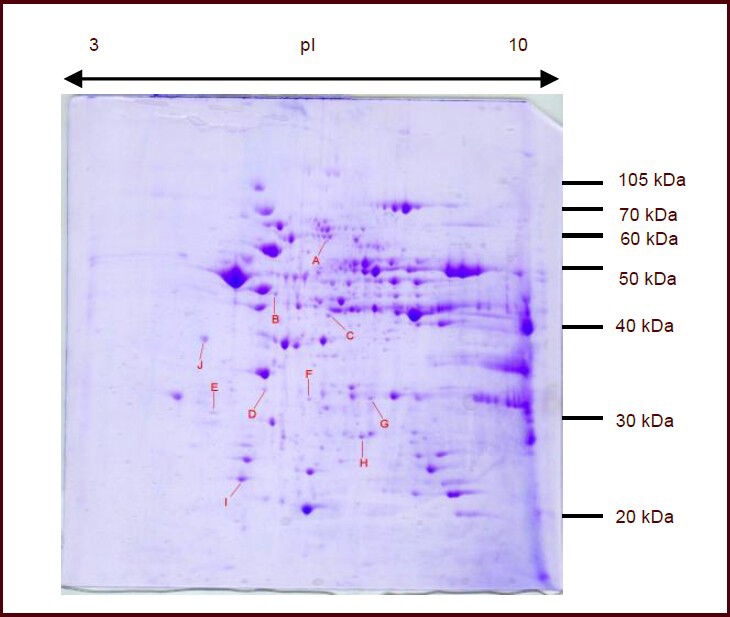
Effect of hypobaric hypoxia on the expression of mitochondrial proteins in rat brain tissue (two-dimensional electrophoresis analysis, Coomassie Blue R-250 staining).
Ten significantly changed protein spots were selected and subjected to matrix-assisted laser desorption/ionizationtime of flight mass spectrometry for protein identification. A–J refers to the serial number of protein spots. The expression of A–E and G proteins was down-regulated, while the expression of F, H, I and J proteins was upregulated after hypoxia. pI: Isoelectric point.
Effect of hypobaric hypoxia on the identification of differential mitochondrial proteins in the rat brain
Ten protein spots in two-dimensional electrophoresis gels were isolated and subjected to matrix-assisted laser desorption/ionization-time of flight mass spectrometry. The peptide mass peaks were compared with those in the NCBI database. These proteins were identified as dihydropyrimidinase-related protein 2, creatine kinase B-type, isovaleryl-CoA dehydrogenase, elongation factor Ts, ATP synthase beta-subunit, 3-mercaptopyruvate sulfurtransferase, electron transfer flavoprotein alpha-subunit, Chain A of 2-enoyl-CoA hydratase, NADH dehydrogenase iron-sulfur protein 8 and tropomyosin beta chain (Table 2, Figures 5, 6).
Figure 5.
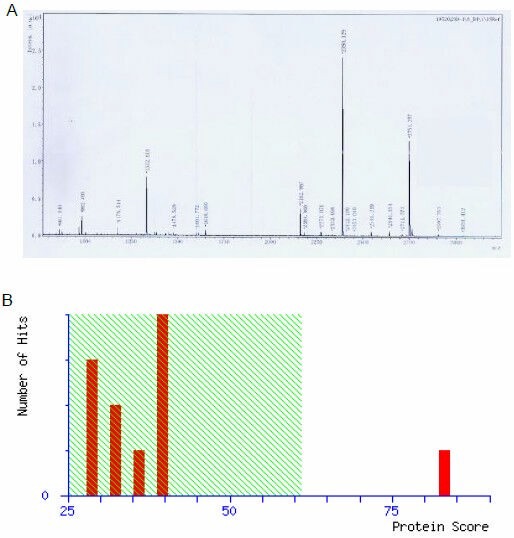
Mass spectrum of spot No. 21 in cerebral mitochondria after rats were exposed to hypobaric hypoxia and analysis results in Mascot software.
(A) Mass spectrum of spot No. 21 (F1-ATPase beta subunit was down-regulated after hypobaric hypoxia) and fourteen peptide-peaks were found. (B) Analysis results in Mascot software. The top score was 82 (P < 0.05) and the F1-ATPase beta subunit was confirmed.
Figure 6.
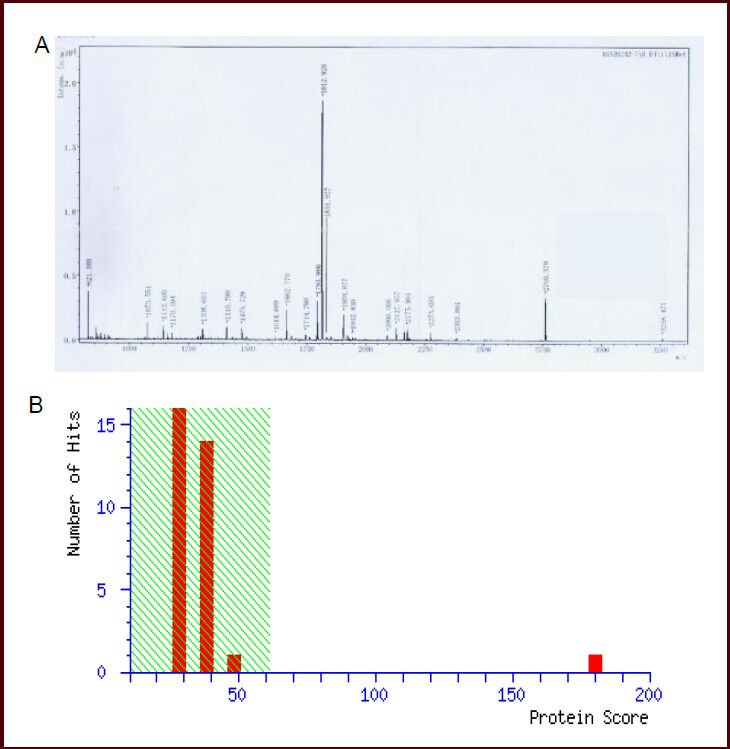
Mass spectrum of spot No. 27 in cerebral mitochondria after rats were exposed to hypobaric hypoxia and analysis results in Mascot software.
(A) Mass spectrum of spot No. 27 (electron transfer flavoprotein alpha subunit was down-regulated after hypobaric hypoxia). Thirty-two peptide-peaks were found. (B) Analysis results in Mascot software. The top score was 180 (P < 0.05) and electron transfer flavoprotein subunit alpha was confirmed.
Additionally, ATP synthase beta-subunit and electron transfer flavoprotein alpha-subunit expression were down-regulated after hypobaric hypoxia, as detected by western blot analysis (Figure 7).
Figure 7.
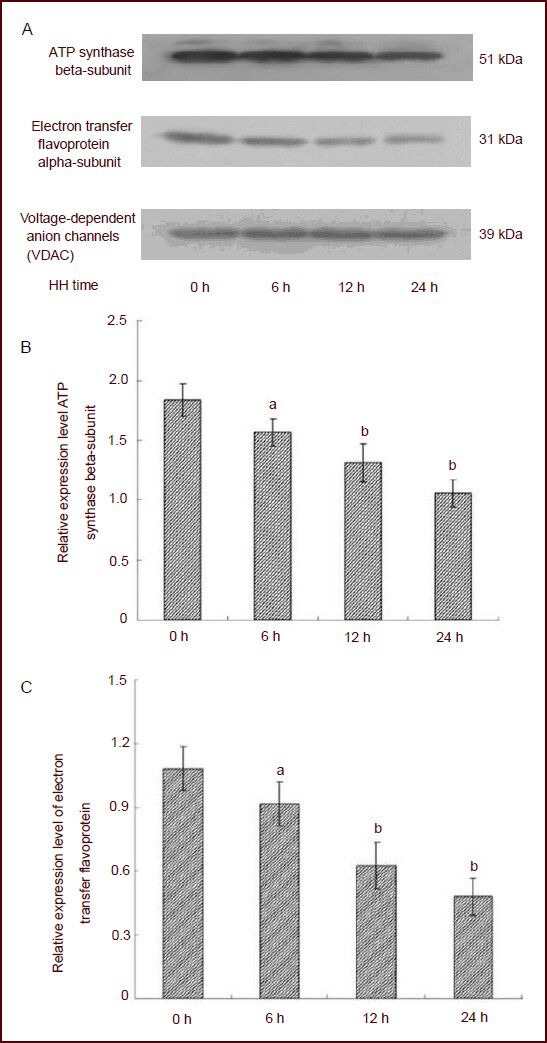
Expression of ATP synthase beta-subunit and electron transfer flavoprotein alpha-subunit in cerebral mitochondria of rats after exposure to hypobaric hypoxia (HH; western blot analysis).
(A) Sodium dodecyl sulfate polyacrylamide gel electrophoresis was used for standardization of proteins from cerebral mitochondria in each group.
(B–C) Quantification of western blot analysis. The Y-axis is the ratio of target protein to internal reference (VDAC) gray values. Compared with the control group (0 hour), ATP synthase beta-subunit and electron transfer flavoprotein alpha-subunit expression were down-regulated after hypobaric hypoxia. aP < 0.05, bP < 0.01, vs. control group (0 hour). Data are expressed as mean ± SD of 15 rats in each group; one-way analysis of variance followed by least significant difference test.
DISCUSSION
High-altitude environments can cause hypobaric hypoxia, which results in severe brain damage[15,16]. Mitochondria act as the power plants in cells[17,18] and play a pivotal role in brain energy metabolism[19,20]. Brain mitochondria are extremely sensitive to hypoxia, which in turn causes the generation of reactive oxygen species, the release of mitochondrial cytochrome C and the opening of the mitochondrial permeability transition pore[21,22]. Thus, mitochondrial dysfunction is critically involved in cerebral hypoxic damage. However, little is currently known about the mechanisms responsible for mitochondrial dysfunction in hypobaric hypoxia-induced brain damage.
To investigate the detailed pathological mechanisms and possible treatment targets of hypobaric hypoxic brain injury, it is essential to identify the expression patterns of cerebral mitochondrial proteins via comparative proteomics.
Therefore, cerebral mitochondria of rats were selected as the research target in this study, and two-dimensional electrophoresis and matrix-assisted laser desorption/ionization time of flight mass spectrometry were used to evaluate the differential expression of cerebral mitochondrial proteins in rats after exposure to hypobaric hypoxia.
After exposure to hypobaric hypoxia, rat nerve cells morphologically changed. Furthermore, the degree of injury in rat nerve cells was enhanced with increasing hypobaric hypoxia duration, suggesting that hypobaric hypoxia can cause severe brain damage in rats. It is generally considered that the brain water content is closely associated with hypoxic brain injury. In this study, a significant increase in brain water content was observed after hypobaric hypoxia, which suggested that cerebral edema is involved in brain injury induced by hypobaric hypoxia.
Moreover, there was evidence of alterations to the expression of some mitochondrial proteins during hypobaric hypoxia. Ten protein spots were found to be differentially expressed in a time-dependent manner. Interestingly, these proteins were closely associated with the function of the mitochondrial respiratory chain and mitochondrial energy production. It is generally considered that mitochondrial dysfunction can cause decreased ATP synthesis, which is mainly associated with the inhibition of the electron transport chain and impairment of ATP synthase[14]. Thus, the F1-ATPase beta-subunit and electron transfer flavoprotein alpha-subunit were selected from the ten protein spots and further validated by western blot analysis. The results were consistent with that of mass spectrometry.
It is well known that F-ATPase can produce ATP from ADP by electron transport complexes and is mainly composed of F1-ATPase complex and F0-ATPase complex[23,24,25]. In this study, we found that F1-ATPase beta-subunit was significantly down-regulated by hypobaric hypoxia, which suggested that hypobaric hypoxia caused the dysfunction of ATP synthase. Moreover, electron transfer flavoprotein serves as a specific electron acceptor and transfer electrons to the mitochondrial respiratory chain for ATP production[26,27,28]. In this study, electron transfer flavoprotein alpha-subunit expression was significantly down-regulated by hypobaric hypoxia, indicating that hypobaric hypoxia causes inhibition of the electron transport chain.
Fatty acid β-oxidation and oxidative phosphorylation are key pathways involved in cellular energetics[29]. The four reactions of the β-oxidation cycle, catalyzed by acyl-CoA dehydrogenases, enoyl-CoA hydratases, L-3-hydroxy- acyl-CoA dehydrogenase, and 3-ketoacyl-CoA thiolase, sequentially remove two carbons until acyl-CoA is converted to acetyl-CoA molecules. Mitochondria are the site of the most important energy generating pathways in humans: oxidative phosphorylation, fatty acid β-oxidation, and the tricarboxylic acid cycle. Two proteins that are related to β-oxidation were identified: Chain A, 2-enoyl- CoA hydratase and isovaleryl-CoA dehydrogenase. The increased expression levels after hypobaric hypoxia exposure indicates increased fat oxidation. Fatty acid metabolism was enhanced following deficits in energy supply induced by hypobaric hypoxia; meanwhile rich unsaturated fatty acids, which were susceptible to oxygen radicals and lipid peroxidation, were present in brain tissue.
The mammalian mitochondrial NADH dehydrogenase (complex I) is the major entry point for the electron transport chain. This large component of the respiratory chain comprises 45 protein subunits, seven of which are encoded by the human mitochondrial genome. Additionally, complex I contains stably bound cofactors, including flavin mononucleotide and eight iron-sulfur clusters. Deficiency in complex I function has been associated with various human diseases including neurodegenerative diseases and the aging process. NADH dehydrogenase [ubiquinone] iron-sulfur protein 8, the core subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (complex I), is a small submit of approximately 23 kDa containing 210 amino-acid residues and is believed to belong to the minimal assembly required for catalysis. In hypobaric hypoxia-induced stress, cells suffer from a displaced NAD/NADH ratio with an overbalance of NADH. Therefore, the decreased level of enzyme production reported in our study can be explained by the detailed mechanism that develops during hypobaric hypoxia-induced stress, which causes damage to the mitochondrial compartment from the excessive levels of proton production and subsequent acidosis.
Creatine kinase is a central controller of cellular energy homeostasis. By reversible interconversion of creatine into phosphocreatine, creatine kinase together with mitochondrial creatine kinase isoenzymes provide temporal and spatial energy buffering of ATP levels to maintain cellular energy homeostasis. Thus, creatine kinase plays a particularly important role in tissues with large and fluctuating energy demands like muscle and the brain, with the mitochondrial isoenzyme of creatine kinase being important for the energetics of oxidative tissue. The activity of mitochondrial isoenzyme of creatine kinase is regulated by ATP/ADP substrate concentrations. Under situations of compromised cellular energy states, which are linked to ischemia, oxidative stress and calcium overload, the function of mitochondrial isoenzyme of creatine kinase is directly impaired by oxidative and radical damage, or by the up-regulation of mitochondrial creatine kinase isoenzyme expression as a compensatory measure for deficits in energy supply. Both of these events may either impair or reinforce the functions of mitochondrial creatine kinase isoenzyme complexes in cellular energy supply and protection of mitochondria from so-called permeability transition, which leads to apoptosis or necrosis. Creatine kinase showed lower levels of expression in the hypobaric hypoxia groups when compared with the control.
The presence of the cytoskeleton protein tropomyosin beta chain was observed in the treatment group. This may be a result of protein recovery during the separation protocol. Even though these skeletal proteins were not of mitochondrial origin, and hence not part of our major scope, they proved to be the primary track for mitochondrial transport, to be attached to mitochondrial membranes, and to regulate intracellular mitochondrial localization as well as proliferation. The expression of tropomyosin beta chain increased in the hypobaric hypoxia groups when compared with the control group, which may suggest that hypobaric hypoxia exposure could affect cytoskeletal and mitochondrial movement, and positioning in cells.
In addition, our previous study found that superoxide dismutase activity was reduced and malondialdehyde content was increased in the brain during hypobaric hypoxia[30], which suggested that hypobaric hypoxia causes the accumulation of oxygen radicals. In conclusion, our study indicates that hypobaric hypoxia can induce the differential expression of several cerebral mitochondrial proteins and that these proteins are regulated to cause the dysfunction of ATP synthesis, the loss of membrane potential and the production of oxygen radicals, resulting in severe brain damage.
MATERIALS AND METHODS
Design
An in vivo, comparative proteomic study.
Time and setting
The experiment was performed at the Logistics College of Chinese People's Armed Police Forces, China from March 2011 to September 2012.
Materials
Sixty adult female Sprague-Dawley rats, weighing 230 ± 20 g, were provided by the Experimental Animal Center of Academy of Military Medical Sciences (Beijing, China, No. SCXK-2007-004). All rats were maintained in the animal house at 23 ± 1°C with a pellet diet and water ad libitum. All animal experiments were performed in accordance with the International Guidelines on the Ethical Use of Animals[31].
Methods
Model of hypobaric hypoxia in rats
The rats in hypobaric hypoxia groups were exposed to a simulated altitude of 7 000 m (barometric pressure –0.06 MPa) in a specially designed decompression chamber (Hangzhou, Zhejiang Province, China; Figure 8). The chamber was flushed with fresh air (0.1 m3/h) to replenish oxygen consumed and remove carbon dioxide produced (pO2 10 ± 2%, temperature 20 ± 3°C and relative humidity 30 ± 5%). The desired altitude was reached at the ascent rate of 450 m/min over approximately 15 minutes.
Figure 8.

Designed decompression chamber for exposure to hypobaric hypoxia (diameter 40 cm, height 85 cm, PaO2 10 ± 2%, temperature 20 ± 3°C and relative humidity 30 ± 5%).
Toluidine blue staining
Following hypobaric hypoxia treatment, the isolated rat brain was fixed with 4% (w/v) paraformaldehyde, and then brain tissue slices were prepared for toluidine blue staining (Nanjing Jiancheng Company, Jiangsu Province, China). Briefly, the slice was incubated with a 0.5% (w/v) solution of toluidine blue at 56°C for 20 minutes. After being dehydrated, cleared and mounted, brain slices of the cerebral cortex were examined under a light microscope (Olympus, Tokyo, Japan).
Brain water content determination
Brain water content was determined using the method of wet-to-dry weight ratio[30]. After exposure to hypobaric hypoxia, the brain of each rat from each group was excised on ice. Following measurement of brain wet weight, the sample was baked at 150°C for 24 hours to obtain dry weight. The brain water content was calculated as follows: brain water content = (wet weight − dry weight)/wet weight × 100%.
Preparation of cerebral mitochondria
In brief, after hypobaric hypoxia treatment, the cerebral cortex of rats was isolated and gently homogenized using a motor-driven glass homogenizer in isolation buffer (0.25 mol/L sucrose, 10 mmol/L HEPES and 1 mmol/L EDTA at pH 7.4; Huashengyuan Company, Tianjin, China). The resultant homogenate was centrifuged at 1 000 × g for 10 minutes at 4°C. Afterwards, the pellet was re-homogenized and centrifuged again at 800 × g for 10 minutes at 4°C. Both supernatants were collected and centrifuged at 20 000 × g for 15 minutes at 4°C. The mitochondria-enriched pellet was homogenized in 3 mL of 25% (v/v) Nycodenz medium containing 1.5 mL of isolation buffer and 1.5 mL of 50% (v/v) Nycodenz medium (50% (v/v) Nycodenz, 10 mmol/L HEPES-NaOH and 1 mmol/L EDTA at pH 7.4) for the second purification. The crude mitochondrial fraction was layered on a discontinuous Nycodenz gradient and centrifuged at 52 000 × g for 90 minutes at 4°C. The major band at the interface of the 25% (v/v) and 30% (v/v) Nycodenz solutions was collected, diluted with the isolation buffer (1:1) and centrifuged twice at 15 000 × g for 15 minutes. The purified mitochondrial pellet was stored at −80°C for subsequent analyses.
Two-dimensional electrophoresis analysis on mitochondrial proteins from the cerebral cortex
Mitochondrial proteins were assessed using the Bradford assay[32]. Protein samples were loaded onto immobilized pH gradient strips (17 cm, pH 3–10, linear), and isoelectric focusing was performed using a Protean IEF cell system (Bio-Rad, Hercules, CA, USA). Briefly, the gels were rehydrated at 20°C for 18 hours at 50 V followed by voltage ramping steps of 250, 500, 1 000, 4 000, 8 000 V until 78 000 V/hour, and the process was limited by a maximum current of 50 μA per gel. In the second dimension of electrophoresis, the proteins were resolved on 12.5% (w/v) sodium dodecyl sulfate-polyacrylamide gels using a Protean II XL system (Bio-Rad), and then the gels were fixed and stained with silver nitrate. Afterwards, the stained gels were dried and scanned on a UMAX Power Look III scanner (resolution 300 DPI, GE Healthcare, Solon, OH, USA), and the images were analyzed using Image Master 2D Platinum software 5.0 (GE Healthcare). The analysis process included pixel detection, background reduction, normalization processing and protein point matching.
Mass spectrometry for protein identification
The mitochondrial protein spots, which were found to be expressed in a time-dependent manner by two-dimensional electrophoresis analysis, were selected and excised from the stained gels. The gel fragments were destained and dehydrated with 25 mmol/L NH4HCO3 in 50% (v/v) acetonitrile for 30 minutes and then dried at 37°C for 20 minutes. Each gel particle was incubated in 10 μL of 25 mmol/L NH4HCO3 with trypsin for 3 hours. Total peptides were extracted twice with 50% (v/v) acetonitrile (Sigma, St. Louis, MO, USA) and 5% (v/v) trifluoroacetic acid (Sigma). Afterwards, the peptides were added to the buffer (0.3% (v/v) trifluoroacetic acid, 50% (v/v) acetonitrile; Sigma), and the mixture was added to saturate 4-hydroxycinnamic acid (Sigma-Aldrich). Finally, a total of 0.3 μL of this mixture was subjected to mass spectrometric analysis (GE Healthcare). The peptide fingerprinting was analyzed using the mascot database provided by Matrixscience (www.matrixscience.com). In the database, the protein was identified when the matching score was more than 61.
Western blot analysis
A total of 50 μg of mitochondrial protein from the cerebral cortex was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% (w/v) nonfat dry milk in tris buffered saline containing 0.05% (v/v) Tween 20 for 1 hour at 37°C. The membrane was incubated with rabbit anti-rat monoclonal antibody (1:1 000; Abcam, Cambridge, UK) overnight at 4°C, and with horseradish peroxidase-conjugated goat anti-rabbit polyclonal antibody (1:8 000; Abcam) for 1 hour at room temperature. Immunoreactivity was visualized by autoradiography with luminol (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and quantified by Image Quant 5.2 (Molecular Dynamics). VDAC was chosen as the reference gene.
Statistical analysis
All results were expressed as mean ± SD. Statistical differences were evaluated by one-way analysis of variance followed by least significant difference test. The differences were considered to be statistically significant when P < 0.05.
Research background: Cerebral mitochondrial dysfunction may be the contributing factor of high-altitude hypoxia, especially acute hypoxic brain injury; however, the molecular mechanisms of its pathogenesis are not clear.
Research frontiers: The majority of studies addressing the expression of a series of functional brain proteins or pathological factors induced by hypoxia focus on some “star” proteins in the brain, which can miss other important target proteins, thus affecting the comprehensive demonstration of the pathological mechanism associated with acute hypoxic brain injury.
Clinical significance: This study investigated the pathogenic molecules of brain mitochondria during acute hypoxia using proteomics technology, and revealed the pathogenic mechanism, thus providing a new target for therapy.
Academic terminology: Matrix assisted laser desorption ionization time of flight mass spectrometry is mainly composed of two parts: matrix-assisted laser desorption/ionization is a soft ionization technique applied to determine the mixtures and biological macromolecules, while time of flight analyzer can be used in the determination of ion matter-to-charge the ratio. Matrix-assisted laser desorption/ionization-time of flight mass spectrometry is characterized by high sensitivity, high accuracy and high resolution.
Peer review: This study is the first proteomics experiment to investigate the expression of subcellular organelle-mitochond- rial proteins under acute hypoxia conditions; the first to explore hypoxic brain injury and mitochondrial differential expression during different hypoxia periods; and has preliminary screened mitochondrial proteins differentially expressed in patients with acute hypoxic brain injury. Our findings will provide targets for the development of drugs for the prevention and treatment of acute hypoxia injury.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, No. 81073152; the Key Science Foundation of Tianjin in China, No. 10JCZDJC21100; the Natural Science Foundation of Tianjin in China, No.10JCYBJC14700, No.13JCQNJC13200; the Science Foundation of Tianjin Key Laboratory in China, No. WHTD 201303-2.
Conflicts of interest: None declared.
Ethical approval: This study was approved by the Ethics Committee of Logistics College of Chinese People's Armed Police Forces, Tianjin, China.
(Reviewed by Dwakarla S, Raye W, Tu QY, Chen HS)
(Edited by Wang J, Yang Y, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Andrews G, Ainslie PN, Shepherd K, et al. The effect of partial acclimatization to high altitude on loop gain and central sleep apnoea severity. Respirology. 2012;17(5):835–840. doi: 10.1111/j.1440-1843.2012.02170.x. [DOI] [PubMed] [Google Scholar]
- [2].Shrestha P, Basnyat B, Küpper T, et al. Cerebral venous sinus thrombosis at high altitude. High Alt Med Biol. 2012;13(1):60–62. doi: 10.1089/ham.2011.1043. [DOI] [PubMed] [Google Scholar]
- [3].Imray C, Wright A, Subudhi A, et al. Acute mountain sickness: pathophysiology, prevention, and treatment. Prog Cardiovasc Dis. 2010;52(6):467–484. doi: 10.1016/j.pcad.2010.02.003. [DOI] [PubMed] [Google Scholar]
- [4].Chen XQ, Kong FP, Zhao Y, et al. High-altitude hypoxia induces disorders of the brain-endocrine-immune network through activation of corticotropin-releasing factor and its type-1 receptors. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2012;28(6):481–487. [PubMed] [Google Scholar]
- [5].Babbar R, Agarwal S. A new approach to hypobaric hypoxia induced cognitive impairment. Indian J Med Res. 2012;136(3):365–367. [PMC free article] [PubMed] [Google Scholar]
- [6].Goodman MD, Makley AT, Huber NL, et al. Hypobaric hypoxia exacerbates the neuroinflammatory response to traumatic brain injury. J Surg Res. 2011;165(1):30–37. doi: 10.1016/j.jss.2010.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Semenov DG, Samoilov MO, Lazarewicz JW. Preconditioning reduces hypoxia-evoked alterations in glutamatergic Ca2+ signaling in rat cortex. Acta Neurobiol Exp. 2008;68(2):169–179. doi: 10.55782/ane-2008-1686. [DOI] [PubMed] [Google Scholar]
- [8].Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292(2):C641–657. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- [9].Chen L, Zhang X, Zhou W, et al. The interactive effects of cytoskeleton disruption and mitochondria dysfunction lead to reproductive toxicity induced by microcystin-LR. PLoS One. 2013;8(1):e53949. doi: 10.1371/journal.pone.0053949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Contreras-Shannon V, Heart DL, Paredes RM, et al. Clozapine-induced mitochondria alterations and inflammation in brain and insulin-responsive cells. PLoS One. 2013;8(3):e59012. doi: 10.1371/journal.pone.0059012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kang S, Seo JH, Heo TH, et al. Batten disease is linked to altered expression of mitochondria-related metabolic molecules. Neurochem Int. 2013;62(7):931–935. doi: 10.1016/j.neuint.2013.03.007. [DOI] [PubMed] [Google Scholar]
- [12].Matsuzaki H, Fujimoto T, Tanaka M, et al. Tespal is a novel component of mitochondria-associated endoplasmic reticulum membranes and affects mitochondrial calcium flux. Biochem Biophys Res Commun. 2013;433(3):322–326. doi: 10.1016/j.bbrc.2013.02.099. [DOI] [PubMed] [Google Scholar]
- [13].Chen LF, Liu JZ, Li B. Characteristics of adenine nucleotide translocator in mitochondria of rat cerebral cortex during hypobaric hypoxia exposure. Acta Physiology Sinica. 2006;58(1):29–33. [PubMed] [Google Scholar]
- [14].Wang N, Qian HY, Zhou XQ, et al. Mitochondrial energy metabolism dysfunction involved in reproductive toxicity of mice caused by endosulfan and protective effects of vitamin E. Ecotoxicol Environ Saf. 2012;82(8):96–104. doi: 10.1016/j.ecoenv.2012.05.014. [DOI] [PubMed] [Google Scholar]
- [15].Zhao de J, Zhang ZY, Harrison J, et al. Genome-wide analysis of transcriptional changes in the thoracic muscle of the migratory locust, Locusta migratoria, exposed to hypobaric hypoxia. J Insect Physiol. 2012;58(11):1424–1431. doi: 10.1016/j.jinsphys.2012.08.006. [DOI] [PubMed] [Google Scholar]
- [16].Boos CJ, Hodkinson P, Mellor A, et al. The effects of acute hypobaric hypoxia on arterial stiffness and endothelial function and its relationship to changes in pulmonary artery pressure and left ventricular diastolic function. High Alt Med Biol. 2012;13(2):105–111. doi: 10.1089/ham.2012.1009. [DOI] [PubMed] [Google Scholar]
- [17].Rafalski VA, Brunet A. Energy metabolism in adult neural stem cell fate. Prog Neurobiol. 2011;93(2):182–203. doi: 10.1016/j.pneurobio.2010.10.007. [DOI] [PubMed] [Google Scholar]
- [18].Runswick MJ, Bason JV, Montgomery MG, et al. The affinity purification and characterization of ATP synthase complexes from mitochondria. Open Biol. 2013;3(2):120160. doi: 10.1098/rsob.120160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huang S, Shingaki-Wells RN, Taylor, et al. The rice mitochondria proteome and its response during development and to the environment. Front Plant Sci. 2013;4(6):16. doi: 10.3389/fpls.2013.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Silva FS, Ribeiro MP, Santos MS, et al. Acitretin affects bioenergetics of liver mitochondria and promotes mitochondrial permeability transition: potential mechanisms of hepatotoxicity. Toxicology. 2013;4(6):93–100. doi: 10.1016/j.tox.2013.01.020. [DOI] [PubMed] [Google Scholar]
- [21].Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp Diabetes Res 2012. 2012 doi: 10.1155/2012/703538. 703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ellinas H, Frost EA. Mitochondrial disorders--a review of anesthetic considerations. Middle East J Anesthesiol. 2011;21(2):235–242. [PubMed] [Google Scholar]
- [23].Matthies D, Haberstock S, Joos F, et al. Cell-free expression and assembly of ATP synthase. J Mol Biol. 2011;413(3):593–603. doi: 10.1016/j.jmb.2011.08.055. [DOI] [PubMed] [Google Scholar]
- [24].Kawai Y, Yoshida K, Kaidoh M, et al. Shear stress-mediated F1/F0 ATP synthase- dependent CO2 gas excretion from human pulmonary arteriolar endothelial cells. J Cell Physiol. 2012;227(5):2059–2068. doi: 10.1002/jcp.22937. [DOI] [PubMed] [Google Scholar]
- [25].Mesbah NM, Wiegel J. The Na(+)-translocating F1F0- ATPase from the halophilic, alkalithermophile Natranaerobius thermophilus. Biochim Biophys Acta. 2011;1807(9):1133–1142. doi: 10.1016/j.bbabio.2011.05.001. [DOI] [PubMed] [Google Scholar]
- [26].Hao XL, Yao HF, Cheng YZ, et al. Homology cloning, sequence characterization, and expression analysis of cDNA encoding electron transfer flavoprotein beta polypeptide in mud crab. Genet Mol Res. 2012;11(4):4316–4322. doi: 10.4238/2012.November.12.11. [DOI] [PubMed] [Google Scholar]
- [27].Hirokawa S, Shimanuki T, Kitajima H, et al. Knockdown of electron transfer flavoprotein β subunit reduced TGF-β- induced α-SMA mRNA expression but not COL1A1 in fibroblast-populated three-dimensional collagen gel cultures. J Dermatol Sci. 2012;68(3):179–186. doi: 10.1016/j.jdermsci.2012.09.012. [DOI] [PubMed] [Google Scholar]
- [28].Lam AK, Silva PN, Altamentova SM, et al. Quantitative imaging of electron transfer flavoprotein autofluorescence reveals the dynamics of lipid partitioning in living pancreatic islets. Integr Biol. 2012;4(8):838–846. doi: 10.1039/c2ib20075a. [DOI] [PubMed] [Google Scholar]
- [29].Wang Y, Mohsen AW, Mihalik SJ, et al. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J Biol Chem. 2010;285(39):29834–29841. doi: 10.1074/jbc.M110.139493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li JY, Qi YT, Guo P, et al. Research on brain damage and expressions of aquaporin1/4 in rats exposed to acute high-altitude hypoxia. Zhongguo Zhongxiyi Jiehe Jijiu Zazhi. 2012;19(6):279–282. [Google Scholar]
- [31].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006 Sep 30; [Google Scholar]
- [32].Kim N, Lee Y, Kim H, et al. Potential biomarkers for ischemic heart damage identified in mitochondrial proteins by comparative proteomics. Proteomics. 2006;6(3):1237–1249. doi: 10.1002/pmic.200500291. [DOI] [PubMed] [Google Scholar]