

Abstract
Transient brain ischemia has been shown to induce hyperphosphorylation of the microtubule-associated protein tau. To further determine the mechanisms underlying these processes, we investigated the interaction between tau, glycogen synthase kinase (GSK)-3β and protein phos-phatase 2A. The results confirmed that tau protein was dephosphorylated during brain ischemia; in addition, the activity of GSK-3β was increased and the activity of protein phosphatase 2A was decreased. After reperfusion, tau protein was hyperphosphorylated, the activity of GSK-3β was decreased and the activity of protein phosphatase 2A remained low. Importantly, the interaction of tau with GSK-3β and protein phosphatase 2A was altered during ischemia and reperfusion. Lithium chloride could affect tau phosphorylation by regulating the interaction of tau with GSK-3β and protein phosphatase 2A, and improve learning and memory ability of rats after transient brain ischemia. The present study demonstrated that it was the interaction of tau with GSK-3β and protein phosphatase 2A, rather than their individual activities, that dominates the phosphorylation of tau in transient brain ischemia. Hyperphosphorylated tau protein may play an important role in the evolution of brain injury in ischemic stroke. The neuroprotective effects of lithium chloride partly depend on the inhibition of tau phosphorylation during transient brain ischemia.
Keywords: neural regeneration, brain injury, brain ischemia, reperfusion, microtubule-associated protein tau, phosphorylation, glycogen synthase kinase 3β, protein phosphatase 2A, lithium chloride, grants-supported paper, neuroregeneration
Research Highlights
-
(1)
The present study shows for the first time that the interaction of tau with glycogen synthase kinase (GSK)-3β and protein phosphatase 2A is altered during transient brain ischemia.
-
(2)
The neuroprotective function of lithium chloride may depend partly on the altered phosphorylation of tau, by regulating the associations between tau, GSK-3β and protein phosphatase 2A during cerebral ischemia.
INTRODUCTION
Cell death is one of the most serious consequences of brain ischemia[1,2]. Despite the existence of numerous agents that can prevent the cascade of events leading to ischemic neuronal death in animal models, clinical trials with such agents have proved disappointing[3,4,5]. Mounting evidence indicates that stroke has many similarities to some neurodegenerative diseases including Alzheimer's disease, providing clues for studying and treating ischemic stroke[6,7]. Intracellular neurofibrillary tangles in the brain are the hallmark pathological feature of Alzheimer's disease and are composed of bundles of straight and paired helical filaments, the major protein component of which is abnormally hyperphosphorylated tau[8,9]. Hyperphosphorylation of tau reduces its ability to bind to microtubules[8,9] and it has therefore been proposed to lead to microtubule destabilization, appearance of neurofibrillary tangles, and neurodegeneration in Alzhei-mer's disease brain. Hyperphosphorylation of tau is a physiologically reversible response of the brain to some stressful conditions, such as heat shock, starvation or ischemia[10]. Ischemia causes a shortage of glucose and oxygen to the brain and results in death of neurons. Reduced glucose metabolism is observed in certain regions of the brain in people with probable and preclinical Alzheimer's disease[11]. These data, combined with a previous observation that brain ischemia induces tau hyperphosphorylation at many paired helical filament-tau epitopes[12], suggest that a common mechanism of tau hyperphos-phorylation might be involved in ischemia and Alzheimer's disease.
Phosphorylation of tau can be regulated by many protein kinases and phosphatases. An increasing number of studies have shown that glycogen synthase kinase 3 (GSK-3) is the most likely candidate for the protein kinase responsible for the abnormal phosphorylation state of tau in Alzheimer's disease brain[13,14]. GSK-3 is a serine/threonine protein kinase that was first isolated and purified as an enzyme capable of phosphorylating and inactivating glycogen synthase[15,16]. It was recognized as a multifunctional enzyme involved in a broad range of biological processes[17]. GSK-3 is inhibited by phosphorylation of a serine residue (Ser9 in GSK-3β and Ser21 in GSK-3α) located in the N-terminal domain. GSK-3 was shown to phosphorylate tau both in vitro and in intact cells on multiple sites, some of which are abnormally hyperphosphorylated in Alzheimer's disease brain[17,18]. Protein phosphatase 2A (PP2A) has been shown to be the main tau phosphatase[19]. PP2A is a family of serine/threonine phosphatases and is ubiquitously expressed in all kinds of tissue and cells. PP2A accounts for as much as 1% of total cellular proteins and for the major portion of serine/threonine phosphatase activity[20]. In response to growth factors or insulin, it has been shown that PP2A is phosphorylated at Tyr307 and results in inactivation of the enzyme[21].
In this study, we hypothesized that GSK-3β (the main tau kinase) and PP2A (the main tau phosphatase) may be involved in the phosphorylation of tau protein during transient brain ischemia. Therefore, the phosphorylation of tau and its interaction with GSK-3β and PP2A were investigated after ischemic insult followed by reperfusion. To further confirm our hypothesis, we administered lithium chloride, a selective inhibitor of GSK-3β, to rats and investigated its effect on this interaction and tau phosphorylation as well as on the behavioral and histological outcomes in rats exposed to cerebral ischemia and reperfusion.
RESULTS
Quantitative analysis of experimental animals
A total of 136 adult male Sprague-Dawley rats were randomly divided into the following groups: sham-operated group, brain ischemia group (four-vessel occlusion), brain ischemia-reperfusion group (four-vessel occlusion for 15 minutes and then reperfusion) and drug treatment group (lithium chloride treatment + brain ischemia or brain ischemia and reperfusion). Each group was subdivided to give eight rats per time point. A total of 136 rats were involved in the final analysis.
Time course of phosphorylation of tau protein in rat hippocampus during brain ischemia and reperfusion
Western blot assay was used to examine serine phosphorylation of tau in brain ischemia and reperfusion. The level of phosphorylated tau at Ser202 and Ser396/404 rapidly decreased during brain ischemia (P < 0.05; Figure 1A, B), indicating that tau was becoming dephosphorylated. After 30 minutes of reperfusion, tau was rephosphorylated, resulting in a level of phosphorylated tau significantly higher than that in the sham-operated group (P < 0.05; Figure 1C, D); however, after 12 hours of reperfusion, the level of phosphorylated tau returned to normal.
Figure 1.
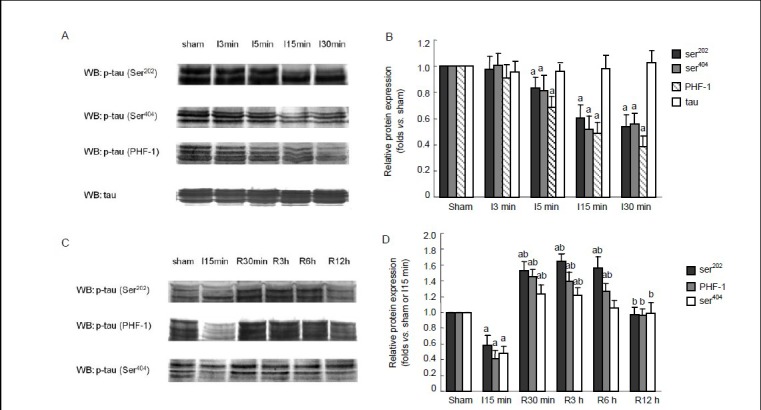
Time course of the protein levels of phosphorylated tau in rat hippocampus during brain ischemia and reperfusion. The protein levels were measured by western blot assay.
(A) Brain ischemia caused a decrease in the levels of phosphorylated tau at Ser202, Ser404 and paired helical filament-1 (PHF-1) sites. (B) Semiquantitative representation of phosphorylated tau at different time points during brain ischemia. (C) After 15 minutes (min) of brain ischemia, followed by 30 minutes, 3 hours (h), 6 hours and 12 hours of reperfusion, tau was rephosphorylated at Ser202, Ser404 and PHF-1 sites. (D) Semiquantitative representation of phosphorylated tau at different time points during reperfusion. (B, D) Data are expressed as the mean ± SD from eight rats at each time point. aP < 0.05, vs. sham-operated group; bP < 0.05, vs. 15-minute ischemia group. I: Ischemia; R: reperfusion; WB: western blot.
Time course of phosphorylated GSK-3β and total GSK-3β protein levels in rat hippocampus during brain ischemia and reperfusion
Considering that GSK-3β is the major tau kinase, its activity was investigated in brain ischemia and reperfusion to explore the underlying mechanisms of tau phosphorylation. The level of phosphorylation decreased rapidly after 3 minutes of ischemia and continued to decrease as the duration of ischemia was prolonged to 5, 15 and 30 minutes (P < 0.05; Figure 2A, B). This indicates that the activity of GSK-3β was increased. During reperfusion, the serine phosphorylation of GSK-3β increased significantly to a level higher than that of the sham-operated group (P < 0.05; Figure 2C, D), indicating that the activity of GSK-3β was inhibited.
Figure 2.
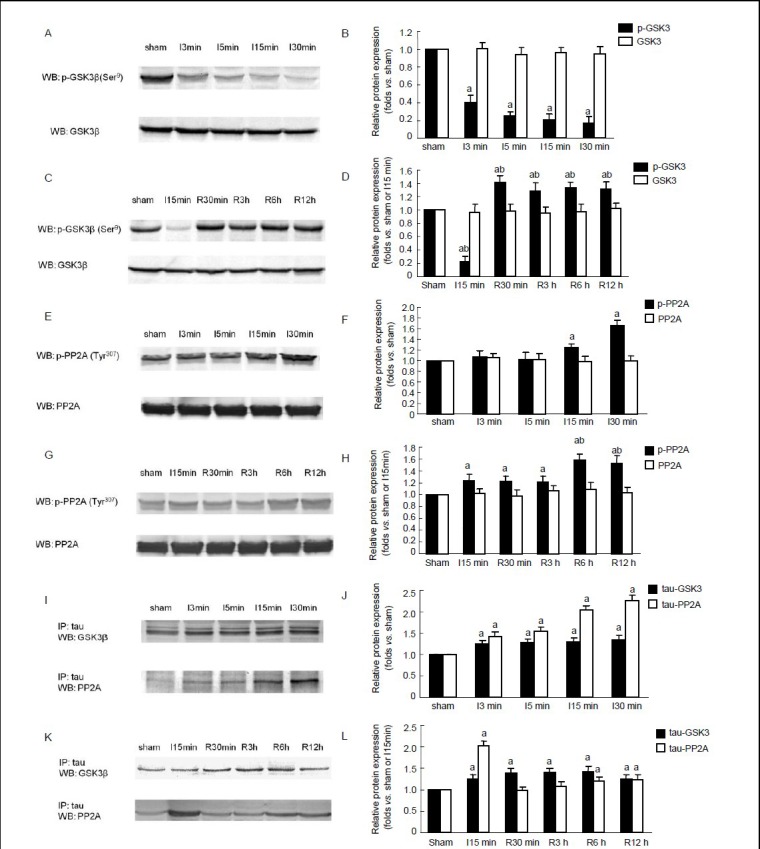
Time course of protein levels of phosphorylated glycogen synthase kinase 3 (p-GSK-3β), GSK-3β, phosphorylated protein phosphatase 2A (p-PP2A) and PP2A in rat hippocampus, and the interaction between tau, GSK-3β and PP2A during brain ischemia and reperfusion.
(A, C) Western blot assay of p-GSK-3β and total protein levels of GSK-3β in brain ischemia (A) and during reperfusion (C). (E, G) Western blot assay of the protein levels of p-PP2A and PP2A in brain ischemia (E) and during reperfusion (G). (I, K) Interaction between tau, GSK-3β and PP2A in brain ischemia (I) and during reperfusion (K).
(B, D, F, H, J, L) Semiquantitative representation of protein levels of p-GSK-3β, GSK-3β, p-PP2A and PP2A and the interaction between tau, GSK-3β and PP2A in A, C, E, G, I and K. Data are expressed as mean ± SD at each time point. aP < 0.05, vs. sham-operated group; bP < 0.05, vs. 15-minute ischemia group, n = 8. I: Ischemia; R: reperfusion; IP: immunoprecipitation; WB: western blot; min: minute; h: hour.
Time course of phosphorylated PP2A and total PP2A protein levels in rat hippocampus during brain ischemia and reperfusion
As PP2A is a major tau phosphatase, the time course of PP2A activity was examined in brain ischemia and reperfusion. Phosphorylation of PP2A at Tyr307 increased after 15–30 minutes of ischemia (P < 0.05; Figure 2E, F), indicating that the activity of PP2A was decreased. After 3 hours of reperfusion, PP2A phosphorylation was significantly increased (P < 0.05; Figure 2G, H) and was significantly higher than that in the sham-operated and ischemia groups, suggesting that the activity of PP2A was largely inhibited.
Interaction of tau with GSK-3β and PP2A during brain ischemia and reperfusion
To further study the roles of GSK-3β and PP2A in tau phosphorylation, the interaction of tau with GSK-3β and PP2A was examined by immunoprecipitation. As shown in Figure 2, the interaction between the two proteins was unchanged during brain ischemia (Figure 2I, J), but was significantly increased after reperfusion (P < 0.05; Figure 2K, L). The interaction between tau and PP2A was notably increased in brain ischemia (P < 0.05; Figure 2I, J), but was decreased after reperfusion (P < 0.05; Figure 2K, L). Therefore, the dephosphorylation of tau may be induced by the increased association between PP2A and tau in brain ischemia. Moreover, PP2A may override GSK-3β in tau dephosphorylation. During reperfusion, both the increased interaction between GSK-3β and tau and the decreased association between PP2A and tau contributed to the increased phosphorylation of tau, notwithstanding the decreased activity of GSK-3β.
Effects of lithium chloride on the phosphorylation of tau in the rat hippocampus during brain ischemia and reperfusion
Pretreatment with lithium chloride not only inhibited dephosphorylation of tau in brain ischemia (P < 0.05; Figure 3A, B), but also inhibited the rephosphorylation of tau in reperfusion (P < 0.05; Figure 3C, D).
Figure 3.
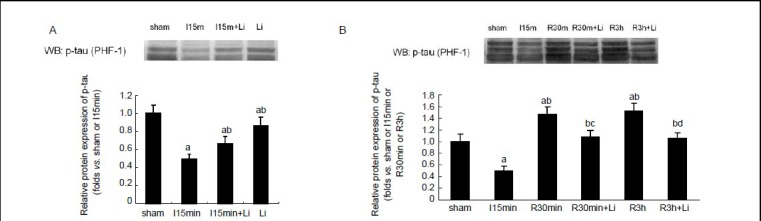
Effects of lithium chloride on the phosphorylation of tau in hippocampus during brain ischemia and reperfusion.
(A) Western blot of phosphorylated tau protein in tissue from sham-operated rats and those subjected to 15 minutes (min) of brain ischemia with or without lithium; (B) western blot of tau phosphorylation after 30 minutes and 3 hours (h) of reperfusion following 15 minutes of brain ischemia.
Data are expressed as mean ± SD from eight rats at each time point in each group. aP < 0.05, vs. sham-operated group; bP < 0.05, vs. 15-minute ischemia group; cP < 0.05, vs. 30-minute I/R group; dP < 0.05, vs. 3-hour I/R group. I: Ischemia; R: reperfusion; WB: western blot.
Effects of lithium chloride on the activities of GSK-3β and PP2A in rat hippocampus during brain ischemia and reperfusion
Pretreatment with lithium chloride resulted in increased serine phosphorylation of GSK-3β in brain ischemia and reperfusion (P < 0.05; Figure 4A, B), which suggested that the activity of GSK-3β was inhibited by lithium chloride. However, lithium chloride had no effects on the activity of PP2A, as the levels of phosphorylated PP2A at Tyr307 were not altered by lithium chloride during brain ischemia and reperfusion.
Figure 4.
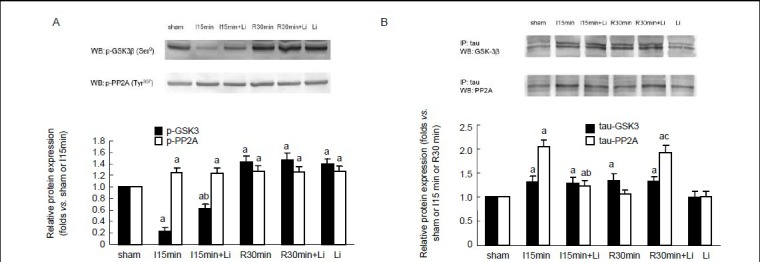
Effects of lithium chloride on the activities of glycogen synthase kinase 3 beta (GSK-3β) and protein phosphatase 2A (PP2A) and the interaction between tau, GSK-3β and PP2A during brain ischemia and reperfusion.
(A) Western blot assay of the protein levels of p-GSK-3β and p-PP2A in rat hippocampus. (B) Immunoprecipitation analysis of the interaction between tau, GSK-3β and PP2A. Data are expressed as mean ± SD from eight rats at each time point. aP < 0.05, vs. sham-operated group; bP < 0.05, vs. 15-minute ischemia group; cP < 0.05, vs. 30 minute I/R group. I: Ischemia; R: reperfusion; WB: western blot; IP: immunoprecipitation; min: minute; h: hour.
Effects of lithium chloride on the interaction of tau with GSK-3β and PP2A during brain ischemia and reperfusion
Lithium chloride had no significant effects on the interaction between tau and GSK-3β in brain ischemia and reperfusion (Figure 4C, D). However, lithium chloride could inhibit not only the increased association between tau and PP2A in ischemia, but also the decreased association between tau and PP2A in reperfusion (P < 0.05).
Effects of lithium chloride on the spatial learning and memory of rats subjected to ischemia and reperfusion
In the hidden platform swimming test, rats subjected to ischemia and reperfusion showed a longer latency to find the platform compared with sham-operated rats. On the first test day, lithium chloride-treated rats with ischemia/reperfusion injury showed a trend to take a shorter time to find the hidden platform than ischemia/reperfusion rats that had not received lithium chloride, but this did not reach statistical significance. After the second test day, lithium chloride-treated ischemia/reperfusion rats spent a significantly shorter time finding the platform compared with ischemia/reperfusion rats without lithium chloride treatment (Figure 5A).
Figure 5.
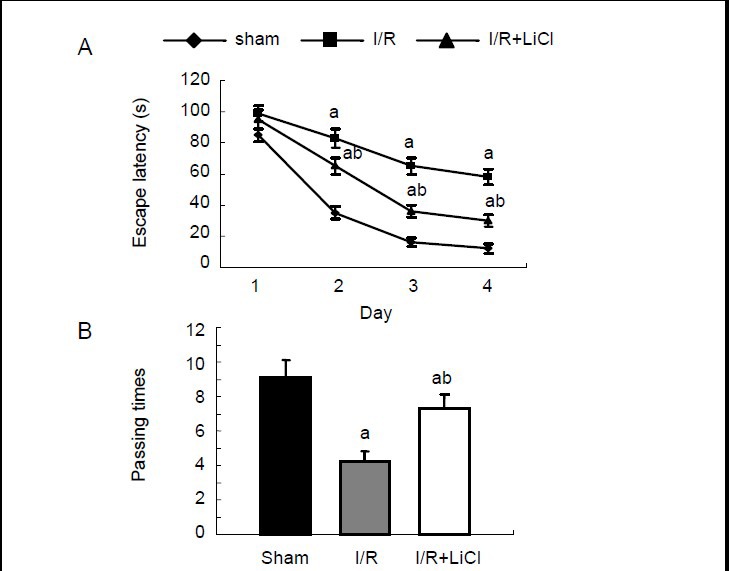
Effects of lithium chloride on the spatial learning and memory ability of rats using the Morris water maze test.
Rats were assigned to three groups: sham-operated group, ischemia/reperfusion group (I/R), lithium chloride-treated I/R group (I/R + LiCl). (A) Time spend to find the submerged platform. (B) Platform-passing times in the probe trial test. Data are expressed as mean ± SD from eight rats in each group. aP < 0.05, vs. sham-operated group; bP < 0.05, vs. I/R group.
In the probe trial test, ischemia/reperfusion rats that had not received lithium chloride showed significantly fewer platform-passing times than sham-operated rats. Lithium chloride-treated ischemia/reperfusion rats displayed an improved ability in learning and memory and showed more platform-passing times than ischemia/reperfusion rats without lithium chloride treatment (Figure 5B). There was no speed difference among all groups (data not shown).
Effects of lithium chloride on the morphology of pyramidal neurons in ischemia/reperfusion rats
To verify further the neuroprotective effects of lithium chloride against ischemic injury, the effects of lithium chloride on the survival of CA1 pyramidal neurons were examined in rat hippocampus after 7 days of reperfusion. There was no obvious difference in neuronal survival between sham-operated and 15-minute ischemia groups (Figure 6A-D). Fifteen minutes of cerebral ischemia followed by 7 days of reperfusion resulted in severe cell death (Figure 6E, F). Treatment with lithium chloride notably suppressed reperfusion-induced neuronal death, which was shown by a greater neuronal density (Figure 6G-I).
Figure 6.
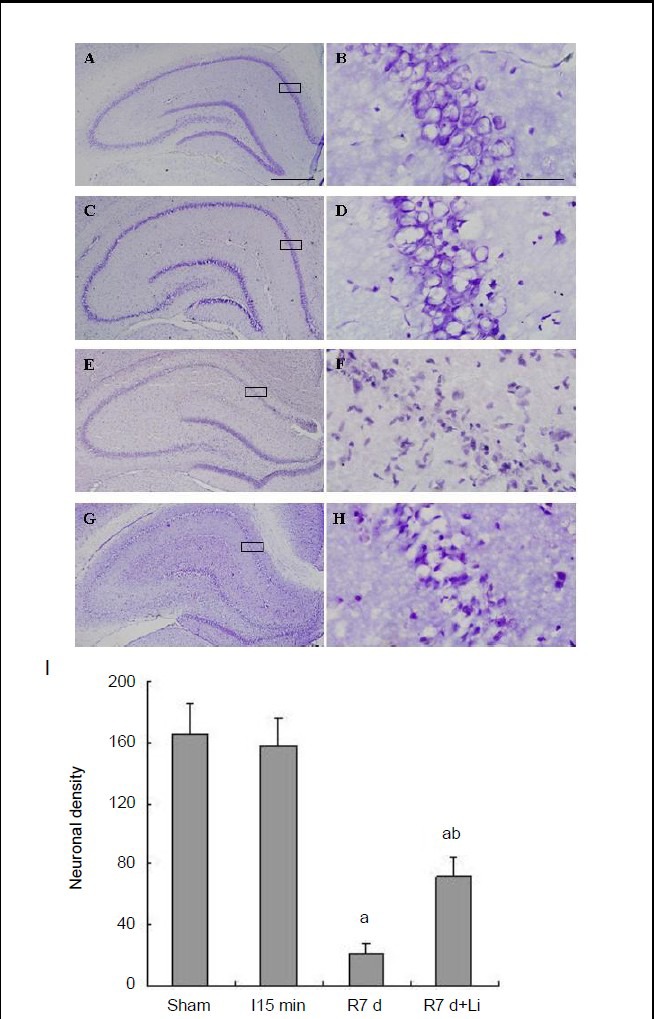
Effects of lithium chloride on the morphology of CA1 pyramidal neurons in ischemia/reperfusion rats.
Representative sections stained with cresyl violet in sham-operated rats (A, B) and rats subjected to 15-minute (min) ischemia (C, D), 7 days (d) of reperfusion following 15 minutes of ischemia (E, F) and 7 days of reperfusion following 15 minutes of ischemia pretreated with lithium chloride (G, H). Boxed areas in left column are shown at higher magnification in right column. Scale bar: 500 μm (A, C, E, G), 80 μm (B, D, F, H). (I) Number of neurons in 1 mm2 hippocampal CA1. Data are expressed as mean ± SD from eight rats in each group. aP < 0.05, vs. sham-operated group; bP < 0.05, vs. 7 days of reperfusion.
DISCUSSION
Hyperphosphorylation of tau is a common feature of neurodegenerative tauopathies including Alzheimer's disease. Therefore, much research has focused on the identification of the protein kinases and protein phosphatases that regulate tau phosphorylation. The proline-directed serine/threonine kinases Cdk5 and GSK-3 have been identified as major candidates mediating tau hyperphosphorylation at sites characteristic for neurodegenerative tauopathies. However, Plattner and colleagues[22] reported recently that Cdk5 did not directly affect tau hyperphosphorylation in vivo and their results established GSK-3 as a key mediator of tau phosphorylation at disease-associated sites.
It was also reported that PP2A was the major tau phosphatase that regulates its phosphorylation at multiple sites and it accounted for 71% of the total tau phosphatase activity in human brain[19]. The abnormal hyperphosphorylation of tau is partially due to a downregulation of PP2A activity in Alzheimer's disease brain. Therefore, the present study focused on the effect of GSK-3β and PP2A in tau phosphorylation during transient brain ischemia.
Previous studies confirmed that cerebral ischemia could result in a rapid dephosphorylation of tau, and tau phosphorylation was differentially recovered after 24 hours of reperfusion[12]. However, the underlying mechanism was not examined. Here, consistent with previous studies, we found that tau was dephosphorylated in brain ischemia and rephosphorylated after reperfusion. We hypothesized that the major tau kinase GSK-3β would be inactivated in ischemia and activated after reperfusion. Contrary to our expectation, we observed that GSK-3β was activated in ischemia and inhibited after reperfusion. Moreover, the activity of PP2A was mildly inhibited in ischemia and intensively inhibited after reperfusion. These results demonstrated that activation of GSK-3β is not obligatory for tau hyperphosphorylation. Next, we investigated the association between GSK-3β, tau and PP2A. We found that the interaction of tau with GSK-3β was always increased in ischemia and reperfusion. The interaction of tau with PP2A was increased in ischemia and decreased after reperfusion. These results reveal a possible mechanism for the dephosphorylation and rephosphorylation of tau protein during transient cerebral ischemia. During brain ischemia, dephosphorylation of tau may be due to the increased association between tau and PP2A despite the elevated activity of GSK-3β. After reperfusion, the increased association between tau and GSK-3β and the decreased association between tau and PP2A may contribute jointly to tau hyperphosphorylation. Our results suggest that it is the associations between GSK-3β, tau and PP2A rather than the activities of GSK-3β and PP2A that dominate the phosphorylation of tau in transient brain ischemia. In the regulation of tau phosphorylation, PP2A may be central and override the role of GSK-3β. To further confirm our hypothesis, we used lithium chloride to inhibit GSK-3β. We found that lithium chloride could inhibit the activation of GSK-3β in ischemia and further inhibit its activity after reperfusion. However, lithium chloride had no obvious effects on the activity of PP2A in ischemia and reperfusion. Lithium chloride had no effects on the interaction between tau and GSK-3β, but it inhibited not only the increased association between tau and PP2A in ischemia, but also the decreased association between PP2A and tau after reperfusion. Thus, lithium chloride not only inhibited dephosphorylation of tau in brain ischemia, but also inhibited the rephosphorylation of tau in reperfusion.
Ischemic stroke is the leading cause of serious, long-term disability and death among elderly people. Reperfusion after an ischemic episode can significantly aggravate injury to the brain. However, the precise mechanism underlying ischemia and reperfusion injury is not clear. There are as yet no clinically effective therapeutic protocols for protection of the brain from damage induced by ischemia and reperfusion. According to the present results, we can speculate that hyperphosphorylation of tau during early reperfusion is a possible reason for ischemia/reperfusion brain injury. Therefore, measures aimed at inhibiting tau hyperphosphorylation may be useful in the treatment of ischemic stroke.
Lithium chloride is an agent widely used to treat bipolar mood disorder. Mounting evidence indicates that lithium chloride has neuroprotective effects on ischemia-induced neuronal death in vivo and in vitro[23,24,25,26]. Nevertheless, the underlying protective mechanisms of lithium chloride in brain ischemia are not well understood. Consistent with previous studies, our results confirmed that lithium chloride is neuroprotective against brain ischemic injury. In the Morris water maze experiment, we found that pretreatment with lithium chloride could also rescue the memory impairment of rats subjected to brain ischemia and reperfusion. Additionally, we provided a possible mechanism for the effects of lithium chloride. As a selective inhibitor of GSK-3β, lithium chloride can suppress the activity of GSK-3β in brain ischemia and reperfusion. More importantly, the associations between GSK-3β, tau and PP2A can be regulated by lithium chloride. Thus, lithium chloride can regulate the state of tau phosphorylation.
In conclusion, we have demonstrated for the first time a mechanism of tau hyperphosphorylation after transient brain ischemia. Inhibition of PP2A could override the inhibition of GSK-3β and lead to tau hyperphosphorylation in reperfusion following transient brain ischemia. According to our data, we can hypothesize that hyperphosphorylation of tau plays an important role in the evolution of brain injury in ischemic stroke. The neuroprotective function of lithium chloride may partly depend on the altered phosphorylation of tau by regulating the association among tau, GSK-3β and PP2A. Strategies addressing tau pathology may be promising in the treatment of stroke. Further studies are required to elucidate the mechanism of lithium chloride in the regulation of the association among GSK-3β, tau and PP2A in stroke and other tauopathies.
MATERIALS AND METHODS
Design
A parallel controlled, comparative, in vivo experiment.
Time and setting
Experiments were performed from May 2009 to July 2011 in School of Life Sciences, Tsinghua University, China.
Materials
A total of 136 adult male Sprague-Dawley rats (Vital River Corporation, Beijing; license No. SCXK (Jing) 2006-2009) aged 7 weeks and weighing 250–300 g were used. All protocols were in strict accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China[27].
Methods
Establishment of brain ischemia/reperfusion models
Transient brain ischemia was induced by four-vessel occlusion method, as described by Pulsinelli et al[28]. Briefly, rats were anesthetized by intraperitoneal injection of 300 mg/kg chloral hydrate and immobilized in stereotaxic apparatus (RWD Life Science Co., Ltd., Shenzhen, Guangdong Province, China). The bilateral vertebral arteries were electrocauterized with a bipolar coagulator. The next day, the bilateral common carotid arteries were occluded with aneurysm clips to induce brain ischemia. After 15 minutes of occlusion, the aneurysm clips were removed for 7 days of reperfusion. Sham-operated rats received the same surgical procedures, but the carotid arteries were not occluded. Body temperature of rats was maintained at 37°C using a heating pad. Rats that did not exhibit loss of their righting reflex during brain ischemia were excluded from the subsequent experiment.
Drug treatments
Lithium chloride (Sigma, St. Louis, MO, USA) was dissolved in saline at a concentration of 10 mmol/L. Rats were treated daily by intraperitoneal injection with lithium chloride (1 mmol/kg; n = 10) or vehicle (0.9% NaCl; n = 10) for 14 days before transient brain ischemia[25]. Drug treatment was continued until the day rats were sacrificed.
Morris water maze for learning and memory abilities of rats
The Morris water maze was used to test spatial learning and memory[29]. The water maze test was performed in a black circular pool (diameter 1.5 m, height 60 cm, filled with water of 22–23°C to a height of 30 cm). In the hidden-platform trials, a platform (10 cm diameter) was placed 2.0 cm below the water line in the southeastern quadrant of the pool. The rat was placed in the water facing the wall at one random start location of four quadrants. Each rat was allowed 120 seconds to find the submerged platform and rest on it for 20 seconds. If the rat failed to find the hidden platform within 120 seconds, it was placed on it for 20 seconds. Two sessions of four trials were conducted on the first testing day, with an interval of 4 hours. The first session was considered as the training procedure. One session of four trials was conducted daily on the next 3 days of test. Four hours after the last trial, a probe trial was given within 120 seconds, in which the platform was removed from the pool. The escape latency, escape distance (data not shown), swimming speed, and swimming pattern of the mice were monitored by a camera directly above the pool.
Immunoprecipitation and western blot assay for GSK-3β, tau and PP2A protein and their phosphorylation
Rats were decapitated immediately after their allocated ischemia and/or reperfusion time. The hippocampi were quickly separated, dipped into liquid nitrogen then stored at –80°C. Hippocampi were homogenized in ice-cold homogenization buffer consisting of 50 mmol/L 3-(N-morpholino) propanesulfonic acid (pH 7.4), 100 mmol/L KCl, 320 mmol/L MgCl2, 0.2 mmol/L dithiothreitol, phosphatase and protease inhibitors (20 mmol/L h-glycerophosphate, 20 mmol/L sodium pyrophosphate, 50 mmol/L NaF, 0.5 mmol/L ethylene diamine tetraacetic acid, 1 mmol/L ethylene glycol bis (2-aminoethyl) tetraacetic acid, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L benzamidine and 5 μg/mL each of aprotinin, leupeptin and pepstatin A). The homogenates were centrifuged at 800 × g for 15 minutes at 4°C, the supernatant was collected and the protein concentrations were determined by the Lowry method[30].
For immunoprecipitation, samples (400 μg of protein) were diluted fourfold with 50 mmol/L N-[2-hydroxyethyl]-piperazine-N’-[2-ethanesulfonic acid] buffer (pH 7.4) containing 10% glycerol, 150 mmol/L NaCl, 1% Triton X-100, 0.5% Nonidet P-40 and 1 mmol/L each of ethylene diamine tetraacetic acid, ethylene glycol bis (2-aminoethyl) tetraacetic acid, phenylmethylsulfonyl fluoride and Na3VO4. Diluted samples were preincubated for 1 hour with 20 μL protein A, and then centrifuged to remove any protein adhered nonspecifically to protein A. The supernatants were incubated with 1–2 μg mouse anti-rat tau monoclonal antibody (1:100; Cell Signaling, Beverly, MA, USA) for 4 hours at 4°C. Then protein A was added to the reaction system and the incubation continued for 2 hours. Samples were centrifuged at 10 000 × g and the pellets were washed three times with N-[2-hydroxyethyl]-piperazine-N′-[2-ethanesulfonic acid] buffer. Bound proteins were eluted by adding 4 × sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer (10 μL) and boiled at 100°C for 10 minutes, then centrifuged at 10 000 × g for 2 minutes. The supernatants were used for immunoblots.
For western blot assay, proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were electrotransferred onto nitrocellulose membrane (pore size 0.45 μm). The membrane was probed with rabbit anti-rat phospho-GSK-3β (Ser9) monoclonal antibody (1:1 000; Cell Signaling); rabbit anti-rat GSK-3β monoclonal antibody (1:1 000; Cell Signaling); mouse anti-rat tau monoclonal antibody (1: 1 000; Cell Signaling); mouse anti-rat phospho-tau (Ser202) monoclonal antibody (1:1 000; Stressgen, Kampenhout, Belgium); mouse anti-rat phospho-tau (Ser404) monoclonal antibody (1:1 000; Santa Cruz Biotechnology); mouse anti-rat paired helical filament-1 monoclonal antibody, which recognizes tau phosphorylated at Ser396/404 (1: 1 000; Albert Einstein College of Medicine, Bronx, NY, USA); rabbit anti-rat phospho-PP2A (Tyr307) monoclonal antibody (1:1 000; Epitomics, Burlingame, CA, USA); and rabbit anti-rat PP2A monoclonal antibody (1:1 000; Epitomics) overnight at 4°C. Detection was performed using alkaline phosphatase-conjugated goat-anti-rabbit IgG (1:10 000; Sigma) or goat anti-mouse IgG (1:10 000; Sigma). The bands on the membrane were scanned and analyzed by an image analyzer (Labworks Software, UVP Inc., Upland, CA, USA). The absorbance values of the bands were determined. The results were expressed as fold-change versus sham-operated group.
Histological staining for the survival of hippocampal CA1 pyramidal neurons
Rats were deeply anesthetized by chloral hydrate and perfused transcardially with chilled physiological saline for 3 minutes and then 4% paraformaldehyde. Brains were removed and stored in the same paraformaldehyde solution overnight. Frozen sections (10 μm) were cut coronally and stained with cresyl violet (Sigma). The sections were examined with light microscopy (Olympus, Tokyo, Japan) and the survival of hippocampal CA1 pyramidal cells per 1 mm length was counted as the neuronal density.
Statistical analysis
Data are expressed as mean ± SD from eight independent animals. Statistical analysis of the results was conducted using Origin 7.0 software (OriginLab Corporation, Northampton, MA, USA), and one-way analysis of variance followed by least significant difference test or Newman-Keuls test. A value of P < 0.05 was considered statistically significant.
Acknowledgments
We are grateful to Dr. Davies P from Albert Einstein College of Medicine, USA for providing paired helical filament-1 antibody.
Footnotes
Funding: This work was supported by the National High Technology Research and Development Program of China (863 Program), No. 2012AA020905; the Biological Industry Development Funds of Shenzhen, No. JC201005260093A; the National Natural Science Foundation of China/Research Grants Council Joint Research Scheme, No. 81161160570; the National Natural Science Foundation of China, No. 81171143; and the Tsinghua-Yue-Yuen Medical Sciences Fund.
Conflicts of interest: None declared.
Ethical approval: The study was approved by the Animal Ethics Committee of Tsinghua University, Beijing, China.
(Reviewed by Slone-Murphy J, Robens J, Ma R, Zhang L)
(Edited by Wang LM, Qiu Y, Li CH, Song LP, Liu WJ, Zhao M)
REFERENCES
- [1].Macrae IM. Preclinical stroke research--advantages and disadvantages of the most common rodent models of focal ischaemia. Br J Pharmacol. 2011;164(4):1062–1078. doi: 10.1111/j.1476-5381.2011.01398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kalra L. Stroke rehabilitation 2009: old chestnuts and new insights. Stroke. 2010;41(2):e88–90. doi: 10.1161/STROKEAHA.109.572297. [DOI] [PubMed] [Google Scholar]
- [3].O’Collins VE, Macleod MR, Cox SF, et al. Preclinical drug evaluation for combination therapy in acute stroke using systematic review, meta-analysis, and subsequent experimental testing. J Cereb Blood Flow Metab. 2011;31(3):962–975. doi: 10.1038/jcbfm.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Haelewyn B, Risso JJ, Abraini JH. Human recombinant tissue-plasminogen activator (alteplase): why not use the ‘human’ dose for stroke studies in rats? J Cereb Blood Flow Metab. 2010;30(5):900–903. doi: 10.1038/jcbfm.2010.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fisher M, Feuerstein G, Howells DW, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40(6):2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vermeer SE, Prins ND, den Heijer T, et al. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348(13):1215–1222. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- [7].Román GC, Kalaria RN. Vascular determinants of cholinergic deficits in Alzheimer disease and vascular dementia. Neurobiol Aging. 2006;27(12):1769–1785. doi: 10.1016/j.neurobiolaging.2005.10.004. [DOI] [PubMed] [Google Scholar]
- [8].Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- [9].Noble W, Pooler AM, Hanger DP. Advances in tau-based drug discovery. Expert Opin Drug Discov. 2011;6(8):797–810. doi: 10.1517/17460441.2011.586690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Castro-Alvarez JF, Gutierrez-Vargas J, Darnaudéry M, et al. ROCK inhibition prevents tau hyperphosphorylation and p25/CDK5 increase after global cerebral ischemia. Behav Neurosci. 2011;125(3):465–472. doi: 10.1037/a0023167. [DOI] [PubMed] [Google Scholar]
- [11].Liu F, Shi J, Tanimukai H, et al. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain. 2009;132(Pt 7):1820–1832. doi: 10.1093/brain/awp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mailliot C, Podevin-Dimster V, Rosenthal RE, et al. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2000;20(3):543–549. doi: 10.1097/00004647-200003000-00013. [DOI] [PubMed] [Google Scholar]
- [13].Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8(3):126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- [14].Spittaels K, Van den Haute C, Van Dorpe J, et al. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275(52):41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- [15].Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107(2):519–527. [PubMed] [Google Scholar]
- [16].Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5) Biochim Biophys Acta. 1984;788(3):339–347. doi: 10.1016/0167-4838(84)90047-5. [DOI] [PubMed] [Google Scholar]
- [17].Hanger DP, Hughes K, Woodgett JR, et al. Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147(1):58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- [18].Mandelkow EM, Drewes G, Biernat J, et al. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314(3):315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- [19].Liu F, Grundke-Iqbal I, Iqbal K, et al. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22(8):1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- [20].Cohen PT. Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem Sci. 1997;22(7):245–251. doi: 10.1016/s0968-0004(97)01060-8. [DOI] [PubMed] [Google Scholar]
- [21].Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257(5074):1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- [22].Plattner F, Angelo M, Giese KP. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006;281(35):25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- [23].Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci U S A. 1999;96(15):8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9(9):2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- [25].Kim YR, van Meer MP, Tejima E, et al. Functional MRI of delayed chronic lithium treatment in rat focal cerebral ischemia. Stroke. 2008;39(2):439–447. doi: 10.1161/STROKEAHA.107.492215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xu J, Culman J, Blume A, et al. Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke. 2003;34(5):1287–1292. doi: 10.1161/01.STR.0000066308.25088.64. [DOI] [PubMed] [Google Scholar]
- [27].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions (Instructions) for the Care and Use of Laboratory Animals 2006-09-30 [Google Scholar]
- [28].Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10(3):267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- [29].Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11(1):47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- [30].Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]