

Abstract
We previously found that the K141N mutation in heat shock protein B8 (HSPB8) was responsible for Charcot-Marie-Tooth disease type 2L in a large Chinese family. The objective of the present study was to generate a transgenic mouse model bearing the K141N mutation in the human HSPB8 gene, and to determine whether this K141NHSPB8 transgenic mouse model would manifest the clinical phenotype of Charcot-Marie-Tooth disease type 2L, and consequently be suitable for use in studies of disease pathogenesis. Transgenic mice overexpressing K141NHSPB8 were generated using K141N mutant HSPB8 cDNA cloned into a pCAGGS plasmid driven by a human cytomegalovirus expression system. PCR and western blot analysis confirmed integration of the K141NHSPB8 gene and widespread expression in tissues of the transgenic mice. The K141NHSPB8 transgenic mice exhibited decreased muscle strength in the hind limbs and impaired motor coordination, but no obvious sensory disturbance at 6 months of age by behavioral assessment. Electrophysiological analysis showed that the compound motor action potential amplitude in the sciatic nerve was significantly decreased, but motor nerve conduction velocity remained normal at 6 months of age. Pathological analysis of the sciatic nerve showed reduced myelinated fiber density, notable axonal edema and vacuolar degeneration in K141NHSPB8 transgenic mice, suggesting axonal involvement in the peripheral nerve damage in these animals. These findings indicate that the K141NHSPB8 transgenic mouse successfully models Charcot-Marie-Tooth disease type 2L and can be used to study the pathogenesis of the disease.
Keywords: nerve regeneration, peripheral nerve injury, axonal injury, animal models, Charcot-Marie-Tooth disease type 2L, gene mutation, pronuclear injection, transgenic model, small heat shock protein B8, NSFC grant, neural regeneration
Introduction
Charcot-Marie-Tooth disease is the most common inherited peripheral neuropathy, with an estimated prevalence of 17–40/10,000[1,2]. Charcot-Marie-Tooth is characterized by distal muscle weakness and atrophy, distal sensory loss, depressed tendon reflexes, and pes cavus. There are two main forms: the demyelinating type 1 (Charcot-Marie-Tooth disease type 1) and the axonal type 2 (Charcot-Marie-Tooth disease type 2). Autosomal-dominant inheritance is most common, but X-linked and autosomal-recessive forms are also seen[1,2,3]. There is considerable genetic heterogeneity, and more than 40 genes have been identified (http://neuromuscular.wustl.edu/time/hmsn.html); however, the underlying pathogenetic mechanisms remain unclear[4,5].
Tang et al.[6] identified an autosomal dominant familial form of Charcot-Marie-Tooth disease type 2 in a Chinese family, linked to 12q24 by genome-wide screening, and designated Charcot-Marie-Tooth disease type 2L (OMIM Number 608673). A positional candidate cloning study showed that the G423T (K141N) mutation in the small heat shock protein B8 (HSPB8) gene co-segregated with the Charcot-Marie-Tooth disease type 2L phenotype, and that it was the causative mutation[7]. The expression of K141NHSPB8 in SHSY5Y cells results in intracellular aggregates that are mainly distributed in the cytoplasm and that are colocalized with HSPB1 and neurofilament light polypeptide NEFL[8,9].
To provide insight into the molecular pathogenesis of the disease, in the present study, the transgenic plasmid pCAGGS-HA- K141NHSPB8 was constructed, and K141NHSPB8 transgenic mice were generated by pronuclear injection. Behavioral, electrophysiological, and pathological analyses were performed on K141NHSPB8 transgenic mice to determine whether they manifest the clinical phenotype of Charcot-Marie-Tooth disease type 2L and would be suitable for studies on disease pathogenesis.
Results
Generation of K141NHSPB8 transgenic mice
We used PCR combined with sequencing for genotyping. The amplification yielded a 976-bp genomic fragment with primers CAG1 and CAG2, and a 516-bp genomic fragment with primers CAG1 and HSP (Figure 1A–C). Sequencing of the 976-bp amplification product showed the presence of the G423T mutation when aligned with the HSPB8 sequence (Figure 1C). Western blot assay revealed extensive expression of the HA-tagged 22-kDa human HSPB8 in heart and gastrocnemius muscle of founder mice (Figure 1D). Three founder males were generated and bred normally to establish lines of K141NHSPB8 transgenic mice.
Figure 1.
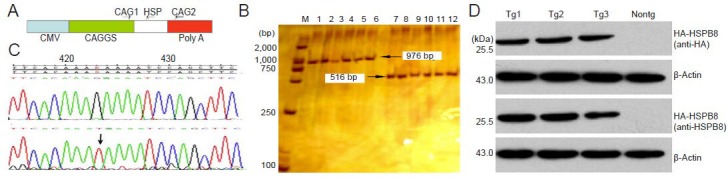
Genotyping of K141NHSPB8 transgenic (Tg) mice.
(A) Relative position of primers used to genotype K141NHSPB8 Tg mice on the linearized pCAGGS- K141NHSPB8 plasmid. (B) Electrophoresis of PCR products showing the 976-bp and 516-bp genomic fragments. M: DL2000 marker; lanes 1–6: Tg1–6 genome amplified using CAG1 + CAG2; lanes 7–12: Tg1–6 genome amplified using CAG1 + HSP. (C) Sequence analysis of K141NHSPB8 Tg mice. The arrow shows the G423T mutation in the HSPB8 gene. (D) Western blot assay of the gastrocnemius muscle samples from K141NHSPB8 Tg mice (1–3) and a wild type C57BL mouse (Nontg) using anti-HA and anti-HSPB8 antibodies. HSPB8: Heat shock protein B8; HA: a nine amino acid peptide sequence (YPYDVPDYA); CMV: cytomegalovirus enhancer; CAG: primer.
Quantitative analysis of the experimental mice
Nine 6-month-old K141NHSPB8 transgenic mice, and nine age-matched C57BL mice, as control, were used for behavioral assessments, and data from all mice were included in the final analyses. Three of the nine 6-month-old K141NHSPB8 transgenic mice and three age-matched C57BL mice were used for sciatic nerve electrophysiological and histopathological studies.
Impaired motor performance in K141NHSPB8 transgenic mice
All K141NHSPB8 transgenic mice were normal at birth and showed normal weaning and grooming behavior. The frequency at birth of the various genotypes exhibited a normal Mendelian inheritance pattern. At 6 months of age, the mice displayed an unsteadiness of gait, reminiscent of the “steppage” gait in Charcot-Marie-Tooth disease patients. Tremor and seizures were absent. When walking over a flat surface in a straight line, the transgenic mice moved clumsily and displayed outward positioning of their hind paw with a significantly more open hind paw angle than wild type mice (P < 0.05; Figure 2A–C). In the fixed-bar test, all the wild type mice balanced and supported their body weight easily, and moved agilely on the bar. In contrast, the transgenic mice either remained motionless or crawled with difficulty using only their forelimbs, dragging their hind limbs behind. Two of the transgenic mice dropped off the bar in all eight consecutive trials within 120–240 seconds. In the rotarod test, the K141NHSPB8 transgenic mice remained on the rotating roller for a significantly shorter period of time than wild type mice (P < 0.05; Figure 3). However, in the pain threshold experiment involving plantar electric shocks, there were no significant differences between K141NHSPB8 mice and age-matched wild type mice.
Figure 2.

Peripheral neuropathy in K141NHSPB8 transgenic (Tg) mice.
(A) At around 6 months of age, K141NHSPB8 Tg mice developed an unsteady gait and signs of muscle weakness of the hind limbs. The left is a Nontg mouse and the right is a Tg mouse. (B) K141NHSPB8 Tg mice showed a slightly outward positioning of the hind paws in footprint analysis, with the pattern of the Nontg animals (upper panel) slightly different from that of Tg animals (lower panel). (C) Analysis of the footprint patterns of the Nontg (n= 9) and Tg (n= 9) mice. The mean hind paw angle ± SEM is shown (P < 0.05, two sample t-test). HSPB8: Heat shock protein B8; Nontg: wild type.
Figure 3.
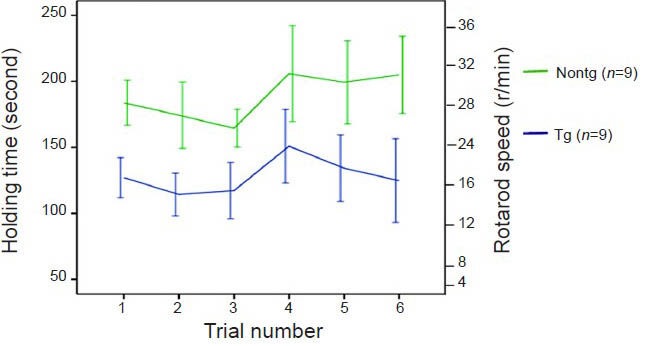
Decreased motor performances in K141NHSPB8 transgenic (Tg) mice. A rotarod test demonstrated motor deficits in K141NHSPB8 Tg mice.
In a series of six consecutive trails, the cumulative time in which animals were able to stay on a rotating rod was recorded. Holding time was significantly lower in Tg than in Nontg mice (F= 69.41, P < 0.01; two-way analysis of variance followed by Bonferroni's post hoc test). Data are expressed as mean ± SEM. HSPB8: Heat shock protein B8; Nontg: wild type.
Electrophysiological alterations in the proximal and distal sciatic nerve in K141NHSPB8 transgenic mice
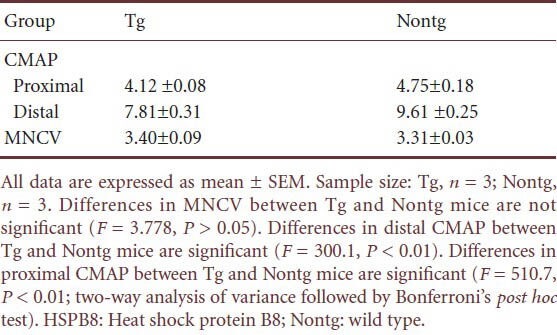
K141NHSPB8 transgenic mice showed a significant decrease in the peak-to-peak amplitude of compound motor action potentials in both proximal and distal sciatic nerve at the age of 6 months, confirming the axonal basis of the motor neuropathy (P < 0.01; Figure 4, Table 1). Motor nerve conduction velocity was normal, indicating that the myelin sheath remained basically intact (Table 1).
Figure 4.
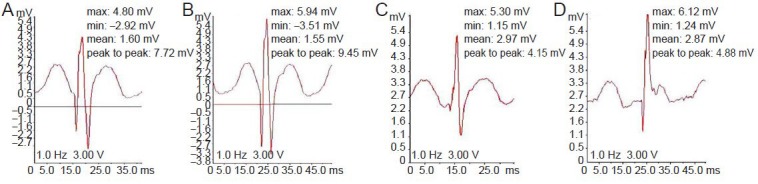
Sciatic nerve compound motor action potential (CMAP) recordings in K141NHSPB8 transgenic (Tg) mice.
Representative traces of sciatic nerve CAMPs recorded from an intrinsic foot muscle after proximal and distal stimulation. Compared to 6-month-old Nontg mice (B, distal CMAP; D, proximal CMAP), age-matched K141NHSPB8 Tg mice showed a significant decrease in both distal (A) and proximal CMAP (C). X-axis represents time (ms); Y-axis represents wave amplitude of the CMAP (mV). HSPB8: Heat shock protein B8; min: minimum; max: maximum.
Table 1.
Compound motor action potential (CMAP) (mV) and motor nerve conduction velocity (MNCV) (m/s) in K141NHSPB8 transgenic (Tg) mice
Histopathological alterations in the distal portion of the sciatic nerve in K141NHSPB8 transgenic mice
Semithin sections of the distal portion of the sciatic nerve from 6-month-old K141NHSPB8 transgenic mice revealed a decrease in the number of axons compared with wild type control mice (P < 0.05; Figure 5A–C). No signs of demyelination or remyelination were observed. Under the electron microscope, ultrathin sections of the sciatic nerve showed the presence of notable axonal edema and degeneration, although the myelin sheath remained relatively intact (Figure 5D, E).
Figure 5.
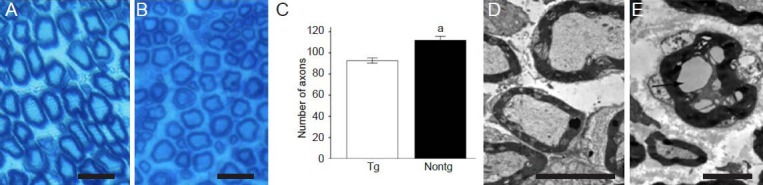
Peripheral axonal deficits in K141NHSPB8 transgenic (Tg) mice.
(A) Normal axonal density and myelin sheath in the sciatic nerve of 6-month-old control Nontg mice (toluidine blue staining). (B) Toluidine blue staining of semithin sciatic nerve sections of 6-month-old K141NHSPB8 Tg showed a decrease in the number of axons. No signs of demyelination or remyelination were visible. (C) Quantification of the number of axons in distal parts of the sciatic nerve. Data are expressed as mean ± SEM. aP < 0.05, vs. Tg (two sample t-test). (D) Electron micrographs of ultrathin sciatic nerve sections of age-matched Nontg mice showed normal axonal and myelin sheath structure. (E) Electron micrographs of ultrathin sciatic nerve sections of 6-month-old K141NHSPB8 Tg mice showed axonal edema and vacuolation (arrow). The myelin sheath remained intact. Scale bars: A and B, 40 μm; D, 5 μm; E, 2.5 μm. HSPB8: Heat shock protein B8; Nontg: wild type.
Discussion
In the present study, we created a transgenic mouse model of Charcot-Marie-Tooth disease type 2L by overexpressing human HSPB8 containing the K141N mutation. The ubiquitous-expression pCAGGS plasmid, driven by the human cytomegalovirus expression system, has proven to be suitable for the creation of transgenic mouse models. Therefore, we chose this plasmid for the generation of the K141NHSPB8 transgenic mice[10,11,12]. PCR and western blot assays on the K141NHSPB8 transgenic founder mice confirmed that the human G423T (K141N) mutant HSPB8 gene had successfully integrated into the mouse chromosome and was widely expressed.
Footprint analysis, a behavioral assessment, mainly reflects the motor function status of the hind limbs. Because the outward positioning of the hind paw helps to control the forward momentum generated during walking, hind paw angle was used as a specific index to evaluate the state of muscle relaxation of the hind limbs[13,14]. The increased angle of paw placement in K141NHSPB8 mice (6 months of age, equivalent to human 16.1 years) suggested motor nerve impairment in the hind limbs. In both the fixed-bar and rotarod tests, the K141NHSPB8 mice showed notable impairment in motor coordination. However, the pain threshold experiment involving plantar electric shocks did not show a significant difference between K141NHSPB8 mice and age-matched wild type mice. This suggests that sensory function in K141NHSPB8 mice was not significantly affected.
Electrophysiological study is mainly used to examine the physiological status of large-diameter nerve fibers. Demyelination of peripheral nerve results in a substantial reduction in conduction velocity, and other typical changes in motor nerve conduction, including a notable increase in motor nerve latency and conduction block. However, axonal deficit is characterized by notable decreases in the amplitude of compound motor action potentials[15,16]. The electrophysiological study of the peripheral nerve in K141NHSPB8 mice revealed a significantly decreased amplitude of compound motor action potentials, and a relatively normal nerve conduction velocity. This suggests that the phenotype of K141NHSPB8 transgenic mice is caused by axonal deficits, rather than myelin abnormalities. Histopathological study of the peripheral nerve in K141NHSPB8 transgenic mice showed reduced numbers of myelinated fibers, axonal vacuoles and degeneration. There was no thickening of the myelin sheath or onion-bulb like structures, which further suggests that peripheral neuropathy is an axonal neuropathy rather than a demyelinating one. The behavioral, electrophysiological and histopathological characteristics of the transgenic mice are similar to the features in patients with familial Charcot-Marie-Tooth disease type 2L, in whom the onset of disease occurs at young or middle age. In these patients, impairment mainly involves the lower limbs, and electrophysiological and histopathological studies show an axonal neuropathy.
The G423T (K141N) mutation in HSPB8 causes Charcot-Marie-Tooth disease type 2L. Interestingly, the K141N and K141E mutations in HSPB8 were also identified in families with distal hereditary motor neuropathy (dHMN)[17,18]. The dHMNs comprise a heterogeneous group of diseases that share the common feature of a length-dependent predominant motor neuropathy. Many forms of dHMN have minor sensory abnormalities and/or a significant upper motor neuron component, and there is often an overlap with the axonal forms of Charcot-Marie-Tooth disease type 2 and with juvenile forms of amyotrophic lateral sclerosis and hereditary spastic paraplegia[19]. Patients in the Charcot-Marie-Tooth disease type 2L family had mild sensory impairments, including deficits in pain and touch, but no evidence of painless injury or ulceration. While both start in adulthood, distal HMN type II progresses more rapidly, and complete paralysis of all distal muscles of the lower extremities occurs within 5 years, while Charcot-Marie-Tooth disease type 2L progresses slowly and all patients remain ambulant. It remains unclear how the K141N mutation in HSPB8 produces different phenotypes in the two different diseases. In the present study, the K141NHSPB8 transgenic mice developed slowly progressing hind limb weakness, and had no obvious sensory impairment at the age of 6 months, indicating relatively mild disease progression, with little or no sensory disturbance.
We consider the K141NHSPB8 transgenic mice that we generated to be a suitable model of human Charcot-Marie-Tooth disease type 2L. However, the degree of sensory involvement in the peripheral nerves needs to be further investigated. Further study on dorsal root ganglion neurons is needed to clarify whether the sensory system is affected to varying degrees in individual transgenic mice[20,21].
In a previous study, combined expression of HSPB8 K141N and HSPB8 K141E mutant proteins in motor neurons resulted in neurite degeneration, manifested by a reduction in number of neurites per cell and a reduction in average length of neurites[22]. In early passage, primary fibroblast cultures derived from dHMN patient skin biopsies, HSPB8 protein aggregates were present and mitochondrial membrane potential was reduced[23]. Cytoplasmic aggregates were observed when K141NHSPB8 was transiently expressed in cultured cells, and cell viability was impaired after heat shock treatment[24]. Overexpression of mutant HSPB8 was found to result in autophagosomes that colocalized with protein aggregates, but failed to colocalize with lysosomes[25]. HSPB8 and Bag3 form a chaperone complex that stimulates degradation of protein substrates by macroautophagy. Thus, defects in HSPB8-mediated autophagy are likely to be pathogenic[26,27,28,29,30].
We anticipate that the novel K141NHSPB8 transgenic mouse model, coupled with cell and tissue culture systems, will be a valuable research tool for elucidating the cellular and molecular pathogenesis of Charcot-Marie-Tooth disease type 2, and should advance the development of novel therapeutic strategies for this neurological disorder.
Materials and Methods
Design
Establishment of a transgenic mouse model.
Time and setting
Major components of experiments were performed at the State Key Laboratory of Medical Genetics and Third Xiangya Hospital, Central South University, China; parts of experiments were performed at Institute of Neuroscience, Shanghai Institute for Biological Sciences and Kunming Institute of Zoology, Chinese Academy of Sciences, China from March 2008 to August 2012.
Materials
A total of nine clean K41NHSPB8 transgenic mice (four females, five males), aged 6 months, weighing 23–28 g, and nine clean wild type C57BL mice (four females, five males), aged 6 months, weighing 23–28 g, as controls, were provided by the Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences in China (license No. SCXK (Jing) 2005-0013). Animals were housed at the Laboratory in the Animal Facility of Third Xiangya Hospital, Central South University in China. Animals were reared in 17 cm × 26 cm cages, each containing two or three mice, under a 12-hour light/dark cycle (lights off between 18:00–06:00), at an average temperature of 22°C, with free access to food and water. This study was approved by the Animal Ethics Committee, Third Xiangya Hospital, Central South University, China.
Methods
Creation and genotyping of K141NHSPB8 transgenic mice
The pEGFPN1- K141NHSPB8 vector constructed in our previous study was used as a template to clone HA-tagged K141NHSPB8 cDNA using the EcoRI restriction endonuclease into the pCAGGS plasmid[8]. An expression vector driven by a human cytomegalovirus immediate-early enhancer linked to the chicken β-actin promoter was used[8]. We excised a 2,896-bp fragment from the pCAGGS-HA- K141NHSPB8 vector with the restriction endonucleases SalI and HindIII, and then with BsaXI. We purified the fragment from an agarose gel with the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany), dialyzed it against injection buffer, and diluted it to a concentration of 2 ng/μL. After preparing C57BL female mice for ovulation and egg fertilization, a 0.5-μL aliquot of DNA was microinjected into the fertilized eggs. Injected zygotes were maintained overnight and transferred into pseudo-pregnant C57BL females. The mice were placed in individually ventilated cages. The tail tips of 1-week-old pups were collected (1–2 cm/pup), and the phenol-chloroform method was used to extract DNA. Genotyping of transgenic animals was performed with two primer sets: forward primer (CAG1), 5′-GCC ACC ATG TAC CCA TAC G-3′ and reverse primer (CAG2), 5′-GCA GGA GGC TGT TTC ATA-3′ to amplify the transgenic construct including the whole HSPB8 gene; forward primer CAG1 and reverse primer (HSP), 5′-TGG GGA AAG TGA GGC AAA TA-3′ to amplify the transgenic construct including part of the HSPB8 gene to confirm that HSPB8 had inserted into the mouse genome. PCR products were separated by 8% polyacrylamide gel electrophoresis, and DNA sequencing was performed using an ABI PRISM 3100 sequencer (Perkin Elmer, Waltham, MA, USA). Sequencing results were analyzed with DNASTAR Lasergene.v7.1 software (DNASTAR. Inc, Madison, WI, USA). The transgenic founders were identified and subsequently transferred to the animal facility of Third Xiangya Hospital, Central South University in China.
Western blot assay for HA-tagged HSPB8 in K141NHSPB8 founder mice
Three 10-month-old K141NHSPB8 founder mice and one wild type C57BL control were killed by cervical dislocation, and flash-frozen tissue samples were maintained at −80 °C. Frozen tissues from heart and gastrocnemius muscles were homogenized in radio immunoprecipitation assay buffer (containing 50 mmol/L Tris, 150 mmol/L NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate and phosphatase inhibitors). Protein concentrations were determined using the microBCA kit (Pierce Chemical Co., Rockford, IL, USA) according to the manufacturer's instructions. Western blot assay was performed as described before[8]. Goat anti-HSPB8 antibody (1:500; Sigma-Aldrich, Milwaukee, WI, USA), mouse anti-HA-Tag antibody (1:2,000; Sigma-Aldrich) and mouse anti-β-actin antibody (1:500; Sigma-Aldrich) were used as primary antibodies and incubated with the blots at 4°C for 2 hours. Rabbit anti-goat IgG (1:2,000; Sigma-Aldrich) and goat anti-mouse lgG (1:6,000 or 1:10,000; Sigma-Aldrich) were the secondary antibodies, and were incubated with the blots at 4°C for 2 hours. The target bands were visualized using an enhanced chemiluminescence detection kit (BioRad, Hercules, CA, USA) and then exposed to X-ray film in a dark room. The X-ray film signal was scanned on an imaging analysis system (Bio-Rad), and absorbance values were analyzed.
Behavioral assessments of K141NHSPB8 transgenic mice
For each behavioral experiment, each mouse was tested twice a day for 4 days in a row to ensure the repeatability and reliability of the data. Footprint analysis was performed as described by Sereda et al.[13]. Waterproof black ink was applied to the hind paws and footprints were taken on watercolor paper. The animal was allowed to walk forward in a 12-cm-wide lane. The footprints were then scanned and the angle of deviation of the median foot axis with regard to the movement axis (i.e., the hind paw angle) was measured for 10 clearly visible footprints per mouse.
Fixed-bar test was performed as described by Norreel et al.[31,32]. For the fixed-bar test, a round wooden bar (diameter: 1.5 cm; length: 50 cm) was positioned 40 cm above the cage floor. Mice were placed on the middle of the bar. The movements and the time (in seconds) that the animals remained on the bar were monitored using videos. A maximum of 5 minutes per trial was allowed. Motor coordination was further assessed with the rotarod test, as described by Zhao et al.[33]. The apparatus (DXP-2, Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing, China), consisting of a base platform and a rotating rod with a diameter of 2.5 cm, was subdivided into four equal sections. The mice were trained to reach their performance baseline with 5 sessions of 2-minute periods of walking at 4 r/min per day for two days before the test. After training, each mouse was placed on the rod and allowed to rest for 30 seconds, and then the rod was rotated at 2 r/min. The rotation speed was increased every 30 seconds to 4, 8, 12, 16, 24, 28, 32 and 36 r/min. In a series of six trials per animal, the time (in seconds) that the mouse remained on the rod was measured. Pain threshold test was performed as previously described[34,35]. Mice were placed in a Threshold Activity Monitoring system (Med Associates Inc., Pittsburgh, PA, USA). Electric shocks began at 0.2 mA, 0.5 seconds, and were increased by 0.1 mA in 1-min intervals until the mouse had the first pain response (either limb flick or jump). The amplitude of the current at which the mouse had the first pain response was recorded.
Nerve electrophysiology in K141NHSPB8 transgenic mices
Following anesthesia with pentobarbital (240 mg/kg), mice were fixed on a thermostat-controlled heating plate at 37°C. The sciatic nerve on one side was carefully isolated. The sciatic and posterior tibial nerves were stimulated with needle electrodes inserted alongside the nerves at the sciatic notch and at the hock. The distance between the two points was 1 cm. The square pulses (1 Hz, 3 mV) were delivered five times per site using the BL-420E+ biological and functional experimental system (Pclab, Chengdu, China). Compound motor action potentials were recorded from an intrinsic foot muscle using a concentric needle electrode. The latencies of compound motor action potentials, elicited by stimulation at both proximal (hip) and distal (hock) sites, were measured from the stimulus artifact to the first negative deflection. Amplitudes were determined as the maximum peak-to-peak voltage. Motor nerve conduction velocity was calculated using the distance between the two points of stimulation (1 cm) and the difference in the latencies between the two points (conduction time).
Sciatic nerve histopathology in K141NHSPB8 transgenic mice
After nerve electrophysiology, anesthetized mice were perfused through the left ventricle with saline followed by 4% paraformaldehyde in cacodylate buffer (pH 7.2). The sciatic nerves on the other side were carefully isolated and dissected from the region of the sciatic notch to the hock. Nerve specimens were placed in either 4% paraformaldehyde (0.05 mol/L PBS, pH 7.2) or 2.5% glutaraldehyde. Specimens fixed in 4% paraformaldehyde were embedded in paraffin, subjected to semi-thin (1-μm) sectioning, stained with 1% toluidine blue at 80°C for 30–45 seconds, then observed with an Olympus CX31 light microscope (Olympus Corporation, Tokyo, Japan). The number of axons was counted on four slides from each mouse. Specimens in 2.5% glutaraldehyde were embedded in porous rubber bodies, subjected to ultrathin (50-nm) sectioning, double stained with uranyl acetate and lead nitrate, and then observed with an H-7500 transmission electron microscope (Hitachi, Tokyo, Japan) and photographed.
Statistical analysis
Data were expressed as mean ± SEM, and processed with SPSS 19.0 software (SPSS, Chicago, IL, USA). Differences between repeated measures of different genotypes were compared using two-way analysis of variance followed by Bonferroni's post hoc test. Comparisons of means were made using a two-sample t-test. A value of P < 0.05 was considered statistically significant.
Footnotes
Funding: This project was funded by the National Natural Science Foundation of China, No. 81071001, 30900805.
Conflicts of interest: None declared.
Peer review: Although progress in molecular genetics research is significant, the pathological mechanism of Charcot-Marie-Tooth still waits to be unveiled. In this study, the K141NHSPB8 transgenic mouse model is successfully generated, and can be used as a model to study the pathogenesis of Charcot-Marie-Tooth disease type 2L.
Copyedited by Patel B, Fitting S, Yu J, Qiu Y, Li CH, Song LP, Zhao M
References
- [1].Dyck PJ, Thomas PK. 4th ed. Philadelphia: Elsevier Saunders; 2005. Peripheral Neuropathy. [Google Scholar]
- [2].Patzkó A, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep. 2011;11(1):78–88. doi: 10.1007/s11910-010-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gentil BJ, Cooper L. Molecular basis of axonal dysfunction and traffic impairments in CMT. Brain Res Bull. 2012;88(5):444–453. doi: 10.1016/j.brainresbull.2012.05.003. [DOI] [PubMed] [Google Scholar]
- [4].Siskind CE, Panchal S, Smith CO, et al. A review of genetic counseling for Charcot Marie Tooth disease (CMT) J Genet Couns. 2013;22(4):422–436. doi: 10.1007/s10897-013-9584-4. [DOI] [PubMed] [Google Scholar]
- [5].Miller LJ, Saporta AS, Sottile SL, et al. Strategy for genetic testing in Charcot-Marie-disease. Acta Myol. 2011;30(2):109–116. [PMC free article] [PubMed] [Google Scholar]
- [6].Tang BS, Luo W, Xia K, et al. A new locus for autosomal dominant Charcot-Marie-Tooth disease type 2 (CMT2L) maps to chromosome 12q24. Hum Genet. 2004;114(6):527–533. doi: 10.1007/s00439-004-1102-1. [DOI] [PubMed] [Google Scholar]
- [7].Tang BS, Zhao GH, Luo W, et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum Genet. 2005;116(3):222–224. doi: 10.1007/s00439-004-1218-3. [DOI] [PubMed] [Google Scholar]
- [8].Zhang RX, Tang BS, Zi XH, et al. The construction of pEGFPN1-wtHSP22 and pEGFPN1-mtHSP22 vectors and their expressions in human neuroblastoma cells. Zhonghua Yi Xue Za Zhi. 2006;86(25):1780–1782. [Google Scholar]
- [9].Zhang RX, Tang BS, Zi XH, et al. Study on aggregate formation mechanism of HSPB8 gene mutation resulting in CMT2L. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2006;23(6):601–604. [PubMed] [Google Scholar]
- [10].Ray PS, Martin JL, Swanson EA, et al. Transgene overexpression of alphaB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB J. 2001;15(2):393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- [11].Ahmad S, Cesana F, Lamperti E, et al. Attenuation of angiotensin II-induced hypertension and cardiac hypertrophy in transgenic mice overexpressing a type 1 receptor mutant. Am J Hypertens. 2009;22(12):1320–1325. doi: 10.1038/ajh.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tang L, Yu CY, Feng R, et al. Establishment of PD-L1 transgenic mouse model and recovery of the motion after spinal cord injury. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2011;27(4):357–359. 363. [PubMed] [Google Scholar]
- [13].Sereda M, Griffiths I, Pühlhofer A, et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16(5):1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- [14].d’Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. doi: 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- [15].Meyer Zu Hörste G, Nave KA. Animal models of inherited neuropathies. Curr Opin Neurol. 2006;19(5):464–473. doi: 10.1097/01.wco.0000245369.44199.27. [DOI] [PubMed] [Google Scholar]
- [16].Patzkó A, Bai Y, Saporta MA, et al. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain. 2012;135(Pt 12):3551–3566. doi: 10.1093/brain/aws299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Irobi J, Van Impe K, Seeman P, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36(6):597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- [18].Timmerman V, De Jonghe P, Simokovic S, et al. Distal hereditary motor neuropathy type II (distal HMN II): mapping of a locus to chromosome 12q24. Hum Mol Genet. 1996;5(7):1065–1069. doi: 10.1093/hmg/5.7.1065. [DOI] [PubMed] [Google Scholar]
- [19].Rossor AM, Kalmar B, Greensmith L, et al. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6–14. doi: 10.1136/jnnp-2011-300952. [DOI] [PubMed] [Google Scholar]
- [20].Nobbio L, Mancardi G, Grandis M, et al. PMP22 transgenic dorsal root ganglia cultures show myelin abnormalities similar to those of human CMT1A. Ann Neurol. 2001;50(1):47–55. doi: 10.1002/ana.1034. [DOI] [PubMed] [Google Scholar]
- [21].Melli G, Höke A. Dorsal Root Ganglia Sensory Neuronal Cultures: a tool for drug discovery for peripheral neuropathies. Expert Opin Drug Discov. 2009;4(10):1035–1045. doi: 10.1517/17460440903266829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Irobi J, Almeida-Souza L, Asselbergh B, et al. Mutant HSPB8 causes motor neuron-specific neurite degeneration. Hum Mol Genet. 2010;19(16):3254–3265. doi: 10.1093/hmg/ddq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Irobi J, Holmgren A, De Winter V, et al. Mutant HSPB8 causes protein aggregates and a reduced mitochondrial membrane potential in dermal fibroblasts from distal hereditary motor neuropathy patients. Neuromuscul Disord. 2012;22(8):699–711. doi: 10.1016/j.nmd.2012.04.005. [DOI] [PubMed] [Google Scholar]
- [24].Li SJ, Tang BS, Zhao GH, et al. The effect of HSPB8 gene mutation on cell viability in Charcot-Marie-Tooth disease type 2L. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2011;28(5):528–531. doi: 10.3760/cma.j.issn.1003-9406.2011.05.012. [DOI] [PubMed] [Google Scholar]
- [25].Kwok AS, Phadwal K, Turner BJ, et al. HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J Neurochem. 2011;119(6):1155–1161. doi: 10.1111/j.1471-4159.2011.07521.x. [DOI] [PubMed] [Google Scholar]
- [26].Carra S, Seguin SJ, Landry J. HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy. 2008;4(2):237–239. doi: 10.4161/auto.5407. [DOI] [PubMed] [Google Scholar]
- [27].Seidel K, Vinet J, Dunnen WF, et al. The HSPB8-BAG3 chaperone complex is upregulated in astrocytes in the human brain affected by protein aggregation diseases. Neuropathol Appl Neurobiol. 2012;38(1):39–53. doi: 10.1111/j.1365-2990.2011.01198.x. [DOI] [PubMed] [Google Scholar]
- [28].Nivon M, Abou-Samra M, Richet E, et al. NF-êB regulates protein quality control after heat stress through modulation of the BAG3-HspB8 complex. J Cell Sci. 2012;125(Pt 5):1141–1151. doi: 10.1242/jcs.091041. [DOI] [PubMed] [Google Scholar]
- [29].Shemetov AA, Gusev NB. Biochemical characterization of small heat shock protein HspB8 (Hsp22)-Bag3 interaction. Arch Biochem Biophys. 2011;513(1):1–9. doi: 10.1016/j.abb.2011.06.014. [DOI] [PubMed] [Google Scholar]
- [30].Behl C. BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy. 2011;7(7):795–798. doi: 10.4161/auto.7.7.15844. [DOI] [PubMed] [Google Scholar]
- [31].Norreel JC, Jamon M, Riviere G, et al. Behavioural profiling of a murine Charcot-Marie-Tooth disease type 1A model. Eur J Neurosci. 2001;13(8):1625–1634. doi: 10.1046/j.0953-816x.2001.01535.x. [DOI] [PubMed] [Google Scholar]
- [32].Kadotani H, Hirano T, Masugi M, et al. Motor discoordination results from combined gene disruption of the NMDA receptor NR2A and NR2C subunits, but not from single disruption of the NR2A or NR2C subunit. J Neurosci. 1996;16(24):7859–7867. doi: 10.1523/JNEUROSCI.16-24-07859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhao X, Ye J, Sun Q, et al. Antinociceptive effect of spirocyclopiperazinium salt compound LXM-15 via activating peripheral α7 nAChR and M4 mAChR in mice. Neuropharmacology. 2011;60(2-3):446–452. doi: 10.1016/j.neuropharm.2010.10.019. [DOI] [PubMed] [Google Scholar]
- [34].Rodgers RJ. Effects of nicotine, mecamylamine, and hexamethonium on shock-induced fighting, pain reactivity, and locomotor behaviour in rats. Psychopharmacology (Berl) 1979;66(1):93–98. doi: 10.1007/BF00431996. [DOI] [PubMed] [Google Scholar]
- [35].Ma WQ, Wei HM, Dong FT, et al. Establishment of chronic neuropathic pain model by L4/5 spinal nerve ligature in rats. Kunming Yixueyuan Xuebao. 2010;31(6):57–60. [Google Scholar]