

Abstract
Regulating serotonin expression can be used to treat psychotic depression. Mifepristone, a glucocorticoid receptor antagonist, is an effective candidate for psychotic depression treatment. However, the underlying mechanism related to serotonin transporter expression is poorly understood. In this study, we cloned the human brain serotonin transporter into Xenopus oocytes, to establish an in vitro expression system. Two-electrode voltage clamp recordings were used to detect serotonin transporter activity. Our results show that mifepristone attenuates serotonin transporter activity by directly inhibiting the serotonin transporter, and suggests that the serotonin transporter is a pharmacological target of mifepristone for the treatment of psychotic depression.
Keywords: nerve regeneration, psychotic depression, mifepristone, serotonin transporter, Xenopus oocyte, serotonin, depression, two-electrode voltage clamp, current recording, antidepressants, serotonin transmission, NSFC grant, neural regeneration
Introduction
Major depressive disorder is a common psychiatric disease and the leading cause of disability (Kling et al., 2009), while psychotic depression affects up to 20% of hospitalized patients with major depression (Flores et al., 2006). Psychotic depression patients have markedly elevated circulating glucocorticoids, which are highly associated with abnormal regulation of hypothalamic-pituitary-adrenal axis activity (Iijima et al., 2010; Watson et al., 2012). Currently, glucocorticoids and their receptors are pharmacological targets for psychotic depression treatment. Epidemiological studies indicate that with successful antidepressant treatment, decreased hypercortisolemia occurs (Pariante, 2003; Thomson and Craighead, 2008; Wulsin et al., 2010).
As an important glucocorticoid receptor antagonist, mifepristone (RU-486) is known to have antidepressant effects in both human and animal tests (Belanoff et al., 2002; Flores et al., 2006; Watson et al., 2012). In clinical studies, RU-486 relieves symptoms of psychotic depression after a brief treatment period (Belanoff et al., 2002; Flores et al., 2006), and is associated with sustained improvements in spatial working memory (Watson et al., 2012). Additionally, a multisite trial of RU-486 for psychotic depression therapy showed an approximately 50% decrease in brief psychiatric rating scale, observed in patients randomized to 1 week RU-486 treatment (Blasey et al., 2009). In animals, acute RU-486 therapy counteracted depressive-like behavior and increased swimming behavior, consistent with an anti-depressant action (Gallagher et al., 2008; Iijima et al., 2010). Similarly, in rats, chronic blockage of glucocorticoid receptors by RU-486 injections, dramatically improved lipopolysaccharide-induced depressive-like behavior (Wang et al., 2011). Moreover, RU-486 decreases depressive-like behavior by modulating central hypothalamic-pituitary-adrenocortical axis responsiveness to depression (Wulsin et al., 2010). Overall, these results indicate that RU-486 is an effective treatment for psychotic depression, and ameliorates abnormalities related to depressive symptoms.
Modulation of serotoninergic neurotransmission is reported as a treatment for psychotic depression (Butler and Meegan, 2008; Zhang et al., 2012). Serotonin (5-hydroxytryptamine) is functionally implicated in a variety of behavioral responses, and plasma serotonin levels are a promising marker of major depression (Meredith et al., 2005). In the central nervous system, serotonergic transmission is critically regulated by serotonin reuptake through the serotonin transporter (SERT; SLC6A4) (Ni and Watts, 2006). As a crucial pharmacological target of antidepressants, the role of SERT in treatment of major depression is well-established (Gutiérrez et al., 1998; Lehto et al., 2008; Willeit et al., 2008). Considerable research has focused on the pharmacological properties of SERT, and genetic polymorphisms in the SERT promoter region (Way and Taylor, 2010; Mueller et al., 2011). Changes in SERT activity are also an important index for indicating psychotic depression, and glucocorticoids are highly associated with serotonergic neurotransmission, thereby contributing to vulnerability for major depression (Buwalda et al., 2005; Charoenphandhu et al., 2011). Animal studies indicate that serotonergic neurotransmission is affected by the stress hormone axis (Lanfumey et al., 2008). RU-486 administration prevents increases in SERT expression, caused by corticosterone in the rat dorsal raphe nucleus (Zhang et al., 2012). In humans, central SERT levels are related to the stress hormone response in patients suffering from negative mood states (Reimold et al., 2011).
Currently, however, the relationship between RU-486 and serotonin neurotransmission mediated by SERT is unknown, with the direct effect of RU-486 on regulation of SERT function unclear. Therefore, in the present study, we cloned human brain SERT and expressed SERT protein using a Xenopus oocyte expression system. We used two-electrode voltage clamp recordings to monitor SERT activity and determine the effect of RU-486 on human brain SERT function, clarifying its electrophysiological properties.
Materials and Methods
Animals
Six female Xenopus laevis frogs were fed twice weekly with frog brittle. The animal research protocol was approved by the Care and Use Committee of Xinxiang Medical University, China.
Drugs
Glucocorticoids and cortisol (99%, powder) were purchased from Sigma-Aldrich (St. Louis, MO, USA). RU-486 (97%, powder) was purchased from Tocris Bioscience (Bristol, UK). All drugs were dissolved to a stock concentration of 100 mmol/L in dimethyl sulfoxide and stored at −20°C before use.
Cloning of human SERT and expression vector construction
Total RNA from human brain was purchased from Ambion Biotechnology (Austin, TX, USA). cDNA was synthesized using a Script III reverse transcript kit (Invitrogen, Beijing, China). The human brain SERT gene (SLC6A4) was cloned by PCR using primers designed from SLC6A4 mRNA sequence obtained from GenBank. Sequences of the SERT cloning primers were: forward, 5′-ATG GAG ACG ACG CCC TTG AA-3′; reverse, 5′-TTA CAC AGC ATT CAA GCG GAT G-3′. PCR reactions were performed using Phusion polymerase (NEB, Ipswich, MA, USA). PCR cycling conditions were as follows: 1 minute at 98°C, followed by 25 cycles of 20 seconds at 98°C, 20 seconds at 58°C, and 2 minutes at 72°C. PCR products (1.9 kb) were agarose gel purified. SERT PCR products were subcloned into the pGEMHE vector using Fastcloning methods (Li et al., 2011). PCR product (15 ng) was used as the template for the second round of amplification with SERT subcloning primers. Sequences of the SERT subcloning primers were: forward, 5′-GCT CAA CTT TGG CCA TGG AGA CGA CGC CCT TGA A-3′; reverse, 5′-TTC TTG AGG CTG GTT TAC ACA GCA TTC AAG CGG ATG-3′. Primers for amplification of the pGEMHE vector were: Vec-start-reverse, 5′-CAT GGC CAA AGT TGA GCG TTT ATT CTG-3′; Vec-end-forward, 5′-TAA ACC AGC CTC AAG AAC ACC-3′. PCR conditions were: 18 cycles of 98°C for 20 seconds, 58°C for 20 seconds, and 72°C for 150 seconds. SERT (1.9 kb) and pGEMHE vector (3.2 kb) PCR products were checked on agarose gels. Final plasmids were named pGEM-SERT, and the SERT sequence confirmed by DNA sequencing.
SERT mRNA in vitro transcription
pGEM-SERT plasmid was linearized and SERT mRNA transcribed using T7 RNA polymerase from the Ambion mMESSAGE mMACHINE kit (Life Technologies, Beijing, China). RNase-free DNase I (1μL) was added to remove DNA template. mRNA was purified using Qiagen mRNA purification kits (Shanghai, China). SERT mRNA was eluted from columns using DEPC-treated water and confirmed on agarose gels to ensure the presence of a single, non-degraded band of the expected size.
SERT-expressing Xenopus laevis oocytes
Female Xenopus laevis frogs were anesthetized, the ovarian lobes removed and placed in incubation solution (82.5 mmol/L NaCl, 1 mmol/L MgCl2, 2.5 mmol/L KCl, 1 mmol/L CaCl2, 2.5 mmol/L sodium pyruvate, 0.6 mmol/L theophylline, 5 mmol/L HEPES, 50 μg/mL gentamicin, 50 μg/mL streptomycin, and 50 U/mL penicillin, pH 7.5). Oocytes were incubated at 16°C before injection. SERT mRNA (60 nL of 1 ng/nL) was injected into oocytes using an automated microinjector (Nanoject; Drummond Scientific Co., Broomall, PA, USA). After injection, oocytes were further incubated for 3–4 days at 16°C in sterile incubation solution before electrophysiological recordings. Control oocytes were injected with equal volumes of water instead of SERT mRNA.
Electrophysiological recordings to characterize response currents in SERT-expressing oocytes
The two-electrode voltage clamp technique was used to characterize response currents in SERT-expressing oocytes. Oocytes were placed in a chamber and perfused with oocyte Ringer's solution (93 mmol/L NaCl, 2.5 mmol/L KCl, 1 mmol/L CaCl2, 1 mmol/L MgCl2, and 5 mmol/L Hepes, pH 7.5). The open chamber was grounded through an agar-KCl bridge. Two electrodes were inserted into the oocyte and voltage clamping applied using a GeneClamp 900A amplifier (Axon Instruments, Union City, CA, USA), at a holding potential of −70 mV. Current signals were filtered at 20 Hz with a low-pass Bessel filter and digitized at 50 Hz. Data were normalized to the maximum voltage clamp current recorded.
Statistical analysis
Clampfit 9.0(Axon Instruments) and OriginPro 7.5 (OriginLab Corporation, Northampton, MA, USA) were used for data recording and analysis. Values were expressed as mean ± SEM. Each experiment was repeated six times.
Results
Expression of human brain SERT in Xenopus oocytes
The two-electrode voltage clamp method is a safer detection method than measurement of radiolabeled ligands, and earlier work in oocytes suggests that inward currents are detected with co-transportation of positively charged serotonin molecules and sodium ions into cells (Butler and Meegan, 2008).
To confirm human brain SERT expression in oocytes and determine SERT function, we performed two-electrode voltage clamping. Different serotonin concentrations were examined as SERT substrates. Inward currents were detected following serotonin perfusion (Figure 1A), indicating that serotonin was transported into oocytes. Control oocytes (water-injected) had no response when exposed to different serotonin concentrations (Figure 1B). The 5-HT dose response was 3.16–1,000 μmol/L, and data were normalized to the maximum current (Figure 1C). The half-maximal effect (EC50) of serotonin on SERT was observed at a concentration of approximately 35.3 ± 3.6 μmol/L.
Figure 1.
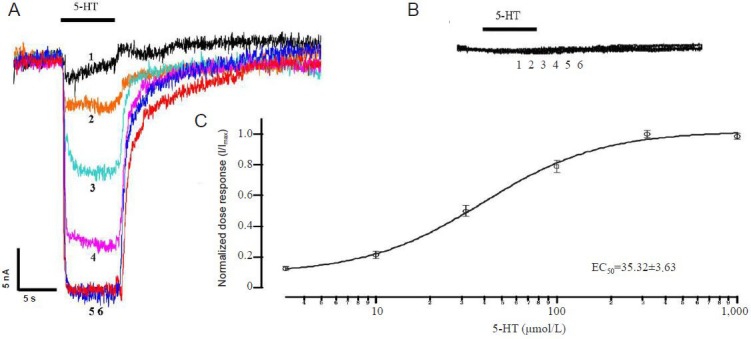
Expression of human brain serotonin transporter (SERT) in Xenopus oocytes.
(A) Dose-response curves for varied serotonin concentrations (3.16–1,000 μmol/L) on SERT channels. (B) No serotonin response in control oo-cytes injected with water. (C) Normalized serotonin dose response on SERT channels. The half-maximal effect (EC50) was observed at a concentra-tion of 35.52 ± 3.63 μmol/L. Data are expressed as mean ± SEM. Each experiment was repeated six times. I: Current intensity; Sweeps 1–6: 3.16, 10, 31.6, 100, 316, and 1,000 μmol/L serotonin (5-HT).
Effect of glucocorticoids and RU-486 on SERT activity
To investigate the effect of glucocorticoids and RU-486 on SERT activity, SERT transport currents were elicited by perfusion of high serotonin concentrations (100 μmol/L), followed by co-application with various drugs (10 μmol/L) and serotonin (100 μmol/L). Serotonin transport did not change when SERT was perfused with cortisol, dexamethasone, or corticosterone (Figure 2). In contrast, RU-486 caused an outward current (Figure 2; trace 5), suggesting RU-486 has a direct effect on SERT, and attenuates SERT channel activity.
Figure 2.
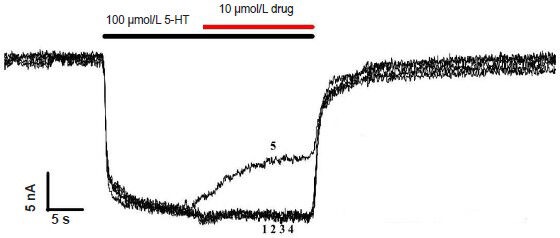
Effect of glucocorticoids and RU-486 on serotonin transporter (SERT) activity.
Response curves of Ringer control, cortisol, dexamethasone, corticos-terone, and glucocorticoid receptor antagonist, mifepristone (RU-486) (10 μmol/L) in 100 μmol/L serotonin (5-HT) activated SERT channels, after the serotonin response reached a steady-state plateau. Each exper-iment was repeated six times. Sweeps 1–5: 100 μmol/L 5-HT + control, cortisol, dexamethasome, corticosterone, or RU-486.
RU-486 blocked serotonin-elicited SERT activity
To further examine RU-486 on SERT activity, RU-486 was diluted into Ringer's solution at different concentrations (1, 3.16, 10, 31.6, and 100 μmol/L) and co-applied with serotonin (50 μmol/L). RU-486 concentration-dependent inhibition of SERT is shown (Figure 3A). RU-486 induced a maximal decrease of 52% (vs. without RU-486) in the serotonin response.
Figure 3.
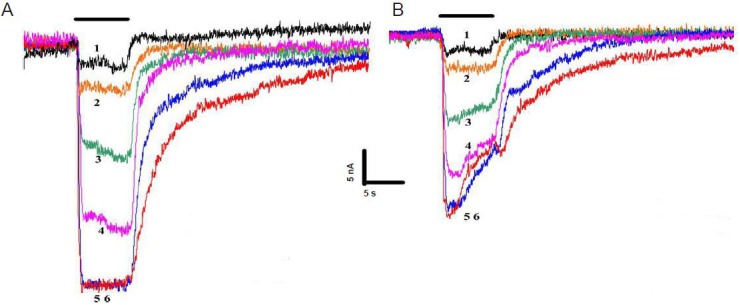
RU-486 blocked serotonin-elicited serotonin transporter (SERT) activity.
(A) Serotonin (50 μmol/L) response in SERT-expressing oocytes, when co-applied with varied glucocorticoid receptor antagonist, mifepristone (RU-486) concentrations. Sweeps 1–6: 3.16, 10, 31.6, 100, 316, and 1,000 μmol/L serotonin. (B) SERT currents were recorded in response to sero-tonin (50 μmol/L). After the serotonin response reached a steady-state plateau, addition of RU-486 (1–100 μmol/L) produced concentration-de-pendent outward currents of SERT. Each experiment was repeated six times. Sweeps 1–6: 3.16, 10, 31.6, 100, 316, and 1,000 μmol/L serotonin + 100 μmol/L RU-486.
To analyze the kinetics of this inhibition, SERT current responses were elicited by serotonin (50 μmol/L). Once channel activation reached a steady-state plateau, variable RU-486 concentrations combined with serotonin (50 μmol/L) were applied. Traces of RU-486 concentration-dependent inhibition of SERT are shown (Figure 3B). SERT concentration-dependent currents were induced by RU-486, suggesting serotonin transport is blocked in the presence of RU-486.
Kinetics of serotonin evoked SERT currents in the absence or presence of RU-486
To study the interaction between RU-486 and SERT, the effect of RU-486 on the serotonin dose-response relationship was examined. Traces of current responses (Figure 4) and serotonin-elicited responses detected with or without application of RU-486 (100 μmol/L) (Figure 5) are shown. Data were normalized to individually saturated serotonin response values, therefore in the presence of RU-486, EC50 values of serotonin on SERT changed from 35.3 ± 3.6 to 23.1 ± 2.6 μmol/L (Figure 5A). When data were normalized to the serotonin maximal response (in the absence of RU-486), application of RU-486 caused a 52% reduction in maximal serotonin efficacy of SERT. These results suggest that RU-486 decreases the efficacy of SERT to transport serotonin, and RU-486 binding to SERT dramatically changes SERT action on serotonin.
Figure 4.
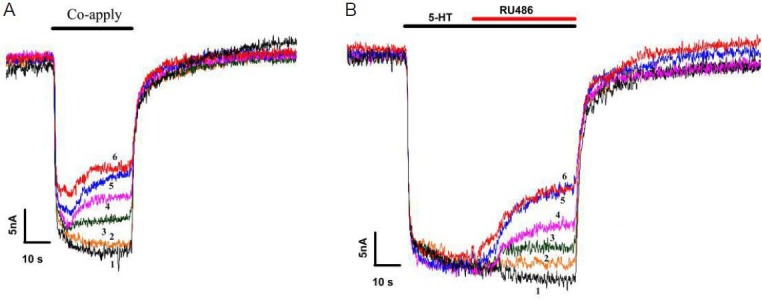
Effect of RU-486 on serotonin (5-HT) dose-response curves.
(A) Serotonin dose-response curves from serotonin transporter (SERT)-expressing Xenopus oocytes. (B) Comparison of serotonin dose-response curves when glucocorticoid receptor antagonist, mifepristone (RU-486) (100 μmol/L) co-applied in the same oocyte. Each experiment was repeated six times. Sweeps 1–6: 50 μmol/L serotonin + 0, 1 3.16, 10, 31.6, and 100 μmol/L RU-486.
Figure 5.
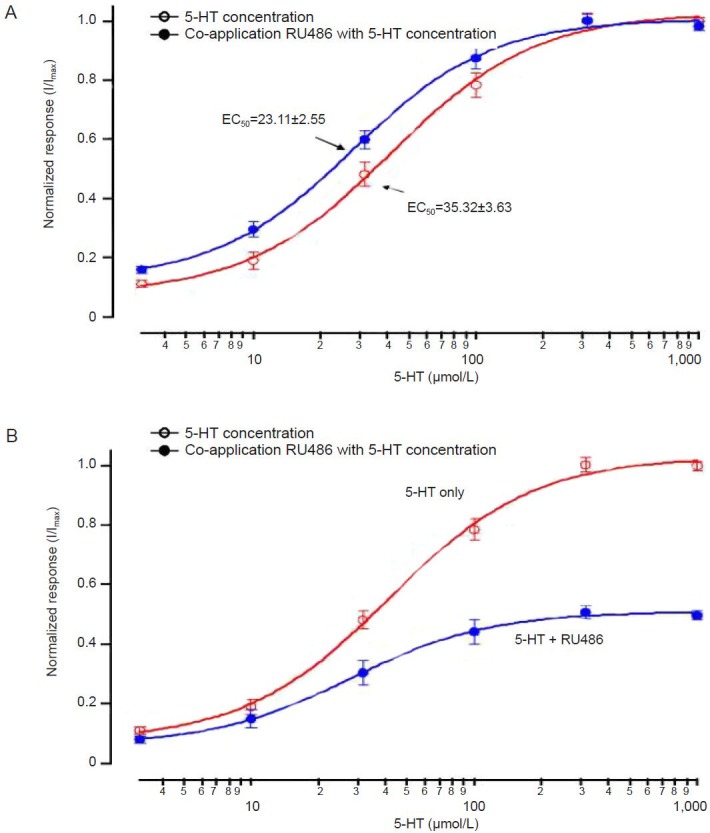
Comparison of normalized serotonin (5-HT) dose-response curves in the absence or presence of 100 μmol/L RU-486.
(A) Data obtained in controls with 100 μmol/L glucocorticoid receptor antagonist, mifepristone (RU-486) were normalized to individually saturated serotonin response values. (B) Data normalized to the maximal response elicited by 1,000 μmol/L serotonin in the absence of RU-486. Data are expressed as mean ± SEM. Each experiment was repeated six times.
Discussion
Radiolabeled serotonin is often used as a ligand for measuring SERT activity in mammalian cells. In this report, Xenopus oocytes were selected as the host for SERT expression, and two-electrode voltage clamping used to monitor serotonin transport activity mediated by SERT. Admittedly, Xenopus oocytes are different to other animal cells, however, as they show no cell membrane expression of ion channels and transporters they are often used for drug screening of ion channels. Moreover, the voltage clamping used in this study offers a safe way to detect serotonin transport without using and generating biohazardous materials, such as radioactive ligands. SERT expression in oocytes caused a serotonin EC50 of approximately 35 μmol/L (Figure 1), higher than the Km values reported with radiotracer uptake in mammalian cells, although this may be partially attributed to the expression properties of Xenopus oocytes.
Numerous studies have reported abnormal circulatory glucocorticoid secretion as a cause of major depression, especially psychotic depression (Yehuda et al., 1993; Carroll and Rubin, 2006; Schuhmacher et al., 2012). Reasons for the abnormality are evident through regulation of the hypothalamic-pituitary-adrenal axis (Iijima et al., 2010), and detected post-mortem by decreased glucocorticoid receptor mRNA (Webster et al., 2002; Perlman et al., 2004). Currently, one strategy for psychotic depression therapy is to block glucocorticoid binding with its receptor, eventually deactivating the function of elevated circulating glucocorticoids (Belmaker, 2008; Nelson, 2012). In clinical studies, treatment with RU-486, a glucocorticoid receptor antagonist, improves psychotic depression compared with a placebo group (Belanoff et al., 2002; DeBattista et al., 2006). RU-486 has also been shown to relieve the symptoms of psychotic depression after a brief period of 4–8 days (Flores et al., 2006; Blasey et al., 2009). Validity of the repeated corticosterone injection-induced depression rat model is further supported by the effect of RU-486 and other glucocorticoid receptor antagonists (Iijima et al., 2010). In contrast, acute treatment with imipramine, a tricyclic antidepressant, and fluvoxamine, a selective serotonin reuptake inhibitor, do not have any effect on regulating depressive-like behavior in animals (Iijima et al., 2010). Overall, this suggests RU-486 is an efficient antidepressant for treating depressive symptoms, with higher efficacy than some tricyclic antidepressants and selective serotonin reuptake inhibitors. In the current study, the effect of RU-486 on serotonin neurotransmission was determined through its effect on SERT activity in vitro. Our findings highlight human brain SERT as a pharmacological target of RU-486. Anti-depressant activity of RU-486 may be partially attributable to direct regulation of SERT, eventually influencing serotonin reuptake.
It is known that alterations in serotoninergic neurotransmission are a cause of depression. As a critical transporter affecting serotoninergic neurotransmission, SERT plays a pharmacological role in affecting depression, and changes in SERT activity are involved in regulation of psychotic depression (Gutiérrez et al., 1998; Lehto et al., 2008; Willeit et al., 2008). Considerable research suggests stress hormones play an essential role during chronic stress (Buwalda et al., 2005; Lanfumey et al., 2008; Way and Taylor, 2010; Reimold et al., 2011), and animal studies indicate that serotonergic neurotransmission may be affected by the stress hormone axis (Lanfumey et al., 2008). SERT protein up-regulation in the dorsal raphe nucleus, caused by a chronic social defeat regimen, is mainly mediated through corticosterone and its receptors (Zhang et al., 2012). In humans, central SERT levels are highly associated with the stress hormone response in patients suffering from negative mood states (Reimold et al., 2011). Moreover, in psychologically healthy samples of young adults, the short/short genotype of the serotonin-transporter-linked polymorphic region is commonly associated with high cortisol reactivity to social threat (Way and Taylor, 2010; Mueller et al., 2011). In the current study, we found no evidence that glucocorticoids have a direct influence on serotonin transport, suggesting the ability of glucocorticoids to regulate serotonin transmission may be through serotonergic gene expression modulation.
SERT is the main target for antidepressants with the objective of achieving selectivity, compared with other monoamine transporters. In this study, we show by recording serotonin-induced SERT currents, that RU-486 has a direct effect on SERT activity and dose-dependent inhibition of SERT. This directly indicates that serotonin transport of SERT is inhibited in the presence of RU-486. RU-486 attenuates SERT activity by directly switching the transporter from the open to the closed phase. RU-486 is often chosen as the receptor antagonist to assess the effect of progesterone or glucocorticoid on serotonergic function mediated by SERT in depression stress (Benmansour et al., 2009; Benmansour et al., 2012; Zhang et al., 2012). These hormones have the ability to regulate the activity of antidepressants, including fluoxetine, fluvoxamine, and citalopram, and these selective serotonin reuptake inhibitors can block SERT function and reduce serotonin clearance (Butler and Meegan, 2008). Our data shows that RU-486 inhibits SERT-induced serotonin transmission, offering a cautionary note to using RU-486 to determine the effects of these hormones on antidepressant drugs focused on serotonin clearance. Identifying the functional chemical group of RU-486 will significantly aid development of new selective serotonin reuptake inhibitors. Our future work will focus on structural analysis to identify the core structure of the RU-486 and SERT interaction.
In conclusion, we report that SERT is a pharmacological target of RU-486, and RU-486 attenuates SERT activity in a concentration-dependent manner. Our data suggests that RU-486 is an efficient SERT blocker, and provides an additional mechanism for evaluating the effect of RU-486 in the treatment of psychotic depression.
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 81100912, 81271376; a grant from Xinxiang Medical University Foundation.
Conflicts of interest: None declared.
Copyedited by James R, Raye W, Yu J, Li CH, Song LP, Zhao M
References
- [1].Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu EE, Schold C, Schatzberg AF. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry. 2002;52:386–392. doi: 10.1016/s0006-3223(02)01432-4. [DOI] [PubMed] [Google Scholar]
- [2].Belmaker RH. The future of depression psychopharmacology. CNS Spectr. 2008;13:682–687. doi: 10.1017/s1092852900013766. [DOI] [PubMed] [Google Scholar]
- [3].Benmansour S, Piotrowski JP, Altamirano AV, Frazer A. Impact of ovarian hormones on the modulation of the serotonin transporter by fluvoxamine. Neuropsychopharmacology. 2009;34:555–564. doi: 10.1038/npp.2008.23. [DOI] [PubMed] [Google Scholar]
- [4].Benmansour S, Weaver RS, Barton AK, Adeniji OS, Frazer A. Comparison of the effects of estradiol and progesterone on serotonergic function. Biol Psychiatry. 2012;71:633–641. doi: 10.1016/j.biopsych.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Blasey CM, DeBattista C, Roe R, Block T, Belanoff JK. A multisite trial of mifepristone for the treatment of psychotic depression: a site-by-treatment interaction. Contemp Clin Trials. 2009;30:284–288. doi: 10.1016/j.cct.2009.03.001. [DOI] [PubMed] [Google Scholar]
- [6].Butler S, Meegan M. Recent developments in the design of anti-depressive therapies: targeting the serotonin transporter. Curr Med Chem. 2008;15:1737–1761. doi: 10.2174/092986708784872357. [DOI] [PubMed] [Google Scholar]
- [7].Buwalda B, Kole MH, Veenema AH, Huininga M, De Boer SF, Korte SM, Koolhaas JM. Long-term effects of social stress on brain and behavior: a focus on hippocampal functioning. Neurosci Biobehav Rev. 2005;29:83–97. doi: 10.1016/j.neubiorev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- [8].Carroll B, Rubin R. Is mifepristone useful in psychotic depression? Neuropsychopharmacology. 2006;31:2793–2797. doi: 10.1038/sj.npp.1301170. [DOI] [PubMed] [Google Scholar]
- [9].Charoenphandhu J, Teerapornpuntakit J, Nuntapornsak A, Krishnamra N, Charoenphandhu N. Anxiety-like behaviors and expression of SERT and TPH in the dorsal raphé of estrogen-and fluoxetine-treated ovariectomized rats. Pharmacol Biochem Behav. 2011;98:503–510. doi: 10.1016/j.pbb.2011.02.023. [DOI] [PubMed] [Google Scholar]
- [10].DeBattista C, Belanoff J, Glass S, Khan A, Horne RL, Blasey C, Carpenter LL, Alva G. Mifepristone versus placebo in the treatment of psychosis in patients with psychotic major depression. Biol Psychiatry. 2006;60:1343–1349. doi: 10.1016/j.biopsych.2006.05.034. [DOI] [PubMed] [Google Scholar]
- [11].Flores BH, Kenna H, Keller J, Solvason HB, Schatzberg AF. Clinical and biological effects of mifepristone treatment for psychotic depression. Neuropsychopharmacology. 2006;31:628–636. doi: 10.1038/sj.npp.1300884. [DOI] [PubMed] [Google Scholar]
- [12].Gallagher P, Watson S, Elizabeth Dye C, Young AH, Nicol Ferrier I. Persistent effects of mifepristone (RU-486) on cortisol levels in bipolar disorder and schizophrenia. J Psychiatr Res. 2008;42:1037–1041. doi: 10.1016/j.jpsychires.2007.12.005. [DOI] [PubMed] [Google Scholar]
- [13].Gutiérrez B, Pintor L, Gastó C, Rosa A, Bertranpetit J, Vieta E, Fañanás L. Variability in the serotonin transporter gene and increased risk for major depression with melancholia. Hum Genet. 1998;103:319–322. doi: 10.1007/s004390050823. [DOI] [PubMed] [Google Scholar]
- [14].Iijima M, Ito A, Kurosu S, Chaki S. Pharmacological characterization of repeated corticosterone injection-induced depression model in rats. Brain Res. 2010;1359:75–80. doi: 10.1016/j.brainres.2010.08.078. [DOI] [PubMed] [Google Scholar]
- [15].Kling MA, Coleman VH, Schulkin J. Glucocorticoid inhibition in the treatment of depression: can we think outside the endocrine hypothalamus? Depress Anxiety. 2009;26:641–649. doi: 10.1002/da.20546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lanfumey L, Mongeau R, Cohen-Salmon C, Hamon M. Corticosteroid–serotonin interactions in the neurobiological mechanisms of stress-related disorders. Neurosci Biobehav Rev. 2008;32:1174–1184. doi: 10.1016/j.neubiorev.2008.04.006. [DOI] [PubMed] [Google Scholar]
- [17].Lehto SM, Tolmunen T, Joensuu M, Saarinen PI, Valkonen-Korhonen M, Vanninen R, Ahola P, Tiihonen J, Kuikka J, Lehtonen J. Changes in midbrain serotonin transporter availability in atypically depressed subjects after oneyear of psychotherapy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:229–237. doi: 10.1016/j.pnpbp.2007.08.013. [DOI] [PubMed] [Google Scholar]
- [18].Li C, Wen A, Shen B, Lu J, Huang Y, Chang Y. FastCloning: a highly simplified, purification-free, sequence-and ligation-independent PCR cloning method. BMC Biotechnol. 2011;11:92. doi: 10.1186/1472-6750-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Meredith EJ, Holder MJ, Chamba A, Challa A, Drake-Lee A, Bunce CM, Drayson MT, Pilkington G, Blakely RD, Dyer MJ. The serotonin transporter (SLC6A4) is present in B-cell clones of diverse malignant origin: probing a potential anti-tumor target for psychotropics. FASEB J. 2005;19:1187–1189. doi: 10.1096/fj.04-3477fje. [DOI] [PubMed] [Google Scholar]
- [20].Mueller A, Armbruster D, Moser DA, Canli T, Lesch KP, Brocke B, Kirschbaum C. Interaction of serotonin transporter gene-linked polymorphic region and stressful life events predicts cortisol stress response. Neuropsychopharmacology. 2011;36:1332–1339. doi: 10.1038/npp.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nelson EB. Psychotic depression--beyond the antidepressant/antipsychotic combination. Curr Psychiatry Rep. 2012;14:619–623. doi: 10.1007/s11920-012-0315-6. [DOI] [PubMed] [Google Scholar]
- [22].Ni W, Watts S. 5-hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT) Clin Exp Pharmacol Physiol. 2006;33:575–583. doi: 10.1111/j.1440-1681.2006.04410.x. [DOI] [PubMed] [Google Scholar]
- [23].Pariante CM. Depression stress and the adrenal axis. J Neuroendocrinol. 2003;15:811–812. doi: 10.1046/j.1365-2826.2003.01058.x. [DOI] [PubMed] [Google Scholar]
- [24].Perlman WR, Webster MJ, Kleinman JE, Weickert CS. Reduced glucocorticoid and estrogen receptor alpha messenger ribonucleic acid levels in the amygdala of patients with major mental illness. Biol Psychiatry. 2004;56:844–852. doi: 10.1016/j.biopsych.2004.09.006. [DOI] [PubMed] [Google Scholar]
- [25].Reimold M, Knobel A, Rapp MA, Batra A, Wiedemann K, Ströhle A, Zimmer A, Schönknecht P, Smolka MN, Weinberger DR, Goldman D, Machulla HJ, Bares R, Heinz A. Central serotonin transporter levels are associated with stress hormone response and anxiety. Psychopharmacology (Berl) 2011;213:563–572. doi: 10.1007/s00213-010-1903-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schuhmacher A, Mössner R, Jessen F, Scheef L, Block W, Belloche AC, Lennertz L, Welper H, Höfels S, Pfeiffer U, Wagner M, Maier W, Schwab S, Zobel A. Association of amygdala volumes with cortisol secretion in unipolar depressed patients. Psychiatry Res. 2012;202:96–103. doi: 10.1016/j.pscychresns.2011.09.007. [DOI] [PubMed] [Google Scholar]
- [27].Thomson F, Craighead M. Innovative approaches for the treatment of depression: targeting the HPA axis. Neurochem Res. 2008;33:691–707. doi: 10.1007/s11064-007-9518-3. [DOI] [PubMed] [Google Scholar]
- [28].Wang DL, Lin WJ, Pan YQ, Kuang XY, Qi XL, Sun H. Chronic blockade of glucocorticoid receptors by RU486 enhances lipopolysaccharide-induced depressive-like behaviour and cytokine production in rats. Brain Behav Immun. 2011;25:706–714. doi: 10.1016/j.bbi.2011.01.011. [DOI] [PubMed] [Google Scholar]
- [29].Watson S, Gallagher P, Porter RJ, Smith MS, Herron LJ, Bulmer S, Young AH, Ferrier IN. A randomized trial to examine the effect of mifepristone on neuropsychological performance and mood in patients with bipolar depression. Biol Psychiatry. 2012;72:943–949. doi: 10.1016/j.biopsych.2012.05.029. [DOI] [PubMed] [Google Scholar]
- [30].Way BM, Taylor SE. The serotonin transporter promoter polymorphism is associated with cortisol response to psychosocial stress. Biol Psychiatry. 2010;67:487–492. doi: 10.1016/j.biopsych.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Webster M, Knable M, O’Grady J, Orthmann J, Weickert CS. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatr. 2002;7:985–994. doi: 10.1038/sj.mp.4001139. 924. [DOI] [PubMed] [Google Scholar]
- [32].Willeit M, Sitte HH, Thierry N, Michalek K, Praschak-Rieder N, Zill P, Winkler D, Brannath W, Fischer MB, Bondy B, Kasper S, Singer EA. Enhanced serotonin transporter function during depression in seasonal affective disorder. Neuropsychopharmacology. 2008;33:1503–1513. doi: 10.1038/sj.npp.1301560. [DOI] [PubMed] [Google Scholar]
- [33].Wulsin AC, Herman JP, Solomon MB. Mifepristone decreases depression-like behavior and modulates neuroendocrine and central hypothalamic–pituitary–adrenocortical axis responsiveness to stress. Psychoneuroendocrinology. 2010;35:1100–1112. doi: 10.1016/j.psyneuen.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yehuda R, Boisoneau D, Mason JW, Giller EL. Glucocorticoid receptor number and cortisol excretion in mood, anxiety and psychotic disorders. Biol Psychiatry. 1993;34:18–25. doi: 10.1016/0006-3223(93)90252-9. [DOI] [PubMed] [Google Scholar]
- [35].Zhang J, Fan Y, Li Y, Zhu H, Wang L, Zhu MY. Chronic social defeat up-regulates expression of the serotonin transporter in rat dorsal raphe nucleus and projection regions in a glucocorticoid‐dependent manner. J Neurochem. 2012;123:1054–1068. doi: 10.1111/jnc.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]