

Abstract
Secondary degeneration occurs commonly in the central nervous system after traumatic injuries and following acute and chronic diseases, including glaucoma. A constellation of mechanisms have been shown to be associated with secondary degeneration including apoptosis, necrosis, autophagy, oxidative stress, excitotoxicity, derangements in ionic homeostasis and calcium influx. Glial cells, such as microglia, astrocytes and oligodendrocytes, have also been demonstrated to take part in the process of secondary injury. Partial optic nerve transection is a useful model which was established about 13 years ago. The merit of this model compared with other optic nerve injury models used for glaucoma study, including complete optic nerve transection model and optic nerve crush model, is the possibility to separate primary degeneration from secondary degeneration in location. Therefore, it provides a good tool for the study of secondary degeneration. This review will focus on the research progress of the mechanisms of secondary degeneration using partial optic nerve transection model.
Keywords: secondary degeneration, partial injury, optic nerve, oxidative stress, excitotoxicity, calcium overload, mitochondrion, macrophage, astrocyte, oligodendrocyte
Introduction
A number of drugs have been screened in worldwide laboratories for neuroprotective effects (Leaver et al., 2006; Fu et al., 2009; Baltmr et al., 2010; Vasudevan et al., 2011; Mi et al., 2012a; Mi et al., 2012b; Ren et al., 2012; Deng et al., 2013; Mi et al., 2013). These drugs were also studied in various eye models, including complete optic nerve transection (CONT), optic nerve (ON) crush, acute and chronic ocular hypertension models and ischemia/reperfusion model (Schwartz, 2004; Yu et al., 2006; Ho et al., 2007, 2009, 2010; Yu et al., 2007; Fu et al., 2009, 2010; Weber et al., 2010; Danesh-Meyer, 2011; Li et al., 2011, 2013; Mi et al., 2012a, 2012b, 2013; Sullivan et al., 2012; Zhang et al., 2012, 2013; Vigneswara et al., 2013; Zhu et al., 2013; Zuo et al., 2013). The aim of these studies was to search for the medicine which could delay secondary degeneration of retinal ganglion cells (RGCs), especially in glaucoma because RGCs in glaucoma patients continued to die even after the intraocular hypertension had been restored to normal by surgery. Secondary degeneration meant the degeneration of neurons and glial cells caused by the noxious factors released from the neurons or glial cells which were damaged by the primary direct events. Actually, “second phase of injury” was defined as “any consequence of the primary insult” because “there is no absolute time when primary damage evolves into delayed effects” after traumatic brain injury and spinal cord injury (LaPlaca et al., 2007). Therefore, it was hard to differentiate secondary degeneration from primary degeneration based on the time-points after injury although the temporal sequence has been used to define secondary degeneration after ON crush (Yoles and Schwartz, 1998). The degeneration of some RGCs would take longer time than others for a variety of causes. In order to clarify this phenomenon, Levkovitch-Verbin and his colleagues established the partial optic nerve transection (PONT) model using monkeys in 2001 and Wistar rats in 2003. Because only the dorsal part of the ON was transected in this model, secondary degeneration occurring in the ventral ON could be separated from primary damage clearly. Damages to RGC axons often led to the retrograde degeneration of the cell bodies in the retinas (Quigley et al., 1977), therefore, PONT model also allowed the separation of secondary degeneration of the cell bodies in the retinas by dye-tracing the cell bodies whose axons were transected after PONT (Fitzgerald et al., 2009a; Li et al., 2013). Although many mechanisms about secondary degeneration have been reviewed after traumatic brain injury and spinal cord injury, and they provide some useful hints for the understanding of secondary degeneration in glaucoma (Farkas and Povlishock, 2007; Oyinbo, 2011), the PONT model was performed in the visual system and more correlated with glaucoma. Therefore, this review will summarily discuss the mechanisms of secondary degeneration after PONT.
Secondary degeneration
The concept of secondary degeneration in the central nervous system (CNS)
The death of neurons and glial cells as an early consequence of the primary pathological events is called primary degeneration. In addition to the primary degeneration, the neurons and glial cells which are not or only partially affected by the primary damage will also die. This kind of degeneration is called secondary degeneration. Secondary degeneration occurs commonly in the CNS after traumatic injuries, acute diseases and chronic neurodegenerative diseases. For example, secondary degeneration emerged after brain trauma (Stoica and Faden, 2010), spinal cord injury (Hausmann, 2003; Oyinbo, 2011), stroke (Guimaraes et al., 2009) and also in chronic neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis (Stewart and Appel, 1988). Except for the degeneration of cell bodies in the gray matter, there is also the degeneration of axons in the white matter in neural tissues (Guimaraes et al., 2009). Secondary degeneration is initiated by the pathological factors released by tissues which are damaged by the primary events. The noxious factors such as calcium dysregulation, excessive free radicals, activation of proteases, overexpression of pro-apoptotic proteins, hydrolytic enzymes and high-level glutamate were involved (Farkas and Povlishock, 2007). Both apoptosis and necrosis were involved in this process (Hausmann, 2003; Farkas and Povlishock, 2007; Guimaraes et al., 2009; Stoica and Faden, 2010; Oyinbo, 2011). In addition, glial cells, such as astrocytes, also take part in secondary degeneration and the dysfunction of astrocytes can lead to glutamate excitotoxicity (Floyd and Lyeth, 2007; Oyinbo, 2011).
Glaucoma is another chronic neurodegenerative disease of CNS and progressive neuropathology persists for a long period of time. Ocular hypertension (OH) is one clear cause for glaucoma and decreasing the OH with surgery has been shown to be the only confirmed effective therapy for glaucoma. But the situation of some patients will further deteriorate even after the OH has been controlled. Therefore, secondary degeneration of RGCs is also believed to occur in glaucoma (Tezel, 2006; Nickells, 2007; Tezel, 2008). The mechanisms of RGC death involved in glaucoma include neurotrophic factor deprivation, axonal transport failure, apoptosis, excitotoxicity, oxidative stress, dysfunction of glial cells and loss of synaptic connectivity, although which mechanisms are specifically related with primary degeneration or secondary degeneration are not known. This part has been well summarized by Almasieh et al. (2012) and will not be included in this review. In addition to RGCs, amacrine cells and photoreceptors will also be affected in glaucoma by trans-synaptic secondary degeneration (Calkins, 2012). Innovative therapy of secondary degeneration presents a new direction for the treatment of glaucoma and other optic neuropathies.
The pathogenesis of secondary degeneration in these pathologic processes of CNS is summarized in Figure 1.
Figure 1.
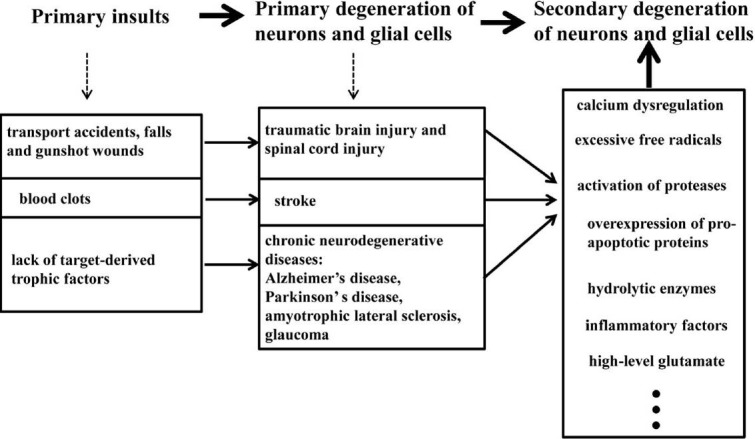
Events in central nervous system degeneration.
Primary insults induce the primary damages of neurons and glial cells in acute neurotrauma, stroke and chronic neurodegenerative diseases; then the noxious factors released by the degenerating cells will cause secondary degeneration.
Secondary degeneration in ON injury models
As discussed above, the prevention of secondary degeneration was a promising direction for the therapy of glaucoma. It was believed that the axonal degeneration was precedent to the death of RGC bodies in glaucoma (Calkins, 2012), therefore, ON injury models were widely used in the study of glaucoma. There are three kinds of ON injury models which have been used for the study of the mechanisms of RGC degeneration and the possible therapy for the loss of RGCs, including CONT model, ON crush model and PONT model. A lot of evidence shows the existence of secondary degeneration in these ON injury models. After CONT, the cell body loss percentages of RGCs were between 14-64% by 1 week after injury although the individual differences exist with regard to the various strains and the distances of cut site from the optic disc (Peinado-Ramon et al., 1996; Koeberle and Ball, 1998; Isenmann et al., 1999; Nakazawa et al., 2002; Ota et al., 2002; Krueger-Naug et al., 2003; Hou et al., 2004; van Adel et al., 2005; Franklin et al., 2006; Kretz et al., 2006; Nakazawa et al., 2006; Fu et al., 2008; Lebrun-Julien et al., 2009). However, in all these studies, most RGCs (more than 80% or 90%) died by 2 weeks (Berkelaar et al., 1994; Peinado-Ramon et al., 1996; Clarke et al., 1998; Koeberle and Ball, 1998; Isenmann et al., 1999; Weise et al., 2000; Nakazawa et al., 2002; Krueger-Naug et al., 2003; Hou et al., 2004; van Adel et al., 2005; Kretz et al., 2006; Nakazawa et al., 2006; Lebrun-Julien et al., 2009) after the injury and nearly all RGCs lost (more than 98%) by 4 weeks (Clarke et al., 1998; Krueger-Naug et al., 2003) after CONT. Although it seems that some RGCs will take a longer time period to die than others after CONT, the occurrence of secondary degeneration after CONT is uncertain because all the axons are transected and the degeneration of axons can lead to the degeneration of RGC bodies. In another model, partial ON crush in Sprague-Dawley (SD) rats, some bodies of RGCs were also shown to die over a protracted period after damage (Yoles and Schwartz, 1998). However, the number of influenced axons after partial crush was uncertain and the intact and damaged axons were mingled together, therefore, it was nearly impossible to distinguish the cells that needed a longer time for degeneration from others in location. In order to clarify this issue, a PONT model has been established using monkeys in 2001 by Levkovitch-Verbin et al. In this model, only the dorsal part of ON was damaged, but cell body degeneration of RGCs whose axons were kept intact after injury occurred in the inferior retinas (Levkovitch-Verbin et al., 2001). The expense of using monkeys for experiments was huge and use of monkeys was limited. Therefore, the Wistar rats were used to repeat this model by the same group and a similar result was obtained in rat PONT model (Levkovitch-Verbin et al., 2003). Convincing information suggested that some of the neighboring RGCs would die from the influence of biochemical events that derived from the original damage in a PONT model. In addition, these RGC bodies that would die from secondary degeneration could be discerned in the location in this model, which was the inferior half retina in Wistar rats. Therefore, a PONT model was believed to provide a good tool for the study of secondary degeneration. This model has been used extensively for studying the mechanisms of secondary degeneration and the possible neuroprotective agents since its establishment (Yoles and Schwartz, 1998; Levkovitch-Verbin et al., 2003, 2010, 2011; Fitzgerald et al., 2009a, 2009b, 2010a, 2010b; Selt et al., 2010; Payne et al., 2011, 2012; Wells et al., 2012; Chu et al., 2013; Cummins et al., 2013; Fitzgerald et al., 2013; Li et al., 2013; Payne et al., 2013; Savigni et al., 2013; Szymanski et al., 2013).
Secondary degeneration after PONT
Location of secondary degeneration of RGCs after PONT
Now the PONT model is performed in three strains of rats, including Wistar rats (Levkovitch-Verbin et al., 2003), PVG hooded rats (Fitzgerald et al., 2009b) and SD rats (Li et al., 2013). The merit of this model is the ability to separate primary degeneration from secondary degeneration accurately in location both in the ON and the retinas. That is, after partial cut in the dorsal ON (about one quarter in Wistar rats and SD rats or one third in PVG hooded rats), the axons in the central and ventral ON would be vulnerable to secondary degeneration (Figure 2) (Levkovitch-Verbin et al., 2003; Fitzgerald et al., 2009a; Fitzgerald et al., 2010b; Li et al., 2013). In the retinas, the localization of primary and secondary degeneration of the cell bodies of RGCs should be based on the topography of the transected axons in the above-mentioned three different strains of rats (Levkovitch-Verbin et al., 2003; Fitzgerald et al., 2009a; Li et al., 2013). If the whole retinas were divided into dorsal (or superior) and ventral (or inferior) halves in PVG hooded rats and SD rats, both primary and secondary degeneration occurred in both halves, however, more cell deaths in the ventral retinas should be attributed to secondary degeneration based on topography of axons demonstrated by the fluorescent lipophilic cationic indocarbocyanine dye DiI tracing (Fitzgerald et al., 2009a; Li et al., 2013). For example, in SD rats used in our group, most degenerating cell bodies (about 3/4) of the RGCs whose axons transected after PONT located in the dorsal retina and a small part of cell bodies (about 1/4) located in the ventral retina and they would die from primary degeneration; other cell bodies of the RGCs whose axons were intact after PONT would die from secondary degeneration (Figure 3). The result indicated that both primary and secondary degeneration occurred in the dorsal and ventral retina, but more secondary degeneration occurred in the ventral retina compared with the dorsal retina. On the other hand, if the retinas of the PVG hooded rats were divided into four quarters in addition to the central area, the primary degeneration was almost limited to central, dorsal and temporal areas and all the loss of the cell bodies in the ventral and nasal zones could be attributed to secondary degeneration (Figure 4) (Fitzgerald et al., 2009a). In SD rats, we only divided the retinas into dorsal and ventral halves; more detailed study of the distribution might be useful in future. In Wistar rats, primary degeneration of the cell bodies of RGCs would be only limited to the dorsal half retinas and all loss of the cell bodies of RGCs in the ventral retinas was attributed to secondary degeneration (Levkovitch-Verbin et al., 2003). In addition, the topography might be influenced by the distance of the cut site from the optic disc (Simon and O’Leary, 1991; Chan and Guillery, 1994).
Figure 2.
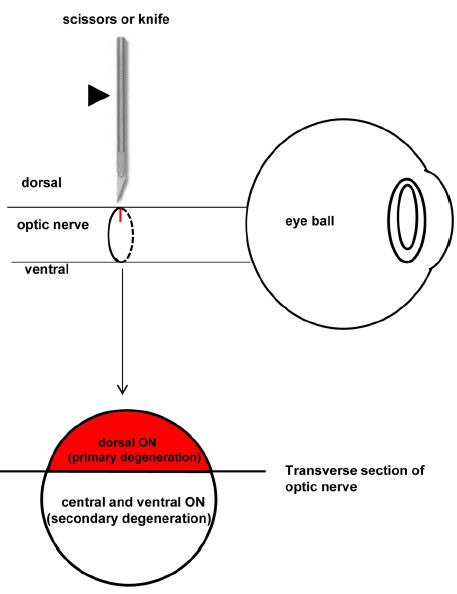
Schematic diagram showing the surgery of partial optic nerve transection (PONT) and the location of primary and secondary degeneration in the optic nerve (ON).
The partial incision in the ON is achieved using a pair of scissors or a diamond knife (indicated by the arrowhead). The axons in the direct damaged sites (dorsal cut site of the ON in the transverse section, red color) will undergo primary degeneration and the axons in the indirect damaged sites (central and ventral areas of the ON in the transverse section, no color) will undergo secondary degeneration.
Figure 3.
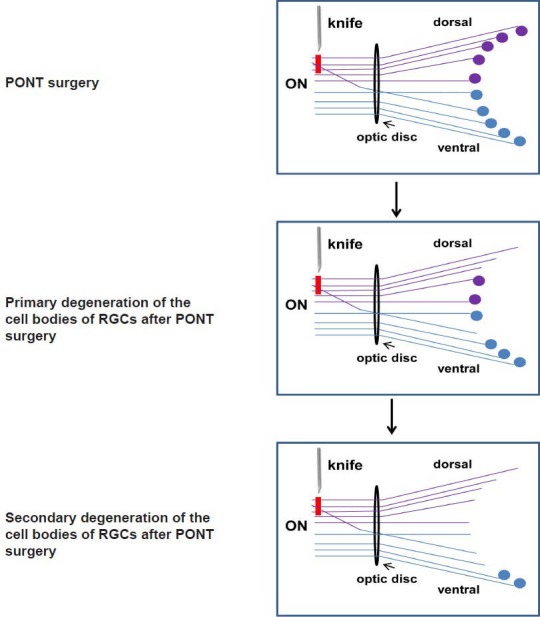
The schematic diagram of the localization of both primary and secondary degeneration of the retinal ganglion cell (RGC) bodies in the retinas in Sprague-Dawley (SD) rats.
The lines indicate the axons in the optic nerve (ON) and the circles indicate the cell bodies of RGCs: the purple structures locate in the dorsal retina and dorsal ON and the blue structures locate in ventral retina and ventral ON. Red line indicates the axons transected after partial optic nerve transection (PONT) in the dorsal part of the ON.
Figure 4.
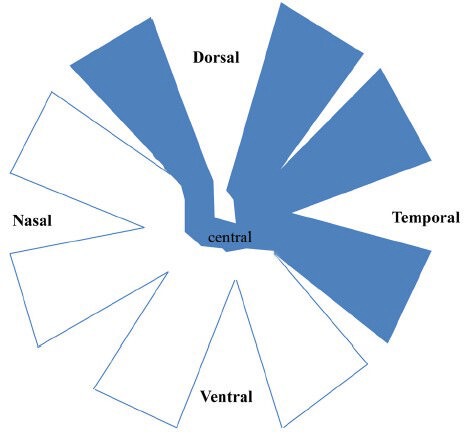
The schematic diagram of the whole-mounted retinas of PVG hooded rats showing the location of the retinal ganglion cell (RGC) bodies whose axons were transected after partial optic nerve transection (PONT).
The retinas were divided into dorsal, ventral, nasal and temporal quarters in addition to the central area. The RGC bodies whose axons were transected after PONT located in the dorsal, temporal and central areas (indicated by blue color), but not in the nasal and ventral areas.
In summary, there are both primary and secondary degeneration after PONT; axons in the ON and cell bodies of RGCs in the retina degenerate in rats. Although more accurate information about axonal topography should be collected for a PONT model, it is still a better model for the study of secondary degeneration than other ON injury models in the visual system, for example, CONT and ON crush. Although some tiny differences exist in the retinotopic graph among these three strains of rats, the ventral retinas and the ventral ON will be indicated as the areas vulnerable to secondary degeneration after PONT in this review.
Mechanisms of secondary degeneration of RGCs after PONT
In some papers reviewed here, the mechanisms underlying primary and secondary degeneration after PONT were studied simultaneously. But the scope of this part will be mainly limited to the mechanisms of secondary degeneration after PONT. However, the mechanisms of secondary degeneration versus primary degeneration after PONT will be summarized and compared at the end of this part.
Apoptosis, necrosis and autophagy: In the retinas, both apoptosis and necrosis contributed to secondary degeneration of the RGC bodies after PONT. Apoptosis of the RGC bodies was confirmed by Hoechst staining (Levkovitch-Verbin et al., 2010) and immunohistochemical staining of caspase-3 (Fitzgerald et al., 2009a). After Hoechst staining, the cells undergoing apoptosis exhibited nuclear condensation and deoxyribonucleic acid (DNA) fragmentation, and therefore could be recognized under fluorescence microscopy. In addition, the involvement of apoptosis was also confirmed by the increasing in the expression of pro-apoptotic genes Bad and Bax (Levkovitch-Verbin et al., 2010). In addition, necrosis was also shown to contribute to secondary degeneration. The necrosis was demonstrated with morphology (nucleic acid staining: Sytox Green) and activation of poly(ADP-ribose) polymerase (PARP) in PVG hooded rats (Fitzgerald et al., 2009a). PARP has been observed to be involved in necrotic RGC death (Kolodziejczyk et al., 2010). In our study using SD rats, terminal-deoxynucleotidyltransferase mediated nick end labeling (TUNEL) positive staining cells increased from 1 week after PONT in inferior retinas. TUNEL staining was believed to detect apoptosis in many studies (Aktas et al., 2013; Deng et al., 2013; Chinskey et al., 2014; Kowluru et al., 2014), but some also argued that it could also detect necrosis and other types of cell death (Grasl-Kraupp et al., 1995). Therefore, both apoptosis and necrosis may be involved in secondary degeneration of RGC bodies after PONT.
In the ON, mitochondrial autophagic profiles were observed in the areas vulnerable to secondary degeneration (Cummins et al., 2013). Therefore, autophagy may be involved in the secondary degeneration of axons in the ON. Whether it is involved in the secondary degeneration of RGC bodies needs further investigation.
Oxidative stress: Oxidative stress, indicated by increased expression of manganese superoxide dismutase (MnSOD) and decreased catalase activity, occurred 5 minutes after PONT in the ventral ON and the immunoreactivity of advanced glycation end product carboxymethyl lysine increased significantly 24 hours after PONT in the ventral ON (Fitzgerald et al., 2010a; Wells et al., 2012). It was hypothesized that activation, accumulation, or redistribution of MnSOD, rather than new protein synthesis, occurred after PONT in the ON because the increase in MnSOD was so fast (5 minutes) after PONT. Western blot analysis of ON vulnerable to secondary degeneration (the ventral ON) may help clarify this issue (Fitzgerald et al., 2010a). In the retinas, the oxidative stress indicated by increasing expression of MnSOD began at 24 hours after PONT in secondary degeneration both in PVG hooded rats and SD rats (Fitzgerald et al., 2010a; Li et al., 2013). Therefore, oxidative stress was involved in the secondary degeneration of both axons and RGC bodies after PONT. The polysaccharides extracted from Lycium barbarum (LBP), lomerizine and 670 nm light treatment could reduce oxidative stress and secondary degeneration of RGCs (Fitzgerald et al., 2009a; Fitzgerald et al., 2009b; Fitzgerald et al., 2010b; Li et al., 2013).
Calcium overload: Nanoscale secondary ion mass spectroscopy showed that the mean intensities of specific areas having the elevated 40Ca/12C in the normal ON in vivo decreased but a punctuate distribution increased at 5 minutes after PONT (Wells et al., 2012). The result might indicate a redistribution of calcium from internal stores to the cytosol. It is known that calcium accumulation can lead to the over production of reactive oxygen species (ROS) in mitochondria (Lewen et al., 2000; Begemann et al., 2010). In addition, the immunoreactivity of glutamate receptor subunit GluR1, a subunit of functional α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, was increased rapidly in the ON vulnerable to secondary degeneration. Increased plasma membrane expression of GluR1 in astrocytes, resulting in increased calcium flux into and across the linked astrocytic network, was thought to lead to the early spread of oxidative stress in neurons (Fitzgerald et al., 2009b).
Mitochondrial change: Although the numbers of mitochondria in the ON vulnerable to secondary degeneration had not changed after PONT, there was alteration in the mitochondrial ultrastructure. Higher accumulation of smaller mitochondrial profiles at day 1 and glial mitochondrial profiles with a more elliptical shape at both days 1 and 7 following injury were observed (Cummins et al., 2013). In addition, the activity of enzymes of the citric acid cycle was dynamically altered during secondary degeneration in the ON (Cummins et al., 2013). As mentioned above, mitochondria are the source of ROS (Lewen et al., 2000). Therefore, changes in mitochondrial morphology and function might be related to the excessive production of ROS.
C-jun: C-jun was shown to be involved in secondary degeneration after PONT in all three strains of rats used for PONT. Western blotting showed that the expression of phospho-c-jun (p-c-jun) increased from 1 day after PONT in the inferior retinas of SD rats (Li et al., 2013). Immunohistochemical staining showed that the number of p-c-jun or c-jun positive cells increased significantly from 3 days after PONT in secondary degeneration in Wistar rats and PVG hooded rats, respectively (Fitzgerald et al., 2010a; Vander and Levkovitch-Verbin, 2012). It seemed that the time-point of increasing c-jun expression was different in SD rats compared with the other two strains. The possible reason was that Western blotting was more sensitive than immunohistochemical staining in the detection of trace increase in the expression of proteins. C-jun was shown to have both pro-apoptotic and anti-apoptotic roles (Levkovitch-Verbin et al., 2006) for RGCs. It was assumed to play an anti-apoptotic role after PONT in Wistar rats (Fitzgerald et al., 2010a) because incidence of apoptotic death was low in this strain (Fitzgerald et al., 2009a). This may be why apoptosis contributed less to the secondary degeneration of RGCs after PONT than necrosis in this strain of rats. But in our study in SD rats, the phosphorylation of c-jun was thought to be pro-apoptotic because the LBP was able to decrease secondary degeneration and phosphorylation of c-jun simultaneously. So it seems that the role of p-c-jun is controversial in vivo in the secondary degeneration, and an in vitro study may be helpful to answer this issue.
Water channel change: Aquaporin 4 (AQP-4) immunointensity in glial fibrillary acidic protein-positive astrocytes increased following PONT (Wells et al., 2012). AQP-4 is the main water channel of the mammalian nervous system and is indicated to co-localize with the inward potassium (K) channel Kir 4.1, as a water-potassium transport complex. Although no change in Kir 4.1 immunoreactivity or K ions was observed, it is possible that the change in the flux of ions in response to AQP-4-mediated astrocytic swelling/hypertrophy results in the spread of altered ionic balances through the astrocytic syncytium and increased secondary degeneration.
Microglia and macrophages: Three days after PONT, the numbers of ionized calcium binding adaptor molecule-1 (Iba-1)- or cluster differentiation 68 (ED-1)-immunoreactive microglia/macrophages increased significantly in the ventral ON but not in the ventral retinas (Fitzgerald et al., 2010a). It seemed that in the ON, the activation of microglia and the infiltration of macrophages were precedent to the secondary loss of axons because the obvious loss of axons began 4 days after PONT (Levkovitch-Verbin et al., 2003). Secondary degeneration occurred in the ventral retinas later than 7 days in PVG hooded rats (Fitzgerald et al., 2009b) after PONT. Therefore, the time points longer than 3 days should be studied in ventral retinas to elucidate whether the activation of microglia/macrophages is precedent to the death of RGC bodies.
The possible roles of microglia and macrophages after injury in CNS are controversial. They can : (1) be a source of oxidative stress; (2) both contribute to and protect against glutamate excitotoxicity, depending on the timing and pathological conditions of infiltration; (3) have pro-inflammatory or anti-inflammatory roles; (4) phagocytize myelin debris to contribute to turnover of myelin; and (5) secret brain-derived neurotrophic factor and glial-derived neurotrophic factor (Benarroch, 2013). In our study, the expression of an oxidative stress marker, anti-hydroxyguanosine 8, increased after PONT in the activated microglia/macrophages in the ON (unpublished data). This result supports that activated microglia/macrophages are the source of ROS after injury. Tumor necrosis factor alpha (TNF-α) was known to be a major molecule involved in inflammation and could be secreted by activated microglia/macrophages. In our study, the expression of TNF-α did not increase in the inferior retinas until 1 week after PONT (Li et al., 2013) and this result showed that inflammation might not contribute to secondary degeneration in the cell bodies of RGCs in the retinas, but whether it is involved in secondary degeneration of axons in the ON needs further study. Other roles of activated microglia/macrophages need further investigation in future.
Astrocytes and oligodendrocytes: Astrocytes became hypertrophic after PONT and the immunoreactivity of oxidative stress markers (MnSOD and advanced glycation end product carboxymethyl lysine) increased in the astrocytes of the ventral ON 5 minutes and 24 hours after PONT, respectively (Fitzgerald et al., 2010a; Wells et al., 2012). Calcium overload is known to contribute to increased oxidative stress. As mentioned above, the ion channel AMPA receptor subunit GluR1 immunointensity and water channel AQP4 immunointensity in astrocytes were significantly increased in the ventral ON 3 hours and 24 hours following PONT, respectively. Change in AQP4 immunoreactivity may indicate its involvement in altered water balance and flux of ions following PONT. These results indicated astrocytes in the spread of calcium and oxidative stress during secondary degeneration (Fitzgerald et al., 2010a; Wells et al., 2012).
Swelling in myelinated axons, but not in unmyelinated axons, was detected after PONT in PVG hooded rats. It implied that the dysfunction of oligodendroglia contributed to axon swelling and damaged axonal transport (Payne et al., 2011). It is known that oligodendrocytes are vulnerable to glutamate excitotoxicity which may be released from the neural tissue after primary injury. But the occurrence of excitotoxicity after PONT has not been investigated yet and the number of oligodendrocytes remains stable after PONT. Therefore, to understand the role of oligodendrocytes in secondary degeneration, the study about excitotoxity should be conducted.
Comparison of the mechanisms between primary and secondary degeneration of RGCs after PONT: In several papers, the comparison of primary and secondary degeneration after PONT was conducted. Levkovitch-Verbin et al found that both primary and secondary degeneration led to caspase 3, growth arrest and DNA damage inducible gene 45α (GADD45α), cyclin-dependent kinase 2(CDK2) and etoposide-induced protein 2.4 homolog (ei24) activation in the retinas, but the activation was longer and more intense in the former. Similarly, both primary and secondary degeneration significantly down-regulated the pro-survival genes Bcl-2 and Bcl-x-L and up-regulated the pro-apoptotic genes Bax, Bad and inhibitor of apoptosis protein-1 (IAP-1), though a delay of the up-regulation of Bax and Bad was seen in secondary degeneration (Levkovitch-Verbin et al., 2010; Levkovitch-Verbin et al., 2011). These results indicated that both primary and secondary degeneration shared some common final mechanisms. However, they also reported that phospho-stress-activated protein kinase/c-jun N-terminal kinase (p-SAPK/JNK) was only activated in primary degeneration but not in secondary degeneration; whereas p-Akt was only activated in secondary areas rather than primary areas (Vander and Levkovitch-Verbin, 2012). The data suggested a difference between primary and secondary degeneration. In addition, they also showed that primary and secondary degeneration was different in their reaction to minocycline (Levkovitch-Verbin et al., 2011). Why minocycline prevents secondary degeneration rather than primary degeneration is unclear, but the reason may be related to specific signaling pathways that take part in secondary degeneration and that can be intervened by minocycline but not or only to a lesser extent in primary degeneration. In addition, the fact that Lycium barbarum (wolfberry) could delay secondary degeneration rather than primary degeneration also indicated the difference in the mechanisms between the two (Li et al., 2013). But which specific signaling pathways play a role in secondary degeneration rather than primary degeneration need further study.
Secondary degeneration of other retinal layers after PONT
In the above paragraphs, the secondary degeneration of RGCs limited to ganglion cell layer has been discussed. What should be noticed is that other retinal layers are also affected after PONT. This point is discussed here as supplementary information. In multifocal electroretinogram (ERG), the P1 component was shown to originate from the outer retinas and maybe ON-bipolar cells in porcine and rhesus monkey (Hood et al., 2002; Ng et al., 2008). In our group, P1 component of SD rats was also shown to originate from the outer retina because it was not affected after inhibiting the inner retinal activity. The amplitude of P1 decreased after PONT in our study indicated that outer retina was affected and secondary degeneration could adversely affect retinal layers beyond retinal ganglion cell level (Chu et al., 2013). This kind of trans-synaptic degeneration has been shown in glaucoma patients. For example, in human glaucomatous patients and monkey models, it showed the decrease of cone opsin message RNA (mRNA)(Pelzel et al., 2006), swelling and patchy loss of cone photoreceptors in the macular region (Nork et al., 2000; Calkins, 2012). In DBA mice, detection with retinal histology showed the attenuation of scotopic a- and b-wave amplitude concurrent with thinning of the inner plexiform layer and of the outer retina layer, respectively (Bayer et al., 2001). In C57 mice, GABAergic types of amacrine cells were reduced by about the same percentages to RGCs (Moon et al., 2005). These data reported the possible degeneration of photoreceptors and amacrine cells in glaucoma. Although the underlying mechanisms of this trans-synaptic degeneration were not clear, the degeneration of other retinal layers than the ganglion cell layer should be assessed when it comes to a neuroprotective agent.
Summary
In neurotrauma and neurodegenerative diseases of the CNS, secondary degeneration of neurons and glial cells occurs. The protection of neurons and glial cells against secondary degeneration is a promising direction for therapy of these diseases. PONT model is a useful tool for studying the mechanisms of secondary degeneration and screening for the neuroprotective drugs in combat against secondary degeneration because it can separate primary degeneration from secondary degeneration in location. Up to now, evidence shows that the mechanisms of secondary degeneration after PONT include apoptosis, necrosis and autophagy. Oxidative stress, calcium overload, mitochondrial dysfunction, c-jun, change of water channel, microglia and macrophages, astrocytes and oligodendrocytes are involved in the secondary degeneration after PONT. The comprehensive understanding of the mechanisms underlying the secondary degeneration of RGCs will provide valuable guidance for the treatment of neurotrauma and CNS neurodegenerative diseases. In addition, it should be noticed that outer retinas will also be affected in glaucoma and after PONT and it should be considered when screening effective drugs for glaucoma.
Footnotes
Funding: The work described in this paper was substantially supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (HKU 776109M). In addition, this work was partially supported by the Fundamental Research Funds for the Central Universities Grant 21609101.
Conflicts of interest: None declared.
References
- [1].Aktas Z, Karaca EE, Gonul II, Hasanreisoglu M, Onol M. Apoptosis in the iris and trabecular meshwork of medically treated and untreated primary open angle glaucoma patients. Int J Ophthalmol. 2013;6:827–830. doi: 10.3980/j.issn.2222-3959.2013.06.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012;31:152–181. doi: 10.1016/j.preteyeres.2011.11.002. [DOI] [PubMed] [Google Scholar]
- [3].Baltmr A, Duggan J, Nizari S, Salt TE, Cordeiro MF. Neuroprotection in glaucoma - Is there a future role? Exp Eye Res. 2010;91:554–566. doi: 10.1016/j.exer.2010.08.009. [DOI] [PubMed] [Google Scholar]
- [4].Bayer AU, Neuhardt T, May AC, Martus P, Maag KP, Brodie S, Lutjen-Drecoll E, Podos SM, Mittag T. Retinal morphology and ERG response in the DBA/2NNia mouse model of angle-closure glaucoma. Invest Ophthalmol Vis Sci. 2001;42:1258–1265. [PubMed] [Google Scholar]
- [5].Begemann M, Grube S, Papiol S, Malzahn D, Krampe H, Ribbe K, Friedrichs H, Radyushkin KA, El-Kordi A, Benseler F, Hannke K, Sperling S, Schwerdtfeger D, Thanhäuser I, Gerchen MF, Ghorbani M, Gutwinski S, Hilmes C, Leppert R, Ronnenberg A, et al. Modification of cognitive performance in schizophrenia by complexin 2 gene polymorphisms. Arch Gen Psychiatry. 2010;67:879–888. doi: 10.1001/archgenpsychiatry.2010.107. [DOI] [PubMed] [Google Scholar]
- [6].Benarroch EE. Microglia: Multiple roles in surveillance, circuit shaping, and response to injury. Neurology. 2013;81:1079–1088. doi: 10.1212/WNL.0b013e3182a4a577. [DOI] [PubMed] [Google Scholar]
- [7].Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14:4368–4374. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res. 2012;31:702–719. doi: 10.1016/j.preteyeres.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chan SO, Guillery RW. Changes in fiber order in the optic nerve and tract of rat embryos. J Comp Neurol. 1994;344:20–32. doi: 10.1002/cne.903440103. [DOI] [PubMed] [Google Scholar]
- [10].Chinskey ND, Zheng QD, Zacks DN. Control of photoreceptor autophagy after retinal detachment: the switch from survival to death. Invest Ophthalmol Vis Sci. 2014;55:688–695. doi: 10.1167/iovs.13-12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chu PH, Li HY, Chin MP, So KF, Chan HH. Effect of lycium barbarum (wolfberry) polysaccharides on preserving retinal function after partial optic nerve transection. PloS One. 2013;8:e81339. doi: 10.1371/journal.pone.0081339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Clarke DB, Bray GM, Aguayo AJ. Prolonged administration of NT-4/5 fails to rescue most axotomized retinal ganglion cells in adult rats. Vision Res. 1998;38:1517–1524. doi: 10.1016/s0042-6989(97)00341-6. [DOI] [PubMed] [Google Scholar]
- [13].Cummins N, Bartlett CA, Archer M, Bartlett E, Hemmi JM, Harvey AR, Dunlop SA, Fitzgerald M. Changes to mitochondrial ultrastructure in optic nerve vulnerable to secondary degeneration in vivo are limited by irradiation at 670 nm. BMC Neurosci. 2013;14:98. doi: 10.1186/1471-2202-14-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Danesh-Meyer HV. Neuroprotection in glaucoma: recent and future directions. Curr Opin Ophthalmol. 2011;22:78–86. doi: 10.1097/ICU.0b013e32834372ec. [DOI] [PubMed] [Google Scholar]
- [15].Deng S, Wang M, Yan Z, Tian Z, Chen H, Yang X, Zhuo Y. Autophagy in retinal ganglion cells in a rhesus monkey chronic hypertensive glaucoma model. PloS One. 2013;8:e77100. doi: 10.1371/journal.pone.0077100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res. 2007;161:43–59. doi: 10.1016/S0079-6123(06)61004-2. [DOI] [PubMed] [Google Scholar]
- [17].Fitzgerald M, Bartlett CA, Harvey AR, Dunlop SA. Early events of secondary degeneration after partial optic nerve transection: an immunohistochemical study. J Neurotrauma. 2010a;27:439–452. doi: 10.1089/neu.2009.1112. [DOI] [PubMed] [Google Scholar]
- [18].Fitzgerald M, Payne SC, Bartlett CA, Evill L, Harvey AR, Dunlop SA. Secondary retinal ganglion cell death and the neuroprotective effects of the calcium channel blocker lomerizine. Invest Ophthalmol Vis Sci. 2009a;50:5456–5462. doi: 10.1167/iovs.09-3717. [DOI] [PubMed] [Google Scholar]
- [19].Fitzgerald M, Bartlett CA, Evill L, Rodger J, Harvey AR, Dunlop SA. Secondary degeneration of the optic nerve following partial transection: the benefits of lomerizine. Exp Neurol. 2009b;216:219–230. doi: 10.1016/j.expneurol.2008.11.026. [DOI] [PubMed] [Google Scholar]
- [20].Fitzgerald M, Bartlett CA, Payne SC, Hart NS, Rodger J, Harvey AR, Dunlop SA. Near infrared light reduces oxidative stress and preserves function in CNS tissue vulnerable to secondary degeneration following partial transection of the optic nerve. J Neurotrauma. 2010b;27:2107–2119. doi: 10.1089/neu.2010.1426. [DOI] [PubMed] [Google Scholar]
- [21].Fitzgerald M, Hodgetts S, Van Den Heuvel C, Natoli R, Hart NS, Valter K, Harvey AR, Vink R, Provis J, Dunlop SA. Red/near-infrared irradiation therapy for treatment of central nervous system injuries and disorders. Rev Neurosci. 2013;24:205–226. doi: 10.1515/revneuro-2012-0086. [DOI] [PubMed] [Google Scholar]
- [22].Floyd CL, Lyeth BG. Astroglia: important mediators of traumatic brain injury. Prog Brain Res. 2007;161:61–79. doi: 10.1016/S0079-6123(06)61005-4. [DOI] [PubMed] [Google Scholar]
- [23].Franklin TB, Murphy JA, Myers TL, Clarke DB, Currie RW. Enriched environment during adolescence changes brain-derived neurotrophic factor and TrkB levels in the rat visual system but does not offer neuroprotection to retinal ganglion cells following axotomy. Brain Res. 2006;1095:1–11. doi: 10.1016/j.brainres.2006.04.025. [DOI] [PubMed] [Google Scholar]
- [24].Fu QL, Hu B, Wu W, Pepinsky RB, Mi S, So KF. Blocking LINGO-1 function promotes retinal ganglion cell survival following ocular hypertension and optic nerve transection. Invest Ophthalmol Vis Sci. 2008;49:975–985. doi: 10.1167/iovs.07-1199. [DOI] [PubMed] [Google Scholar]
- [25].Fu QL, Li X, Yip HK, Shao Z, Wu W, Mi S, So KF. Combined effect of brain-derived neurotrophic factor and LINGO-1 fusion protein on long-term survival of retinal ganglion cells in chronic glaucoma. Neuroscience. 2009;162:375–382. doi: 10.1016/j.neuroscience.2009.04.075. [DOI] [PubMed] [Google Scholar]
- [26].Fu QL, Hu B, Li X, Shao Z, Shi JB, Wu W, So KF, Mi S. LINGO-1 negatively regulates TrkB phosphorylation after ocular hypertension. Eur J Neurosci. 2010;31:1091–1097. doi: 10.1111/j.1460-9568.2010.07127.x. [DOI] [PubMed] [Google Scholar]
- [27].Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- [28].Guimaraes JS, Freire MA, Lima RR, Souza-Rodrigues RD, Costa AM, dos Santos CD, Picanco-Diniz CW, Gomes-Leal W. Mechanisms of secondary degeneration in the central nervous system during acute neural disorders and white matter damage. Rev Neurol. 2009;48:304–310. [PubMed] [Google Scholar]
- [29].Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- [30].Ho YS, Yu MS, Lai CS, So KF, Yuen WH, Chang RC. Characterizing the neuroprotective effects of alkaline extract of Lycium barbarum on beta-amyloid peptide neurotoxicity. Brain Res. 2007;1158:123–134. doi: 10.1016/j.brainres.2007.04.075. [DOI] [PubMed] [Google Scholar]
- [31].Ho YS, Yu MS, Yik SY, So KF, Yuen WH, Chang RC. Polysaccharides from wolfberry antagonizes glutamate excitotoxicity in rat cortical neurons. Cell Mol Neurobiol. 2009;29:1233–1244. doi: 10.1007/s10571-009-9419-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ho YS, Yu MS, Yang XF, So KF, Yuen WH, Chang RC. Neuroprotective effects of polysaccharides from wolfberry, the fruits of Lycium barbarum, against homocysteine-induced toxicity in rat cortical neurons. J Alzheimers Dis. 2010;19:813–827. doi: 10.3233/JAD-2010-1280. [DOI] [PubMed] [Google Scholar]
- [33].Hood DC, Frishman LJ, Saszik S, Viswanathan S. Retinal origins of the primate multifocal ERG: implications for the human response. Invest Ophthalmol Vis Sci. 2002;43:1673–1685. [PubMed] [Google Scholar]
- [34].Hou B, You SW, Wu MM, Kuang F, Liu HL, Jiao XY, Ju G. Neuroprotective effect of inosine on axotomized retinal ganglion cells in adult rats. Invest Ophthalmol Vis Sci. 2004;45:662–667. doi: 10.1167/iovs.03-0281. [DOI] [PubMed] [Google Scholar]
- [35].Isenmann S, Engel S, Gillardon F, Bahr M. Bax antisense oligonucleotides reduce axotomy-induced retinal ganglion cell death in vivo by reduction of Bax protein expression. Cell Death Differ. 1999;6:673–682. doi: 10.1038/sj.cdd.4400538. [DOI] [PubMed] [Google Scholar]
- [36].Koeberle PD, Ball AK. Effects of GDNF on retinal ganglion cell survival following axotomy. Vision Res. 1998;38:1505–1515. doi: 10.1016/s0042-6989(97)00364-7. [DOI] [PubMed] [Google Scholar]
- [37].Kolodziejczyk K, Saab AS, Nave KA, Attwell D. Why do oligodendrocyte lineage cells express glutamate receptors? F1000 Biol Rep. 2010;2:57. doi: 10.3410/B2-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kowluru RA, Zhong Q, Santos JM, Thandampallayam M, Putt D, Gierhart DL. Beneficial effects of the nutritional supplements on the development of diabetic retinopathy. Nutr Metab (Lond) 2014;11:8. doi: 10.1186/1743-7075-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kretz A, Schmeer C, Tausch S, Isenmann S. Simvastatin promotes heat shock protein 27 expression and Akt activation in the rat retina and protects axotomized retinal ganglion cells in vivo. Neurobiol Dis. 2006;21:421–430. doi: 10.1016/j.nbd.2005.08.003. [DOI] [PubMed] [Google Scholar]
- [40].Krueger-Naug AM, Emsley JG, Myers TL, Currie RW, Clarke DB. Administration of brain-derived neurotrophic factor suppresses the expression of heat shock protein 27 in rat retinal ganglion cells following axotomy. Neuroscience. 2003;116:49–58. doi: 10.1016/s0306-4522(02)00582-1. [DOI] [PubMed] [Google Scholar]
- [41].LaPlaca MC, Simon CM, Prado GR, Cullen DK. CNS injury biomechanics and experimental models. Prog Brain Res. 2007;161:13–26. doi: 10.1016/S0079-6123(06)61002-9. [DOI] [PubMed] [Google Scholar]
- [42].Leaver SG, Cui Q, Plant GW, Arulpragasam A, Hisheh S, Verhaagen J, Harvey AR. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006;13:1328–1341. doi: 10.1038/sj.gt.3302791. [DOI] [PubMed] [Google Scholar]
- [43].Lebrun-Julien F, Morquette B, Douillette A, Saragovi HU, Di Polo A. Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol Cell Neurosci. 2009;40:410–420. doi: 10.1016/j.mcn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- [44].Levkovitch-Verbin H, Dardik R, Vander S, Melamed S. Mechanism of retinal ganglion cells death in secondary degeneration of the optic nerve. Exp Eye Res. 2010;91:127–134. doi: 10.1016/j.exer.2009.11.014. [DOI] [PubMed] [Google Scholar]
- [45].Levkovitch-Verbin H1, Spierer O, Vander S, Dardik R. Similarities and differences between primary and secondary degeneration of the optic nerve and the effect of minocycline. Graefes Arch Clin Exp Ophthalmol. 2011;249:849–857. doi: 10.1007/s00417-010-1608-2. [DOI] [PubMed] [Google Scholar]
- [46].Levkovitch-Verbin H, Quigley HA, Kerrigan-Baumrind LA, D’Anna SA, Kerrigan D, Pease ME. Optic nerve transection in monkeys may result in secondary degeneration of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2001;42:975–982. [PubMed] [Google Scholar]
- [47].Levkovitch-Verbin H, Quigley HA, Martin KR, Zack DJ, Pease ME, Valenta DF. A model to study differences between primary and secondary degeneration of retinal ganglion cells in rats by partial optic nerve transection. Invest Ophthalmol Vis Sci. 2003;44:3388–3393. doi: 10.1167/iovs.02-0646. [DOI] [PubMed] [Google Scholar]
- [48].Levkovitch-Verbin H, Dardik R, Vander S, Nisgav Y, Kalev-Landoy M, Melamed S. Experimental glaucoma and optic nerve transection induce simultaneous upregulation of proapoptotic and prosurvival genes. Invest Ophthalmol Vis Sci. 2006;47:2491–2497. doi: 10.1167/iovs.05-0996. [DOI] [PubMed] [Google Scholar]
- [49].Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- [50].Li H, Liang Y, Chiu K, Yuan Q, Lin B, Chang RC, So KF. Lycium barbarum (wolfberry) reduces secondary degeneration and oxidative stress, and inhibits JNK pathway in retina after partial optic nerve transection. PloS One. 2013;8:e68881. doi: 10.1371/journal.pone.0068881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Li SY, Yang D, Yeung CM, Yu WY, Chang RC, So KF, Wong D, Lo AC. Lycium barbarum polysaccharides reduce neuronal damage, blood-retinal barrier disruption and oxidative stress in retinal ischemia/reperfusion injury. PloS One. 2011;6:e16380. doi: 10.1371/journal.pone.0016380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mi X, Chiu K, Van G, Leung JWC, Lo ACY, Chung SK, Chang RC, So K. Effect of Lycium barbarum Polysaccharides on the expression of endothelin-1 and its receptors in an ocular hypertension model of rat glaucoma. Neural Regen Res. 2012a;7:645–651. doi: 10.3969/j.issn.1673-5374.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mi XS, Zhong JX, Chang RC, So KF. Research advances on the usage of traditional Chinese medicine for neuroprotection in glaucoma. J Integr Med. 2013;11:233–240. doi: 10.3736/jintegrmed2013037. [DOI] [PubMed] [Google Scholar]
- [54].Mi XS, Feng Q, Lo AC, Chang RC, Lin B, Chung SK, So KF. Protection of retinal ganglion cells and retinal vasculature by Lycium barbarum polysaccharides in a mouse model of acute ocular hypertension. PloS One. 2012b;7:e45469. doi: 10.1371/journal.pone.0045469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Moon JI, Kim IB, Gwon JS, Park MH, Kang TH, Lim EJ, Choi KR, Chun MH. Changes in retinal neuronal populations in the DBA/2J mouse. Cell Tissue Res. 2005;320:51–59. doi: 10.1007/s00441-004-1062-8. [DOI] [PubMed] [Google Scholar]
- [56].Nakazawa T, Takahashi H, Shimura M. Estrogen has a neuroprotective effect on axotomized RGCs through ERK signal transduction pathway. Brain Res. 2006;1093:141–149. doi: 10.1016/j.brainres.2006.03.084. [DOI] [PubMed] [Google Scholar]
- [57].Nakazawa T, Tomita H, Yamaguchi K, Sato Y, Shimura M, Kuwahara S, Tamai M. Neuroprotective effect of nipradilol on axotomized rat retinal ganglion cells. Curr Eye Res. 2002;24:114–122. doi: 10.1076/ceyr.24.2.114.8162. [DOI] [PubMed] [Google Scholar]
- [58].Ng YF, Chan HH, Chu PH, Siu AW, To CH, Beale BA, Gilger BC, Wong F. Pharmacologically defined components of the normal porcine multifocal ERG. Doc Ophthalmol. 2008;116:165–176. doi: 10.1007/s10633-007-9076-7. [DOI] [PubMed] [Google Scholar]
- [59].Nickells RW. From ocular hypertension to ganglion cell death: a theoretical sequence of events leading to glaucoma. Can J Ophthalmol. 2007;42:278–287. [PubMed] [Google Scholar]
- [60].Nork TM, Ver Hoeve JN, Poulsen GL, Nickells RW, Davis MD, Weber AJ, Vaegan, Sarks SH, Lemley HL, Millecchia LL. Swelling and loss of photoreceptors in chronic human and experimental glaucomas. Arch Ophthalmol. 2000;118:235–245. doi: 10.1001/archopht.118.2.235. [DOI] [PubMed] [Google Scholar]
- [61].Ota T, Hara H, Miyawaki N. Brain-derived neurotrophic factor inhibits changes in soma-size of retinal ganglion cells following optic nerve axotomy in rats. J Ocul Pharmacol Ther. 2002;18:241–249. doi: 10.1089/108076802760116160. [DOI] [PubMed] [Google Scholar]
- [62].Oyinbo CA. Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiol Exp (Wars) 2011;71:281–299. doi: 10.55782/ane-2011-1848. [DOI] [PubMed] [Google Scholar]
- [63].Payne SC, Bartlett CA, Harvey AR, Dunlop SA, Fitzgerald M. Chronic swelling and abnormal myelination during secondary degeneration after partial injury to a central nervous system tract. J Neurotrauma. 2011;28:1077–1088. doi: 10.1089/neu.2010.1665. [DOI] [PubMed] [Google Scholar]
- [64].Payne SC, Bartlett CA, Harvey AR, Dunlop SA, Fitzgerald M. Myelin sheath decompaction, axon swelling, and functional loss during chronic secondary degeneration in rat optic nerve. Invest Ophthalmol Vis Sci. 2012;53:6093–6101. doi: 10.1167/iovs.12-10080. [DOI] [PubMed] [Google Scholar]
- [65].Payne SC, Bartlett CA, Savigni DL, Harvey AR, Dunlop SA, Fitzgerald M. Early proliferation does not prevent the loss of oligodendrocyte progenitor cells during the chronic phase of secondary degeneration in a CNS white matter tract. PloS One. 2013;8:e65710. doi: 10.1371/journal.pone.0065710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Peinado-Ramon P, Salvador M, Villegas-Perez MP, Vidal-Sanz M. Effects of axotomy and intraocular administration of NT-4, NT-3 and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Invest Ophthalmol Vis Sci. 1996;37:489–500. [PubMed] [Google Scholar]
- [67].Pelzel HR, Schlamp CL, Poulsen GL, Ver Hoeve JA, Nork TM, Nickells RW. Decrease of cone opsin mRNA in experimental ocular hypertension. Mol Vis. 2006;12:1272–1282. [PubMed] [Google Scholar]
- [68].Quigley HA, Davis EB, Anderson DR. Descending optic nerve degeneration in primates. Invest Ophthalmol Vis Sci. 1977;16:841–849. [PubMed] [Google Scholar]
- [69].Ren R, Li Y, Liu Z, Liu K, He S. Long-term rescue of rat retinal ganglion cells and visual function by AAV-mediated BDNF expression after acute elevation of intraocular pressure. Invest Ophthalmol Vis Sci. 2012;53:1003–1011. doi: 10.1167/iovs.11-8484. [DOI] [PubMed] [Google Scholar]
- [70].Savigni DL, O’Hare Doig RL, Szymanski CR, Bartlett CA, Lozic I, Smith NM, Fitzgerald M. Three Ca channel inhibitors in combination limit chronic secondary degeneration following neurotrauma. Neuropharmacology. 2013;75C:380–390. doi: 10.1016/j.neuropharm.2013.07.034. [DOI] [PubMed] [Google Scholar]
- [71].Schwartz M. Optic nerve crush: protection and regeneration. Brain Res Bull. 2004;62:467–471. doi: 10.1016/S0361-9230(03)00076-5. [DOI] [PubMed] [Google Scholar]
- [72].Selt M, Bartlett CA, Harvey AR, Dunlop SA, Fitzgerald M. Limited restoration of visual function after partial optic nerve injury; a time course study using the calcium channel blocker lomerizine. Brain Res Bull. 2010;81:467–471. doi: 10.1016/j.brainresbull.2009.11.004. [DOI] [PubMed] [Google Scholar]
- [73].Simon DK, O’Leary DD. Relationship of retinotopic ordering of axons in the optic pathway to the formation of visual maps in central targets. J Comp Neurol. 1991;307:393–404. doi: 10.1002/cne.903070305. [DOI] [PubMed] [Google Scholar]
- [74].Stewart SS, Appel SH. Trophic factors in neurologic disease. Annu Rev Med. 1988;39:193–201. doi: 10.1146/annurev.me.39.020188.001205. [DOI] [PubMed] [Google Scholar]
- [75].Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sullivan TA, Geisert EE, Templeton JP, Rex TS. Dose-dependent treatment of optic nerve crush by exogenous systemic mutant erythropoietin. Exp Eye Res. 2012;96:36–41. doi: 10.1016/j.exer.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Szymanski CR, Chiha W, Morellini N, Cummins N, Bartlett CA, O’Hare Doig RL, Savigni DL, Payne SC, Harvey AR, Dunlop SA, Fitzgerald M. Paranode abnormalities and oxidative stress in optic nerve vulnerable to secondary degeneration: Modulation by 670 nm light treatment. PloS One. 2013;8:e66448. doi: 10.1371/journal.pone.0066448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Tezel G. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res. 2006;25:490–513. doi: 10.1016/j.preteyeres.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tezel G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res. 2008;173:409–421. doi: 10.1016/S0079-6123(08)01128-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].van Adel BA, Arnold JM, Phipps J, Doering LC, Ball AK. Ciliary neurotrophic factor protects retinal ganglion cells from axotomy-induced apoptosis via modulation of retinal glia in vivo. J Neurobiol. 2005;63:215–234. doi: 10.1002/neu.20117. [DOI] [PubMed] [Google Scholar]
- [81].Vander S, Levkovitch-Verbin H. Regulation of cell death and survival pathways in secondary degeneration of the optic nerve - a long-term study. Curr Eye Res. 2012;37:740–748. doi: 10.3109/02713683.2012.673679. [DOI] [PubMed] [Google Scholar]
- [82].Vasudevan SK, Gupta V, Crowston JG. Neuroprotection in glaucoma. Indian J Ophthalmol. 2011;59(Suppl):S102–113. doi: 10.4103/0301-4738.73700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Vigneswara V, Berry M, Logan A, Ahmed Z. Pigment epithelium-derived factor is retinal ganglion cell neuroprotective and axogenic after optic nerve crush injury. Invest Ophthalmol Vis Sci. 2013;54:2624–2633. doi: 10.1167/iovs.13-11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Weber AJ, Viswanathan S, Ramanathan C, Harman CD. Combined application of BDNF to the eye and brain enhances ganglion cell survival and function in the cat after optic nerve injury. Invest Ophthalmol Vis Sci. 2010;51:327–334. doi: 10.1167/iovs.09-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Weise J, Isenmann S, Klocker N, Kugler S, Hirsch S, Gravel C, Bahr M. Adenovirus-mediated expression of ciliary neurotrophic factor (CNTF) rescues axotomized rat retinal ganglion cells but does not support axonal regeneration in vivo. Neurobiol Dis. 2000;7:212–223. doi: 10.1006/nbdi.2000.0285. [DOI] [PubMed] [Google Scholar]
- [86].Wells J, Kilburn MR, Shaw JA, Bartlett CA, Harvey AR, Dunlop SA, Fitzgerald M. Early in vivo changes in calciumions oxidative stress markers, and ion channel immunoreactivity following partial injury to the optic nerve. J Neurosci Res. 2012;90:606–618. doi: 10.1002/jnr.22784. [DOI] [PubMed] [Google Scholar]
- [87].Yoles E, Schwartz M. Degeneration of spared axons following partial white matter lesion: implications for optic nerve neuropathies. Exp Neurol. 1998;153:1–7. doi: 10.1006/exnr.1998.6811. [DOI] [PubMed] [Google Scholar]
- [88].Yu MS, Ho YS, So KF, Yuen WH, Chang RC. Cytoprotective effects of Lycium barbarum against reducing stress on endoplasmic reticulum. Int J Mol Med. 2006;17:1157–1161. [PubMed] [Google Scholar]
- [89].Yu MS, Lai CS, Ho YS, Zee SY, So KF, Yuen WH, Chang RC. Characterization of the effects of anti-aging medicine Fructus lycii on beta-amyloid peptide neurotoxicity. Int J Mol Med. 2007;20:261–268. [PubMed] [Google Scholar]
- [90].Zhang E, Yau SY, Lau BW, Ma H, Lee TM, Chang RC, So KF. Synaptic plasticity, but not hippocampal neurogenesis, mediated the counteractive effect of wolfberry on depression in rats(1) Cell Transplant. 2012;21:2635–2649. doi: 10.3727/096368912X655181. [DOI] [PubMed] [Google Scholar]
- [91].Zhang YK, Wang J, Liu L, Chang RC, So KF, Ju G. The effect of Lycium barbarum on spinal cord injury, particularly its relationship with M1 and M2 macrophage in rats. BMC Complement Altern Med. 2013;13:67. doi: 10.1186/1472-6882-13-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhu J, Zhang J, Ji M, Gu H, Xu Y, Chen C, Hu N. The role of peroxisome proliferator-activated receptor and effects of its agonist pioglitazone on a rat model of optic nerve crush: PPARgamma in retinal neuroprotection. PloS One. 2013;8:e68935. doi: 10.1371/journal.pone.0068935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zuo L, Khan RS, Lee V, Dine K, Wu W, Shindler KS. SIRT1 promotes RGC survival and delays loss of function following optic nerve crush. Invest Ophthalmol Vis Sci. 2013;54:5097–5102. doi: 10.1167/iovs.13-12157. [DOI] [PMC free article] [PubMed] [Google Scholar]