

Abstract
Activation of nuclear factor kappa B (NF-κB) is a hallmark of various central nervous system (CNS) pathologies. Neuron-specific inhibition of its transcriptional activator subunit RelA, also referred to as p65, promotes neuronal survival under a range of conditions, i.e., for ischemic or excitotoxic insults. In macro- and microglial cells, post-lesional activation of NF-κB triggers a growth-permissive program which contributes to neural tissue inflammation, scar formation, and the expression of axonal growth inhibitors. Intriguingly, inhibition of such inducible NF-κB in the neuro-glial compartment, i.e., by genetic ablation of RelA or overexpression of a transdominant negative mutant of its upstream regulator IκBα, significantly enhances functional recovery and promotes axonal regeneration in the mature CNS. By contrast, depletion of the NF-κB subunit p50, which lacks transcriptional activator function and acts as a transcriptional repressor on its own, causes precocious neuronal loss and exacerbates axonal degeneration in the lesioned brain. Collectively, the data imply that NF-κB orchestrates a multicellular program in which κB-dependent gene expression establishes a growth-repulsive terrain within the post-lesioned brain that limits structural regeneration of neuronal circuits. Considering these subunit-specific functions, interference with the NF-κB pathway might hold clinical potentials to improve functional restoration following traumatic CNS injury
Keywords: nuclear factor kappa B, RelA, p65, p50, central nervous system injury, axonal regeneration, neural regeneration
Introduction
Cellular responses induced by traumatic central nervous system (CNS) injury are associated with both destructive and regenerative processes mediated by neurons, glial and immunogenic cell types, and the endothelial compartment. They are orchestrated by defined changes in gene expression, leading to chronological cascades of primary and secondary tissue damage followed by wound closure and scar formation. Activation of the transcription factor nuclear factor kappa B (NF-κB) represents an early marker for cellular stress in numerous tissues including brain. NF-κB consists of five subunits (RelA [also called p65], RelB, c-Rel, p105/50, p100/52), with only Rel proteins possessing transcriptional activator function. Within the CNS, NF-κB signaling encompasses activation of preformed RelA and p50 containing dimers, which are cytoplasmically sequestered by inhibitory IκBα (canonical pathway). Fast kinetics of canonical NF-κB is guaranteed by stimulus-dependent activation of the IκB kinase (IKK) complex leading to serine (Ser) phosphorylation of IκBα by IKK-β subunits, its proteosomal degradation, and the nuclear translocation and DNA binding of RelA/p50 (Vallabhapurapu and Karin, 2009). Activity assays using κB-dependent expression of the reporter enzyme β-galactosidase (β-gal) reveal a constitutive activation of NF-κB in neurons of the developing and, at least in part, of the mature CNS (Bhakar et al., 2002). Thereby, the expression of β-gal correlates well with the presence of both RelA and p50 subunits in κB-binding complexes, as subsequently detected by DNA binding supershift experiments (Schmidt-Ullrich et al., 1996). Contrarily, reporter gene expression is completely absent in brain regions that are sensitive to anti-p50 serum only, thus indicating a transcriptional repressor function of DNA-associated p50 homodimers within the adult CNS. So far, there is only limited knowledge on the signaling pathways that control constitutive activation of NF-κB in developing and mature neurons (Gavalda et al., 2009; Konig et al., 2012; McKelvey et al., 2012) and its necessity for normal neurodevelopment and function is still under debate (Kretz et al., 2013; Listwak et al., 2013) (for more information, see (Gutierrez and Davies, 2011)).
Inducible activation of NF-κB in CNS injury
In addition to the aforementioned constitutive activation under physiological conditions, neuronal NF-κB becomes strong and rapidly activated under metabolic stress. Upstream of NF-κB, tumor necrosis factor (TNF) and TNF-like weak inducer of apoptosis (TWEAK) act as activators of NF-κB signaling under pathological conditions (Potrovita et al., 2004; Pradillo et al., 2005). Functionally, RelA-containing dimers induce the expression of pro-apoptotic target genes bim and noxa in ischemic neurons, and inhibition of this upregulation exerts neuronal protection (Inta et al., 2006). Experiments on rodents with injured optic nerve (ON) or spinal cord (SC) have also been conducted to study κB-dependent gene expression in response to axonal injury. Nerve fiber bundles are punctually disconnected by traumatic impact, and analysis of the degeneration and regeneration of neuronal cell bodies and axonal fibers shows that NF-κB activation occurs at the impact site as early as 0.5 hours post-injury, where it persists for several days (Bethea et al., 1998). Blocking NF-κB by immediate post-lesional application of a single dosage of either the pharmacological inhibitor pyrrolidine dithiocarbamate or synthetic “decoy” oligodeoxynucleotides targeting RelA/p50-binding sequences reduces early expression of pro-inflammatory iNOS and Cox-2. Thereby, SC tissue sparing becomes evident after only 24 hours post-injury (Jimenez-Garza et al., 2005; Rafati et al., 2008). Consequently, using the Basso, Beattie, and Bresnahan (BBB) score test for hind limb function, locomotor recovery is significantly improved as early as the first week after moderate SC injury in rats receiving the inhibitor, with no further locomotor advantage thereafter. Collectively, understanding the molecular and cellular mechanisms of the NF-κB pathway in axonal injury holds clinical potentials to stimulate recovery after traumatic CNS lesions by pharmacological treatment.
NF-κB in neurons
One rationale for this positive effect of blocking NF-κB is its activation in transected nerve fibers and associated cell somata, which may be located at considerable distances from the lesion center. Thereby, NF-κB transduces signals related to peripheral cell damage to the neuronal soma/nucleus. Such intracellular redistribution of NF-κB was first demonstrated using enhanced green fluorescence protein-tagged RelA fusion proteins (EGFP-RelA), where stimulation of hippocampal neurons with glutamate induces retrograde transport of RelA from synapses back to the nucleus (Wellmann et al., 2001). The description of a cell-autonomous NF-κB activation by axotomized neurons originates from studies on retinal ganglion cells (RGCs), which can be easily axotomized in the absence of gray matter damage using the ON crush model (Choi et al., 1998). Thereby, RGC survival at 4 weeks post-injury is significantly enhanced in transgenic relAflox;Nes-Cre mice harboring neuroectodermal deletion of transactivating RelA (own data). Similar to the pro-apoptotic gene expression pattern induced by metabolic stress, inducible NF-κB might trigger a cell death program inside axotomized neurons. As functional recovery following traumatic injury ideally relies on the presence of a large number of surviving neurons, reducing cell death by diminished activation of RelA is surely of advantage for promoting axonal regeneration and network restoration. Notably, subunit-unspecific interference with NF-κB signaling, e.g., by intraretinal application of the IKK inhibitor sulfasalazine, does not reduce but actually accelerates degeneration of axotomized RGCs (Choi et al., 2000). Thus, NF-κB activation within injured neurons – dependent on the involved subunit – also confers cell survival. This might be related to p50-dependent gene expression or to the transcriptional repressor function of p50 homodimers (Nakamura-Yanagidaira et al., 2011) and is in line with the observation that deletion of this subunit promotes neuronal death under various conditions of neurotoxicity (Li et al., 2008).
In vitro studies have emphasized a neuritogenic potential of NF-κB in developing neurons. Thereby, NF-κB signaling either stimulates or inhibits neurite outgrowth in cultured superior cervical ganglion sympathetic neurons or nodose ganglion sensory neurons depending on the cell type's specific phosphorylation status (Ser536) of RelA (Gutierrez et al., 2008). Although these studies were performed on immature neurons derived from neonatal brains, it is plausible that RelA-regulated transcription profiles may become reactivated in the mature CNS to stimulate regrowth of injured axons and to restore the neuronal network. This notion is supported by morphological and functional studies on transgenic IκB/tTA mice that allow for temporarily restricted reactivation of NF-κB in IκBα-dn neurons. In these mice, NF-κB signaling is in a tetracycline-dependent manner inhibited by expression of the super repressor IκBα-dn, but can be restored by doxycycline application (Tet OFF system) (Imielski et al., 2012). Pathophysiologically, IκB/tTA mice suffer from an inherited atrophy of hippocampal mossy fibers due to the absence of κB-dependent protein kinase A and FOXO1 expression in the developing dentate gyrus. Most importantly, reactivation of NF-κB in the mature disordered brain of adult mice by doxycycline treatment initiates the renewal of granule cells and regrowth of mossy fibers in the dentate gyrus to the level of healthy wild-type (WT) animals (Imielski et al., 2012). It remains to be explored whether this signaling pathway likewise holds a potential to stimulate neurogenesis and axonal regrowth in instances of traumatic nerve injuries. Collectively, cell culture approaches combined with in vivo regeneration studies on neuron-specific NF-κB knockouts are highly informative tools for the imperative search for NF-κB target genes involved in adult neurito-/axonogenesis.
NF-κB and tissue inflammation
NF-κB exerts multifunctional roles in traumatic CNS injury and additional mechanisms, particularly those involved in neuro-glial interactions, contributing to the protective outcome of NF-κB inhibition. As revealed by the suppressed upregulation of pro-inflammatory iNOS and COX-2 in decoy- or pyrrolidine dithiocarbamate-treated rats, traumatic tissue damage triggers the expression of κB-dependent chemokines and cytokines by resident and invading inflammatory cell populations. A prototypical source of inflammatory mediators are primary immune cells, such as neutrophils and macrophages, which rapidly become activated to infiltrate the lesion site. Initially required for primary immune responses and for clearance of cell debris, overactivation of immune cells exacerbates secondary tissue damage. Their contribution to neuronal protection or cell loss can be tested by cell type-specific inhibition of NF-κB. Suppression of IKK/NF-κB-dependent activation in myeloid cell-specific IKK-β knockout (ikkβΔmye) mice significantly reduces the infiltration of CD11b-positive neutrophils and macrophages. While the overall migratory capacity of neutrophils from ikkβΔmye mice appears unaltered, they show a more than 80% reduced expression of the neutrophil-attracting chemokine CXCL1 in the injured SC compared to WT mice (Kang et al., 2011). Consequently, attenuated neutrophil recruitment to the lesion site mitigates the neuroinflammatory response. This, in turn, translates to a reduced neuronal loss and significantly improves recovery of motoric functions at 7 days after SC injury (Kang et al., 2011).
Mechanical tissue damage, as it massively emerges during traumatic injuries, is associated with necrotic cell breakage and intracellular material release. Among the resident cell populations of the CNS, microglia representing the primary immune cells of the brain are highly specialized sensors for cell necrosis. Microglial activation is considered to be a particularly important factor that determines the initiation and severity of the inflammatory response, i.e., by attracting blood-derived immune cells. It has been demonstrated that NF-κB signaling in microglia is critically involved in neuronal death induced by amyloid-β peptide and that constitutive inhibition of microglial NF-κB confers neuroprotection (Chen et al., 2005). Future microglia-focused studies might elucidate NF-κB's role in this resident immune cell of the CNS in spreading unintentional tissue damage following traumatic injury.
Activated astrocytes also contribute to the pro-inflammatory milieu within the lesion site. Astrocyte-specific inhibition of NF-κB by overexpression of non-functional IκBα (GFAP-IκBα-dn) reduces the lesion-induced secretion of CXCL10 and CCL2 at an early time point after injury (Bethea et al., 2005). In these mice, altered cytokine expression is accompanied by a significantly enhanced preservation of the peri-lesional white matter substance and a faster recovery of hind limb function within the first weeks. Thus, in parallel with its role in myeloid cells, activation of NF-κB in astrocytes confers tissue destruction by exacerbating neuroinflammation and secondary tissue damage immediately to the lesion event.
NF-κB and axonal growth inhibition
Additionally, NF-κB in glial cells acts beyond the acute phase of traumatic injury and hampers axonal regeneration for long term. In the context of wound healing, formation of a glial scar substitutes for the damaged tissue, but also constitutes a structural barrier that limits regeneration by permanent exposure of axonal growth inhibitors. As implicated by recent data from different laboratories, it is of the highest significance for regeneration research that the upregulation of such molecules in scar-forming astrocytes and oligodendrocytes (ODCs) is orchestrated by NF-κB. Thereby, expression of the astrocyte-derived chondroitin sulfate proteoglycans (CSPGs) neurocan and phosphocan, both essential constituents of the glial scar, has been analyzed in GFAP-IκBα-dn mice. As expected, CSPGs are stably expressed within the lesion core of WT mice, whereas their expression remains low in transgenic mice at 8 weeks after SC injury (Bethea et al., 2005). While functional improvements are consolidated 2 weeks after the lesion in WT mice, GFAP-IκBα-dn mice show a progressive gain-of-function up to 6 weeks after injury (Bethea et al., 2005). Thus, inhibition of astroglial NF-κB not only improves hind limb function at early time points after injury but also extends the period for functional recovery and delays chronic manifestation of complaints. Anterograde tract tracing by intracortical injection of the Alexa-488-labeled marker streptavidin reveals enhanced axonal sparing and sprouting events of descending axonal tracts caudal to the injury site (Brambilla et al., 2009). Thus, plastic reorganization of circuits involved in locomotion contributes to the improved functional recovery after SC injury in GFAP-IκBα-dn mice. Using the ON crush model, the role of NF-κB in axonal regeneration in terms of axonogenesis into and beyond the lesion site – in the absence of axonal sprouting of spared fibers – can be studied. In line with the concept that NF-κB programs scar tissue towards growth inhibition, translesional regrowth of adult RGCs inversely correlates with the expression of CSPGs in the ON of relAflox;Nes-Cre mice that lack transactivating RelA in the neuro-glial compartment including astrocytes (own data). Therefore, inhibiting NF-κB in scar-forming astrocytes establishes a growth-supporting terrain which enables sprouting of spared axonal fibers and translesional regeneration of newly formed axons.
Expression of κB-dependent growth inhibitors is not restricted to astrocytes. Within the ON and other myelinated fiber tracts, RelA is abundantly expressed in ODCs. While its function in ODC development remains unknown (Kretz et al., 2013), it becomes activated in ODCs after ON injury. Functionally, inhibition of oligodendroglial RelA activation by conditional deletion in relAflox;CNP1-Cre mice enhances RGC regeneration (own data). Among the various growth inhibitors found in CNS white matter, myelin basic protein (MBP) is a likely candidate for RelA-dependent upregulation during constitution of the glial scar. Notably, promotors of the mouse and human MBP gene contain conserved NF-κB cis-regulatory elements that specifically bind RelA and induce MBP expression in oligodendroglioma cells following tumor necrosis factor stimulation (Huang et al., 2002). Within the developing peripheral nervous system, RelA is a pivotal factor for the induction of myelin formation in Schwann cells (Nickols et al., 2003). Moreover, NF-κB inhibition in immature Schwann cells promotes regeneration of the adult nervus facialis, probably via reduced Krox-20-dependent myelin formation and compaction (Morton et al., 2012). Similar mechanisms may be expected for remyelination processes within the post-lesional CNS.
Conclusions
In summary, activation of NF-κB triggers a multicellular and growth-permissive genetic program which causes neuronal death and hampers fiber regeneration and restoration of neuronal networks via short- and long-term impacts. Functionally, the timely regulated induction of axonal growth inhibitors at the lesion site by NF-κB might be indispensable to stabilize the surrounding neuro-architecture, i.e., under disturbed tissue homeostasis associated with wound-healing. However, these molecular breaks in neuronal plasticity are unfavorable for axonal regeneration, particularly after the acute phase of the injury has declined. Overall, it is an exciting concept to consider NF-κB as a master regulator of post-lesional gene transcription which orchestrates the cell type-specific expression of neurotoxic and growth inhibitory factors by affected neurons, astrocytes, ODCs, and microglia (Figure 1). While inhibition of NF-κB might hold clinical relevance, the contribution of this transcription factor to reprograming a growth-permissive environment is highly subunit-specific. This is illustrated by injury studies on constitutive p50 knockout mice, in which neuronal survival and axonal regeneration are completely abolished (own data). This might be attributable to the pivotal function of p50 for neuro-axonal maintenance, wherein p50 acts as a permanent transcriptional repressor of Jagged1, a ligand for the neuritogenesis inhibitor Notch (Bonini et al., 2011), and the pro-apoptotic Bcl2-associated X protein (Nakamura-Yanagidaira et al., 2011).
Figure 1.
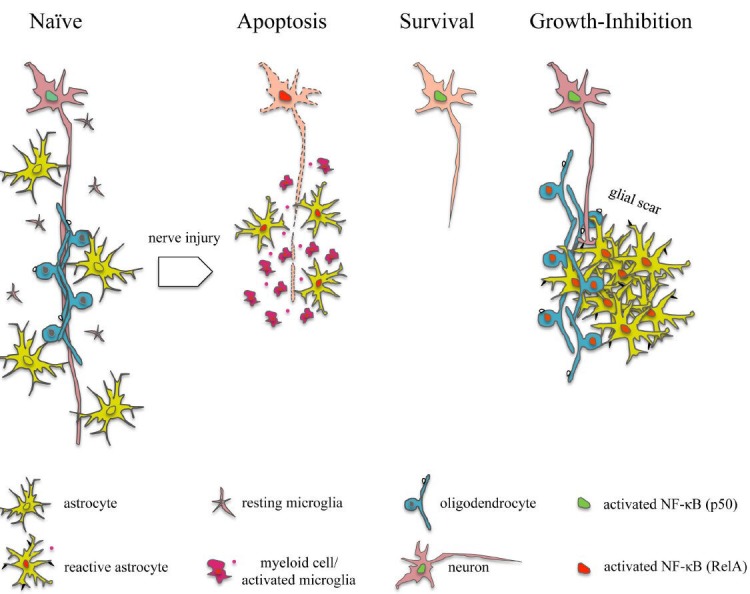
Schematic representation of nuclear factor kappaB (NF-êB) subunit and cell type-specific activation patterns in traumatic nerve injury.
In naïve neurons, p50 is required for survival and axonal maintenance. Nerve injury induces activation of RelA in associated neuronal soma, thereby promoting apoptosis. Shifting the balance between RelA and p50 towards p50, e.g., by conditional deletion of RelA promotes neuronal survival. During the primary phase of injury, RelA likewise becomes activated in myeloid cells/activated microglia and astrocytes at the lesion site. RelA-dependent secretion of pro-inflammatory cytokines and chemokines (red dots) exacerbates neuronal damage. During wound healing and glial scar formation, activation of RelA in astrocytes and oligodendrocytes contributes to the (re-)expression of membrane-associated growth inhibitors (black symbols), thus leading to permanent growth-repulsion of regenerating axons.
In the past, administration of the immune-suppressive drug methylprednisolone at high dose has been the only pharmacological therapy in acute SC injury (Bracken, 2012). The treatment does not, however, allow for full restoration of locomotor movement. Moreover, recent clinical guidelines do not include the recommendation of this drug anymore because high-dose steroids are associated with harmful advers effects, thus further emphasizing the need for more targeted therapies (Hurlbert et al., 2013). With respect to its subunit and cell type-specific functions in axonal injury, pharmacological intervention of the NF-κB pathway to stimulate axonal regeneration can be envisaged for the future. Given its dual properties for neuronal survival, global interference with NF-κB seems unfavourable but requests for specific inhibition of RelA-dependent expression of pro-apoptotic, pro-inflammatory and growth-inhibitory mediators. This might be achieved by antibody therapy targeting the nuclear localization sequence or the phosphorylation site of RelA, both are required to gain its full transcriptional activity. Further, RelA-mediated gene expression can be reduced by activation of the deacatylase sirtuin 1, which targets RelA-Lys310 for deacetylation. Importantly, sirtuin 1 activity is stimulated by the drug resveratrol and post-lesional application of this compound confers neuroprotection in experimental stroke therapy (Lanzillotta et al., 2012). These results indicate that epigenetic therapy of axonal injury might be of great clinical relevance for the future.
Footnotes
Funding: This work was supported by the Leibniz Association, Germany, and the VELUX Foundation, Switzerland.
Conflicts of interest: None declared.
References
- 1.Bethea JR, Bracchi-Ricard V, Brambilla R. Inhibition of astroglial NF-kappa B reduces inflammation and improves functional recovery following spinal cord injury and EAE. Immunology. 2005;116:86–86. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bethea JR, Castro M, Keane RW, Lee TT, Dietrich WD, Yezierski RP. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci. 1998;18:3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J Neurosci. 2002;22:8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonini SA, Ferrari-Toninelli G, Uberti D, Montinaro M, Buizza L, Lanni C, Grilli M, Memo M. Nuclear factor kappaB-dependent neurite remodeling is mediated by Notch pathway. J Neurosci. 2011;31:11697–11705. doi: 10.1523/JNEUROSCI.1113-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bracken MB. Steroids for acute spinal cord injury. Cochrane Database Syst Rev. 2002;(3):CD001046. doi: 10.1002/14651858.CD001046. [DOI] [PubMed] [Google Scholar]
- 6.Brambilla R, Hurtado A, Persaud T, Esham K, Pearse DD, Oudega M, Bethea JR. Transgenic inhibition of astroglial NF-kappa B leads to increased axonal sparing and sprouting following spinal cord injury. J Neurochem. 2009;110:765–778. doi: 10.1111/j.1471-4159.2009.06190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- 8.Choi JS, Sungjoo KY, Joo CK. NF-kappa B activation following optic nerve transection. Korean J Ophthalmol. 1998;12:19–24. doi: 10.3341/kjo.1998.12.1.19. [DOI] [PubMed] [Google Scholar]
- 9.Choi JS, Kim JA, Kim DH, Chun MH, Gwag BJ, Yoon SK, Joo CK. Failure to activate NF-kappaB promotes apoptosis of retinal ganglion cells following optic nerve transection. Brain Res. 2000;883:60–68. doi: 10.1016/s0006-8993(00)02886-9. [DOI] [PubMed] [Google Scholar]
- 10.Gavalda N, Gutierrez H, Davies AM. Developmental switch in NF-kappaB signalling required for neurite growth. Development. 2009;136:3405–3412. doi: 10.1242/dev.035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez H, Davies AM. Regulation of neural process growth, elaboration and structural plasticity by NF-kappaB. Trends Neurosci. 2011;34:316–325. doi: 10.1016/j.tins.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutierrez H, O’Keeffe GW, Gavalda N, Gallagher D, Davies AM. Nuclear factor kappa B signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J Neurosci. 2008;28:8246–8256. doi: 10.1523/JNEUROSCI.1941-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang CJ, Nazarian R, Lee J, Zhao PM, Espinosa-Jeffrey A, de Vellis J. Tumor necrosis factor modulates transcription of myelin basic protein gene through nuclear factor kappa B in a human oligodendroglioma cell line. Int J Dev Neurosci. 2002;20:289–296. doi: 10.1016/s0736-5748(02)00022-9. [DOI] [PubMed] [Google Scholar]
- 14.Hurlbert RJ, Hadley MN, Walters BC, Aarabi B, Dhall SS, Gelb DE, Rozzelle CJ, Ryken TC, Theodore N. Pharmacological therapy for acute spinal cord injury. Neurosurgery. 2013;72(Suppl 2):93–105. doi: 10.1227/NEU.0b013e31827765c6. [DOI] [PubMed] [Google Scholar]
- 15.Imielski Y, Schwamborn JC, Luningschror P, Heimann P, Holzberg M, Werner H, Leske O, Puschel AW, Memet S, Heumann R, Israel A, Kaltschmidt C, Kaltschmidt B. Regrowing the adult brain: NF-kappaB controls functional circuit formation and tissue homeostasis in the dentate gyrus. PLoS One. 2012;7:e30838. doi: 10.1371/journal.pone.0030838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inta I, Paxian S, Maegele I, Zhang W, Pizzi M, Spano P, Sarnico I, Muhammad S, Herrmann O, Inta D, Baumann B, Liou HC, Schmid RM, Schwaninger M. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappaB subunit RelA in cerebral ischemia. J Neurosci. 2006;26:12896–12903. doi: 10.1523/JNEUROSCI.3670-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jimenez-Garza O, Camacho J, Ibarra A, Martinez A, Guizar-Sahagun G. Early effects of modulating nuclear factor-kappaB activation on traumatic spinal cord injury in rats. Ann N Y Acad Sci. 2006;1053:148–150. doi: 10.1196/annals.1344.012. [DOI] [PubMed] [Google Scholar]
- 18.Kang J, Jiang MH, Min HJ, Jo EK, Lee S, Karin M, Yune TY, Lee SJ. IKK-beta-mediated myeloid cell activation exacerbates inflammation and inhibits recovery after spinal cord injury. Eur J Immunol. 2011;41:1266–1277. doi: 10.1002/eji.201040582. [DOI] [PubMed] [Google Scholar]
- 19.Konig HG, Fenner BJ, Byrne JC, Schwamborn RF, Bernas T, Jefferies CA, Prehn JH. Fibroblast growth factor homologous factor 1 interacts with NEMO to regulate NF-kappaB signaling in neurons. J Cell Sci. 2012;125:6058–6070. doi: 10.1242/jcs.111880. [DOI] [PubMed] [Google Scholar]
- 20.Kretz A, Herrmann KH, Fischer S, Engelmann C, Witte OW, Reichenbach JR, Weih F, Haenold R. Dysfunctional NF-kappaB and brain myelin formation. Eur J Hum Genet, 2013. 2013 Oct 30; doi: 10.1038/ejhg.2013.240. doi: 101038/ejhg2013240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanzillotta A, Pignataro G, Branca C, Cuomo O, Sarnico I, Benarese M, Annunziato L, Spano P, Pizzi M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol Dis. 2012;49C:177–189. doi: 10.1016/j.nbd.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Lu Z, Li WL, Yu SP, Wei L. Cell death and proliferation in NF-kappaB p50 knockout mouse after cerebral ischemia. Brain Res. 2008;1230:281–289. doi: 10.1016/j.brainres.2008.06.130. [DOI] [PubMed] [Google Scholar]
- 23.Listwak SJ, Rathore P, Herkenham M. Minimal NF-kappaB activity in neurons. Neuroscience. 2013;250:282–299. doi: 10.1016/j.neuroscience.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKelvey L, Gutierrez H, Nocentini G, Crampton SJ, Davies AM, Riccardi CR, O’Keeffe G W. The intracellular portion of GITR enhances NGF-promoted neurite growth through an inverse modulation of Erk and NF-kappaB signalling. Biol open. 2012;1:1016–1023. doi: 10.1242/bio.20121024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morton PD, Johnstone JT, Ramos AY, Liebl DJ, Bunge MB, Bethea JR. Nuclear factor-kappaB activation in Schwann cells regulates regeneration and remyelination. Glia. 2012;60:639–650. doi: 10.1002/glia.22297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura-Yanagidaira T, Takahashi Y, Sano K, Murata T, Hayashi T. Development of spontaneous neuropathy in NF-kappaBp50-deficient mice by calcineurin-signal involving impaired NF-kappaB activation. Mol Vis. 2011;17:2157–2170. [PMC free article] [PubMed] [Google Scholar]
- 27.Nickols JC, Valentine W, Kanwal S, Carter BD. Activation of the transcription factor NF-kappaB in Schwann cells is required for peripheral myelin formation. Nat Neurosci. 2003;6:161–167. doi: 10.1038/nn995. [DOI] [PubMed] [Google Scholar]
- 28.Potrovita I, Zhang W, Burkly L, Hahm K, Lincecum J, Wang MZ, Maurer MH, Rossner M, Schneider A, Schwaninger M. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J Neurosci. 2004;24:8237–8244. doi: 10.1523/JNEUROSCI.1089-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pradillo JM, Romera C, Hurtado O, Cardenas A, Moro MA, Leza JC, Davalos A, Castillo J, Lorenzo P, Lizasoain I. TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:193–203. doi: 10.1038/sj.jcbfm.9600019. [DOI] [PubMed] [Google Scholar]
- 30.Rafati DS, Geissler K, Johnson K, Unabia G, Hulsebosch C, Nesic-Taylor O, Perez-Polo JR. Nuclear factor-kappaB decoy amelioration of spinal cord injury-induced inflammation and behavior outcomes. J Neurosci Res. 2008;86:566–580. doi: 10.1002/jnr.21508. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt-Ullrich R, Memet S, Lilienbaum A, Feuillard J, Raphael M, Israel A. NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development. 2008;122:2117–2128. doi: 10.1242/dev.122.7.2117. [DOI] [PubMed] [Google Scholar]
- 32.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 33.Wellmann H, Kaltschmidt B, Kaltschmidt C. Retrograde transport of transcription factor NF-kappa B in living neurons. J Biol Chem. 2001;276:11821–11829. doi: 10.1074/jbc.M009253200. [DOI] [PubMed] [Google Scholar]