

Abstract
Transforming growth factor-beta (TGF-β) type II receptor (TβRII) levels are extremely low in the brain tissue of patients with Alzheimer's disease. This receptor inhibits TGF-β1/SMAD signaling and thereby aggravates amyolid-beta deposition and neuronal injury. Dab2, a specific adapter protein, protects TβRII from degradation and ensures the effective conduction of TGF-β1/SMAD signaling. In this study, we used an adenoviral vector to overexpress the Dab2 gene in the mouse hippocampus and investigated the regulatory effect of Dab2 protein on TGF-β1/SMAD signaling in a mouse model of Alzheimer's disease, and the potential neuroprotective effect. The results showed that the TβRII level was lower in APP/PS1 mouse hippocampus than in normal mouse hippocampus. After Dab2 expression, hippocampal TβRII and p-SMAD2/3 levels were significantly increased, while amyloid-beta deposition, microglia activation, tumor necrosis factor-α and interleulin-6 levels and neuronal loss were significantly attenuated in APP/PS1 mouse brain tissue. These results suggest that Dab2 can exhibit neuroprotective effects in Alzheimer's disease by regulating TGF-β1/SMAD signaling.
Keywords: nerve regeneration, transforming growth factor-β1, Dab2, Alzheimer's disease, amyolid-beta, neuron, SMAD2, SMAD3, microglia, neural regeneration
Introduction
Alzheimer's disease is not only the most common neurodegenerative disease, but also the most common cause of dementia, and its prevalence increases with age, affecting approximately 4.4% of the population aged ≥ 65 years[1,2,3,4]. Owing to a lack of understanding of Alzheimer's disease pathogenesis and treatment, there is no effective method to reverse or prevent disease progression[5]. Therefore, recognizing amyloid-beta protein generation and the potential molecular regulatory mechanism underlying the produced neurotoxicity is an important step toward developing novel drugs for Alzheimer's disease[6].
The transforming growth factor-β superfamily mainly consists of transforming growth factor-β1, -β2 and -β3 subtypes[7,8]. Transforming growth factor-ββ exhibits a variety of biological effects, and participates widely in diverse pathophysiological processes, including cell apoptosis and proliferation, stem cell differentiation and embryonic development, extracellular matrix formation, wound repair and inflammatory reactions[9,10,11,12]. Transforming growth factor-β1 is the most important subtype in the central nervous system, being mainly expressed in neurons and microglia, and its level can be increased by stress after nerve injury. Accumulating evidence has demonstrated that transforming growth factor-β1, as a pleiotropic cytokine, exhibits important neuroprotective effects. In invertebrates, transforming growth factor-β1 can regulate synaptic growth and differentiation, neurotransmitter release and synaptic protein distribution and phosphorylation[13].
The Tgfb1–/+ transgenic mouse exhibits a transforming growth factor-β1 mRNA level that is only 50% of the normal level and, compared with normal mice, nerve cells in Tgfb1–/+ transgenic mice show an increased susceptibility to age and excitotoxins[14]. Tgfb1–/– transgenic mice show a wide range of degenerative diseases of the nervous system, accompanied by decreased levels of laminin and synaptophysin and microglial proliferation[14]. Transforming growth factor-β1 exhibits a synergic effect together with many neurotrophic factors, including nerve growth factor, brain-derived nerve growth, and neurotrophin-3, 4, maintains the growth of nerve cells, and protects them from injury[15].
Transforming growth factor-β1 is closely related to Alzheimer's disease and amyloid-beta. The level of transforming growth factor-β1 in the brain and cerebrospinal fluid is increased, but its level in the serum is lower in Alzheimer's disease patients than in unaffected people[16,17]. Wyss-Coray et al.[18,19] reported that, in APP transgenic mice, transforming growth factor-β1 overexpression in microglia can attenuate amyloid-beta deposition in the hippocampus; moreover, transforming growth factor-β1 can also promote amyloid-beta phagocytosis and degradation in vitro. In addition to its effects on amyloid-beta metabolism, transforming growth factor-β1 can protect neurons from amyloid-beta-induced neurotoxicity[20]. Because transforming growth factor-β1/SMADs are inhibited in multiple sites, the neuroprotective effect of transforming growth factor-β1 on Alzheimer's disease cannot function normally[21]. In Alzheimer's disease patients, the TβRII level is decreased and is related to the severity of Alzheimer's disease. Age-related neuronal degeneration and hippocampal amyloid-beta deposition appear in TβRII knockout mice[22]. Lee et al.[23] found that SMAD2 could not be transported into nucleus after phosphorylation, resulting in abnormal intracellular aggregation of phosphorylated SMAD2 (phospho-SMAD2, p-SMAD2) molecules; this abnormal aggregation of p-SMAD2 is related to neurofibrillary tangles and granulovacuolar degeneration. Therefore, ensuring normal conduction of transforming growth factor-β1/SMAD signaling has become a problem to be solved in the treatment of Alzheimer's disease using transforming growth factor-ββ1[24,25].
TβRII expression is regulated by many factors, including microRNAs, ubiquitination, and membrane transport. In a previous study by Wang et al.[26], in APP/PS1 transgenic mice, hippocampal TββRII expression was found to be regulated by microRNA-106b; specifically, microRNA-106b aggravated amyloid-beta-induced neurotoxicity by downregulating the TβRII level. Dab2, a specific adapter protein, can protect TββRII from degradation in vivo[27]. A previous study has reported that Dab2 expression is downregulated in Alzheimer's disease mice[28]. Therefore, it is presumed that loss of Dab2 can lead to downregulation of TββRII expression, block the normal conduction of transforming growth factor-β1/SMAD signaling in Alzheimer's disease, and thereby accelerate the progression of Alzheimer's disease. To test this hypothesis, we used an adenoviral vector to overexpress Dab2 in the APP/PS1 transgenic mouse model of Alzheimer's disease, and investigated the regulatory effect of Dab2 protein on transforming growth factor-β1/SMAD signaling and brain injury in Alzheimer's disease.
Results
Quantitative analysis of experimental animals
Ten BL6/C57 mice were used as negative controls receiving no treatments. Twenty-four APP/PS1 transgenic mice were randomly and evenly divided into Alzheimer's disease, vector and Dab2 groups. The rats in these groups were administered physiological saline, adenovirus-associated virus (AAV)9-RFP plasmids and Dab2 plasmids, respectively, via bilateral hippocampi. In each group, mice at the age of 9 months underwent behavioral tests. All mice were sacrificed at 10 months of age and molecular biological and immunological tests were performed as shown in Figure 1A. No mice died throughout the entire experiment.
Figure 1.
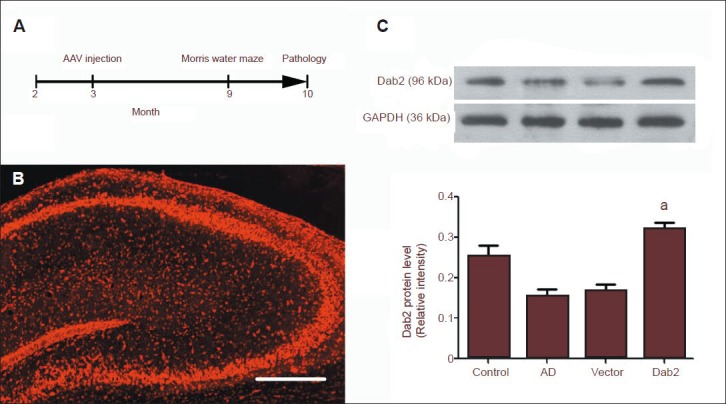
Effect of Dab2 plasmid injection on hippocampal Dab2 expression.
(A) Mice in each group received hippocampal injection of AAV9 plasmid at 3 months of age, underwent behavioral tests at 9 months and were sacrificed at 10 months for molecular biological and pathological tests. (B) By fluorescence microscopy, AAV9 plasmids can be seen to be expressed in the mouse hippocampus, showing red fluorescence. Scale bar: 100 μm. (C) The hippocampal Dab2 level in the Alzheimer's disease group was significantly lower than that in the conrol group, and the hippocampal Dab2 level in the Dab2 group was significantly higher than that in the vector group (aP < 0.01). One-way analysis of variance and the SNK-q test were used to assess the significance of differences between groups. Data are represented as mean ± SEM from at least three independent experiments. The Dab2 level is expressed as the ratio of the target protein absorbance value to that for GAPDH.
Dab2 plasmid injection increased hippocampal Dab2 protein expression
To test whether the Dab2 plasmid is expressed in mouse hippocampus, we observed hippocampal slices under a fluorescence microscope. Expression of the Dab2 plasmid carrying RFP was observed in hippocampal slices (Figure 1B). Western blot analysis showed that the hippocampal Dab2 level was significantly lower in the Alzheimer's disease group than in the control group. After hippocampal injection of AAV9-RFP plasmid (vector group), the hippocampal Dab2 level was similar to that in the Alzheimer's disease group. The hippocampal Dab2 level was significantly higher in the Dab2 group, into which the AAV9-CMV-Dab2 plasmid was injected, than in the vector group (P < 0.01; Figure 1C). These results indicate that AAV can be used as a safe expression vector to overexpress Dab2 gene in the central nervous system.
Dab2 regulated mouse hippocampal transforming growth factor-β1/SMAD signal transduction
To investigate the regulatory effect of Dab2 on hippocampal transforming growth factor-β1/SMAD signal transduction, we detected hippocampal TβRII and phosphorylated SMAD2/5 levels by western blotting. The results showed that the hippocampal TβRII level was significantly lower, while the pSMAD2/3 levels were significantly increased in the Alzheimer's disease group compared with the control group. These findings are consistent with those reported by Tesseur et al.[22] (Figure 2A, B). TβRII and pSMAD2/3 levels in the Dab2 group were significantly higher than in the vector group (Figure 2).
Figure 2.
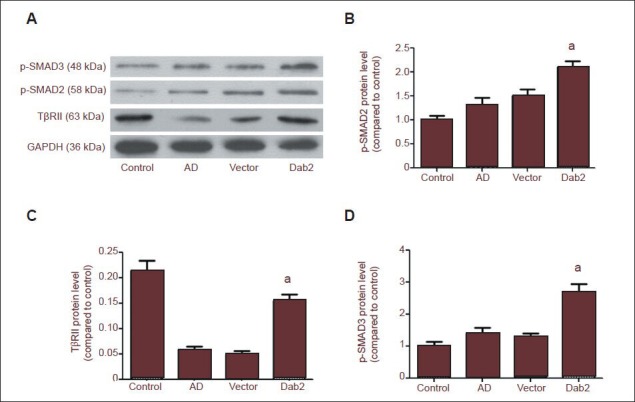
Effect of Dab2 overexpression on hippocampal transforming growth factor-β1 (TGF-β1)/SMAD signaling in Alzheimer's disease mice.
(A) Western blot analysis of hippocampal TβRII and phosphorylated SMAD2/3 levels. (B–D) Hippocampal TβRII and phosphorylated SMAD2/3 levels were significantly higher in the Dab2 group than in the vector group. One-way analysis of variance and the SNK-q test were used to assess the significance of differences between groups. Data are represented as mean ± SEM from at least three independent experiments. TβRII: TGF-β II receptor.
Dab2 overexpression improved the abnormal behaviors of Alzheimer's disease mice
Morris water maze test results showed that the escape latency and swimming distance of mice in the Dab2 group were significantly shorter than those in the vector group (P = 0.039, 0.039; Figure 3A, B). These results demonstrate that learning ability in the Dab2 group was significantly better than that in the vector group. After removal of the platform, the percentage of time that mice spent in quadrant A was significantly greater in the Dab2 group than in the vector group (P = 0.035; Figure 3C).
Figure 3.
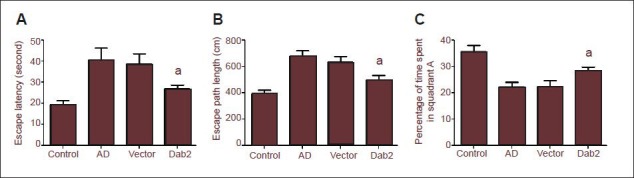
Effect of Dab2 overexpression on the learning and memory abilities of Alzheimer's disease (AD) mice as shown by the Morris water maze test.
(A, B) The escape latency and swimming distance of mice in the Dab2 group were significantly shorter than those of mice in the vector group (aP < 0.05). (C) After removal of the platform, the percentage of time that mice spent in quadrant A was significantly higher in the Dab2 group than in the vector group (aP < 0.05). One-way analysis of variance and the SNK-q test were used to assess the significance of differences between groups. Data are represented as mean ± SEM from at least three independent experiments.
Dab2 overexpression decreased the amyloid-beta level in brain tissue
ELISA results showed that the levels of dissoluble and indissoluble amyloid-beta40 and amyloid-beta42 in mouse brain tissue were not significantly different between the Alzheimer's disease group and the vector group (P > 0.05). Soluble amyloid-beta40 and amyloid-beta42 levels were significantly lower in the Dab2 group than in the vector group (P < 0.05; Figure 4). These results demonstrated that Dab2 can attenuate the neurotoxic effects of dissoluble amyloid-beta on nerve cells and amyloid-beta deposition in brain tissue.
Figure 4.
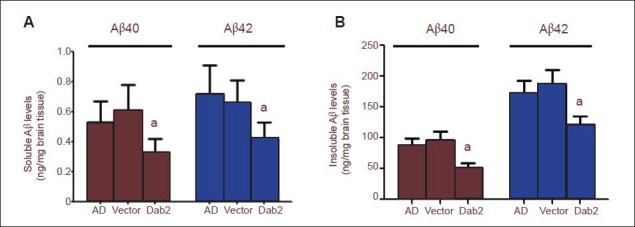
Effects of Dab2 overexpression on amyloid-beta (Aβ) level in the brain tissue of Alzheimer's disease (AD) mice.
The levels of soluble (A) and insoluble Aβ40, 42 (B) in the Dab2 group were significantly lower than those in the Alzheimer's disease group and vector group (aP < 0.05). One-way analysis of variance and the SNK-q test were used to assess the significance of differences between groups. Data are represented as mean ± SEM from at least three independent experiments.
Dab2 reduced microglial activation and inflammatory factor expression in the hippocampi of Alzheimer's disease mice
Through immunofluorescence staining, Ibal-positive microglia with irregular morphology and many neurites were observed in the hippocampi of mice in the Dab2, vector and Alzheimer's disease groups (Figure 5A). Under 100-fold magnification, the number of Iba1-positive microglia was significantly lower in the Dab2 group than in the vector group (P = 0.038) and Alzheimer's disease group (P = 0.019). There was no significant difference in numbers of Iba1-positive cells between the vector group and the Alzheimer's disease group (P = 0.904; Figure 5B).
Figure 5.
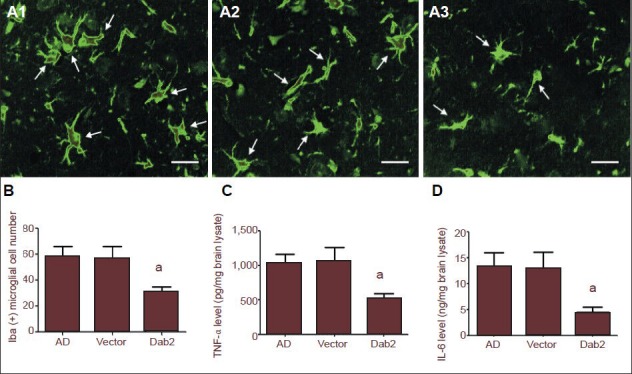
Effects of Dab2 overexpression on microglia activation and inflammatory factor levels in the hippocampi of Alzheimer's disease (AD) mice.
(A1–A3) By fluorescence microscopy, Iba1-positive microglia (arrows, green) with irregular morphology and many neurites were observed in the hippocampi of Alzheimer's disease mice. Scale bars: 10 μm. (B) The number of Iba1-positive microglia was significantly lower in the Dab2 group than in the vector and AD groups (aP < 0.05). (C, D) Tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) levels were significantly lower in the Dab2 group than in the vector and AD groups (aP < 0.05). One-way analysis of variance and the SNK-q test were used to assess the significance of differences between groups. Data are represented as mean ± SEM from at least three independent experiments.
To investigate the effect of Dab2 on inflammatory reactions in the brain tissue of Alzheimer's disease mice, we detected hippocampal tumor necrosis factor-α and interleukin-6 levels by ELISA. The results showed that hippocampal tumor necrosis factor-α and interleukin-6 levels were significantly lower in the Dab2 group than in the vector group (P = 0.029, 0.040), and there were no significant differences in tumor necrosis factor-α and interleukin-6 levels between the vector group and the Alzheimer's disease group (P = 0.886, 0.883; Figure 5D). These results suggest that Dab2 overexpression can reduce hippocampal microglia activation and inflammatory factor release in Alzheimer's disease mice, thereby providing anti-inflammatory effects.
Dab2 inhibited hippocampal neuronal loss
To investigate the effects of Dab2 on hippocampal neurons in Alzheimer's disease mice, we detected hippocampal neuron-specific enolase-positive cells by immunofluorescence staining. The results showed that, in the normal mouse hippocampal tissue, neurons were arranged in an orderly, dense fashion and the cytoplasm was stained green. Compared with normal mice, different degrees of neuronal loss appeared in the hippocampus tissue of mice in the Alzheimer's disease, vector and Dab2 groups (Figure 6A). Under 400-fold magnification, significantly more neuron-specific enolase-positive cells were seen in the Dab2 group than in the vector group (P = 0.035), and there were no significant differences between the Alzheimer's disease and vector groups (P = 0.842; Figure 6B). These results suggest that Dab2 overexpression can attenuate pathogenic factor-induced mouse hippocampal neuronal loss.
Figure 6.
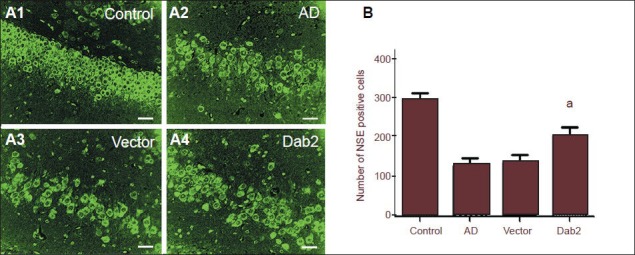
Effect of Dab2 overexpression on hippocampal neurons in Alzheimer's disease (AD) mice.
(A1–A4) By fluorescence microscopy, neuron-specific enolase (NSE)-positive cells exhibited green fluorescence in the cytoplasm. Hippocampal neurons are arranged compactly and orderly in the control group. Hippocampal neurons were reduced in number and arranged in a disorderly fashion in the AD and vector groups. The level of reduction in hippocampal neurons was lower in the Dab2 group compared with the AD group. Scale bars: 10 μm. (B) The number of hippocampal NSE-positive cells in the Dab2 group was significantly higher than the numbers in the vector and AD groups (aP < 0.05). One-way analysis of variance and the SNK-q test were used to assess the significance of differences between groups. Data are represented as mean ± SEM from at least three independent experiments.
Discussion
AAV is a defective, non-pathogenic human parvovirus with a length of approximately 4.7 kb, and, after recombination, the AAV vector retains only 4% of the original viral genes[29,30,31]. Compared with other viral vectors, such as adenovirus and lentivirus, AAV vector is highly safe, has a wide range of host cells, and can be used to express exogenous genes for a long time period in vivo[32,33]. AAV9 is an important AAV vector subtype. Numerous studies have documented that, via direct injection, the genes carried by AAV9 vectors are highly expressed in hippocampal nerve cells and, therefore, the vector can be used for Alzheimer's disease studies[34]. The confocal laser scanning microscopy results in this study showed that, 3 months after AAV9 plasmid injection, RFP expression was still very high in the mouse hippocampus, indicating that AAV9 vectors express exogenous genes in brain tissue for a long time period. However, 7 months after AAV9 plasmid injection, Dab2 protein level was higher in the Dab2 group than in the Alzheimer's disease and vector groups, as shown by western blot analysis. The Dab2 gene carried by AAV9 plasmid can be expressed for a long period of time, which is required by many experiments.
Transforming growth factor-β1 binds to the cell membrane surface receptor TβRII, leading to a conformational change in TβRII, and then the complex binds to a conserved domain enriched in serine and glycine in the TβRI cytoplasmic region, namely, the GS domain, to form a TβRII-TGFβ-TβRI complex[35]. Then, TβRII autophosphorylates and is subsequently activated; thus, TβRI can specifically identify SMAD2/3 protein, phosphorylates it (to form p-SMAD2/3) and then mediates an intracellular signaling cascade[36,37]. A previous study reported that the TβRII level is extremely low in the hippocampus of Alzheimer's disease patients, which hinders the effective conduction of transforming growth factor-β/SMAD signaling, and, after TββRII knockout, a large amount of amyloid-beta deposition is seen in the mouse hippocampus, which accelerates neuronal aging[22]. The results of the present study showed that TβRII expression in the brain tissue of APP/PS1 mice was significantly lower than that in wild-type mice, consistent with the results of a previous report. Our results also showed that, although TβRII expression is decreased, the p-SMAD2/3 level in brain tissue was significantly higher in APP/PS1 mice than in wild-type mice. Combined with previous findings, these findings suggest that, although the p-SMAD2/3 level is increased, the expression of genes involved in intranuclear regulation is blocked; therefore, transforming growth factor-β1/SMAD signal transduction in the brains of APP/PS1 mice is inhibited.
Because TβRII is a key factor in transforming growth factor-β1/SMAD signal transduction and the progression of Alzheimer's disease, understanding the regulation of TβRII expression in an Alzheimer's disease model can help clarify the pathological mechanism of Alzheimer's disease and aid the development of drugs for treating Alzheimer's disease in the clinic. The TβRII post-transcriptional level is affected by various factors, and TββRII membrane transport is an important one[38,39]. Under the physiological or pathological state, TβRII receptors on the cell membrane surface are included in intracellular early endosome antigen 1 [EEA1(+)] by endocytosis, and some intracellular TβRII receptors are recycled to the membrane surface. The remainder are directly degraded in cells, thereby regulating the TβRII level and transforming growth factor-β1/SMAD signaling transduction[39,40]. Dab2, a member of the Dab protein family, mainly regulates the endocytosis of cell surface proteins and is closely related to cellular adhesion and proliferation as well as actin formation[27]. A previous study has shown that Dab2 expression is decreased in Alzheimer's disease mice[28]. Our results also confirm that Dab2 expression is downregulated in the brain tissue of APP/PS1 mice. It is presumed that this decrease in Dab2 expression likely causes a TβRII circulatory disorder and subsequent degradation, leading to a decrease in TβRII level on the cellular membrane surface and blocking of transforming growth factor-β1/SMAD signaling transduction. Our results showed that Dab2 overexpression in hippocampal tissue leads to increased TβRII and p-SMAD2/3 levels, indicating that Dab2 can indeed regulate transforming growth factor-β1/SMAD signal transduction in a mouse model of Alzheimer's disease, exhibiting a neuroprotective effect.
To investigate the neuroprotective effect of Dab2, we further detected its regulatory effect on amyloid-beta levels and neurotoxicity. Amyloid-beta is an initial factor and a key link in the pathological mechanism of Alzheimer's disease[5]. Amyloid-beta overexpression and abnormal accumulation in brain tissue can destroy intracellular calcium homeostasis, lead to excessive oxygen free radical formation, activate inflammatory factors, cause inflammation of focal brain tissue, lead to synapse reduction and neuronal loss, and finally result in an abnormal integrative function of the central nervous system[41,42,43]. There is strong evidence that transforming growth factor-β1 signaling shows an inhibitory effect on either amyloid-beta generation or neurotoxicity[21,44]. The results from this study showed that, when Dab2 protein was overexpressed in the hippocampal tissue of Alzheimer's disease mice, both soluble and insoluble amyloid-betaβ levels are reduced, indicating that Dab2 can reduce amyloid-beta generation. Amyloid-betaβ can induce neuronal apoptosis by abnormally activating the cell cycle, and is closely related to late-state degradation of β-catenin and excessive Tau phosphorylation[45,46,47]. After binding to TβRII, transforming growth factor-β1 activates components of the SMAD and non-SMAD pathways, and thereby inhibits the abnormal activation of the cell cycle[21]. In this study, immunofluorescence analysis revealed a significant reduction in the number of cells expressing neuron-specific enolase in the hippocampi of Alzheimer's disease mice, while the numbers of neurons overexpressing Dab2 were not significantly reduced. Based on this and previous findings, we consider that Dab2 can regulate transforming growth factor-β1/SMAD signaling transduction by increasing the numbers of cell membrane surface TβRII receptors, and thereby attenuates amyloid-beta generation and neurotoxic effects on nerve cells. In this study, we also performed a Morris water maze test, which is a means for studying the mechanism underlying cerebral learning and memory abilities and is widely used in the study of Alzheimer's disease[48,49]. The results from this study showed that, compared with the vector group, escape latency and swimming distance were significantly shortened, and the time spent within the quadrant with the platform removed was significantly prolonged, in the Dab2 group, suggesting that Dab2 overexpression can improve the memory ability and spatial orientation ability of Alzheimer's disease mice, verifying the neuroprotective effect of Dab2.
More attention has been paid to the role of inflammatory reactions in the occurrence of Alzheimer's disease. Increasing evidence has demonstrated that a chronic inflammatory reaction consistently exists in the brain tissue of Alzheimer's disease patients and is possibly a factor involved in inducing the formation and development of other pathological characteristics[50,51,52]. Transforming growth factor-β1, as an inflammatory regulator, can inhibit inflammatory reactions of the central nervous system and microglial activation[14,53]. In various neurodegenerative diseases, inhibition of transforming growth factor-β1/SMAD signaling is related to T cells and microglia-mediated local inflammation[14,54]. Results from this study showed that the numbers of activated microglia and tumor necrosis factor-α and interleukin-6 levels in the brain tissue of Alzheimer's disease mice were significantly greater than in common mice. After Dab2 overexpression in the brain tissue of Alzheimer's disease mice, the number of activated microglia was significantly decreased, and tumor necrosis factor-α and interleukin-6 levels were also significantly decreased compared with the levels in the negative control group. These results indicate that Dab2 can alleviate the inflammatory reaction in the brain tissue of Alzheimer's disease mice. A previous study has confirmed that Dab2 can inhibit inflammatory reactions by regulating nuclear factor-κB signaling[55]. Therefore, we considered that Dab2 inhibits inflammatory reactions in the brain tissue of Alzheimer's disease mice possibly not only by regulating transforming growth factor-β1/SMAD signaling, but also by other mechanisms, such as inhibition of nuclear factor-κB signaling.
In conclusion, we performed Dab2 overexpression in the brain tissue of Alzheimer's disease mice and investigated the role of Dab2 protein in the pathological mechanism of Alzheimer's disease. The results showed that Dab2 protein can regulate transforming growth factor-β1/SMAD signal transduction in the brain tissue of Alzheimer's disease mice and can inhibit amyloid-beta generation and deposition, inflammatory reactions and neuronal loss by enhancing this signal transduction. This study was designed to further clarify the role of transforming growth factor-β1/SMAD signaling in the pathophysiological process of Alzheimer's disease and provide new directions for clinical treatments for Alzheimer's disease.
Materials and Methods
Design
A randomized, controlled molecular biology trial.
Time and setting
This study was performed in the Laboratory of Immunology, Centre for Translational Medicine, First Hospital of Jilin University Bethune Medical Center, China from May 2012 to February 2013.
Materials
Animals
Twenty-four 2-month-old male APP/PS1 double transgenic mice and ten negative control 2-month-old BL6/C57 mice (5 male and 5 female) were purchased from the Animal Research Institute, Nanjing University, China. All animals were housed separately in a 21 ± 2°C and 30–35% relative humidity environment with artificial 12-hour illumination. There was no difference in exposure factors between groups. The experimental protocol was approved by the Animal Ethics Committee of Jilin University, China. After raising for 1 month, animals were used for further experiments.
Plasmids
AAV9-Dab2 plasmids and negative control AAV9-RFP plasmids were synthesized by Jikai Gene Chemical Technology Co., Ltd. (Shanghai, China).
Methods
Hippocampal injection of plasmids
Plasmids were injected into the mouse hippocampus according to a previously described method[56]. After anesthesia by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (20 mg/kg), 3-month-old mice were fixed in a small animal stereotaxic instrument (Ruiwode Life Science Co., Ltd., Shenzhen, China) and the skin was disinfected with 75% alcohol. Physiological saline or AAV plasmid (1 × 109 viral particles (VP)) was injected into the hippocampus via a needle inserted at a depth of 1.8 mm (Hamilton Co, Reno, NV, USA), 2.1 mm posterior to and 1.8 mm left and right lateral to the bregma, 2 µL each side. After injection, the needle was slowly withdrawn, the drill hole was sealed and the skin was sutured; then, mice were housed separately in a constant temperature environment and given free access to water and food.
Behavioral tests
Six months after AAV plasmid injection (i.e., at the age of 9 months), behavioral tests were performed using a Morris water maze test according to a previously described method[57]. Briefly, a cylindrical tank with a diameter of 100 cm and a height of 40 cm contained 23 ± 1°C water to a depth of 25 cm. Four points were marked on the tank wall to divide the tank into four equal quadrants (A, B, C, D). A small platform was placed in the center of quadrant A and hidden 1 cm deep away from the water surface. Initially, mice were directly placed on the platform for 30 seconds to feel the spatial location of the platform. When testing, a random quadrant was designed as the starting area and mice were placed in a water tank, on the top of which, a camera was installed to record the swim path. Then, the platform was taken out and mice were re-placed in the tank for automatic re-recording of the data by a computer. The maximum recording time was 200 seconds. Mouse spatial learning ability and memory ability in each group were measured within 4 days. On day 4, escape latency and escape distance were recorded. The percentage of mice staying in quadrant A after removing the platform in the whole swim path was calculated.
Detection of amyloid-beta, tumor necrosis factor-α and interleukin-6 levels in brain tissue by ELISA
According to Donkin's method[58], amyloid-beta, tumor necrosis factor-α and interleukin-6 levels in brain tissue were detected by ELISA. Briefly, 500 µL of brain tissue, eight volumes of ice-cod bicarbonate buffer (100 mmol/L Na2CO3, 50 mmol/L NaCl pH 11.5) and protease inhibitor were added into a homogenizer for homogenization for 20 seconds. Then, the mixture was centrifuged at 12,000 × g for 45 minutes. Amyloid-beta, tumor necrosis factor-α and interleukin-6 levels in the supernatant were determined according to the instructions provided in the amyloid-beta (Invitrogen, Camarillo, CA, USA), tumor necrosis factor-α and interleukin-6 kits (Abcam, Hong Kong, China). The remaining precipitate was re-suspended with 50 mmol/L Tris-HCl (pH 8.0) containing 5 mol/L guanidine hydrochloride (pH 8.0). Insoluble amyloid-beta level was measured by kit instruction (Invitrogen).
Detection of hippocampal Dab2, TβRII and p-SMAD2/3 levels by western blot analysis
Hippocampal Dab2, TβRII and p-SMAD2/3 protein levels were detected by western blot analysis[59]. A small amount of hippocampal tissue was chopped into small pieces, homogenized, and lysed on ice with lysis buffer here (Beyotime, Shanghai, China) for 30 minutes. The lysate was centrifuged at 12,000 × g for 20 minutes and then the supernatant was taken to determine Dab2, TβRII and p-SMAD2/3 levels. An aliquot (15 µg) of each sample was thoroughly mixed with 4 µL of 5 × loading buffer and denatured in boiling water for 5 minutes. Protein samples were subjected to 10% SDS-PAGE (Beyotime) at 80 V for 40 minutes and at 110 V for 90 minutes, and then transferred onto PVDF membranes at 200 mA for 60 minutes and enveloped with blocking solution for 2 hours. After washes with western cleaning solution (Beyotime), protein samples were incubated with goat anti-IgG polyclonal antibody (1: 200; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. The next day, after washes, sample was incubated with horseradish peroxidase-labeled rabbit anti-goat polyclonal antibody at room temperature for 2 hours. Thereafter, the bands were developed by enhanced chemiluminescence. The absorbance of the scanned bands was determined using Image J (National Institutes of Health, Rockville, MD, USA). The results are expressed as the ratio of the target protein absorbance value to that for GAPDH.
Determination of the numbers of microglia and neurons by immunofluorescence staining
According to a method described previously[60], mouse hippocampal slices were stained for Iba1 and neuron-specific enolase. After washes with 0.01 mol/L PBS, brain tissue was fixed with methanol containing 0.3% hydrogen peroxide for 20 minutes, blocked with 10% bovine serum albumin (Gibco, Grand Island, NY, USA), washed with 0.01 mol/L PBS, treated with rabbit anti-mouse polyclonal antibody IgG (which was diluted with 0.03% Triton-X-100-containing PBS at 1:200; Santa Cruz Biotechnology) overnight at 4°C. The next day, after washes with 0.01 mol/L PBS, brain tissue was treated with 3% bovine serum albumin-diluted (1:200) goat anti-rabbit monoclonal antibody IgG-FITC (Santa Cruz Biotechnology) and incubated for 1 hour at room temperature. After three washes with 0.1 mol/L PBS for 5 minutes each time, brain tissue was mounted with anti-quenching fluorescence mounting medium. Finally, brain tissue was observed under the fluorescence microscope (Olympus, Japan) and photographed. Five fields of view were taken from each of six consecutive slices and Iba1- and neuron-specific enolase-positive cells were counted. The mean value was taken as the final result.
Statistical analysis
All data were statistically processed using SPSS 18.0 software (SPSS, Chicago, IL, USA) and measurement data are expressed as mean ± SEM. One-way analysis of variance and the SNK-q test were used for intergroup comparisons. A level of α = 0.05 was considered statistically significant.
Footnotes
Conflicts of interest: None declared.
Peer review: This study was the first to report that Dab2 can regulate transforming growth factor-β1/SMADs signal pathway in an Alzheimer's disease model and exhibit a neuroprotective effect through this pathway.
Copyedited by McGowan D, Stow A, Xu WH, Xu J, Li CH, Song LP, Zhao M
References
- [1].Casey G. Alzheimer's and other dementias. Nurs N Z. 2012;18:20–24. [PubMed] [Google Scholar]
- [2].Povova J, Ambroz P, Bar M, et al. Epidemiological of and risk factors for Alzheimer's disease: a review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2012;156:108–114. doi: 10.5507/bp.2012.055. [DOI] [PubMed] [Google Scholar]
- [3].Bassil N, Mollaei C. Alzheimer's dementia: a brief review. J Med Liban. 2012;60:192–199. [PubMed] [Google Scholar]
- [4].Singh S, Kushwah AS, Singh R, et al. Current therapeutic strategy in Alzheimer's disease. Eur Rev Med Pharmacol Sci. 2012;16:1651–1664. [PubMed] [Google Scholar]
- [5].Walsh DM, Teplow DB. Alzheimer's disease and the amyloid beta-protein. Prog Mol Biol Transl Sci. 2012;107:101–124. doi: 10.1016/B978-0-12-385883-2.00012-6. [DOI] [PubMed] [Google Scholar]
- [6].Sagare AP, Bell RD, Zlokovic BV. Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer's disease. J Alzheimers Dis. 2013;33(Suppl 1):S87–100. doi: 10.3233/JAD-2012-129037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Roberts AB, Anzano MA, Wakefield LM, et al. Type beta transforming growth factor: a bifunctional regulator of cellular growth. Proc Natl Acad Sci U S A. 1985;82:119–123. doi: 10.1073/pnas.82.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brown JP, Twardzik DR, Marquardt H, et al. Vaccinia virus encodes a polypeptide homologous to epidermal growth factor and transforming growth factor. Nature. 1985;313:491–492. doi: 10.1038/313491a0. [DOI] [PubMed] [Google Scholar]
- [9].Araujo-Jorge TC, Waghabi MC, Bailly S, et al. The TGF-beta pathway as an emerging target for Chagas disease therapy. Clin Pharmacol Ther. 2012;92:613–621. doi: 10.1038/clpt.2012.102. [DOI] [PubMed] [Google Scholar]
- [10].Sakaki-Yumoto M, Katsuno Y, Derynck R. TGF-beta family signaling in stem cells. Biochim Biophys Acta. 2013;1830:2280–2296. doi: 10.1016/j.bbagen.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Beyer TA, Narimatsu M, Weiss A, et al. The TGFbeta superfamily in stem cell biology and early mammalian embryonic development. Biochim Biophys Acta. 2013;1830:2268–2279. doi: 10.1016/j.bbagen.2012.08.025. [DOI] [PubMed] [Google Scholar]
- [12].Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chin J, Angers A, Cleary LJ, et al. Transforming growth factor beta1 alters synapsin distribution and modulates synaptic depression in Aplysia. J Neurosci. 2002;22:RC220. doi: 10.1523/JNEUROSCI.22-09-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brionne TC, Tesseur I, Masliah E, et al. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- [15].Krieglstein K, Strelau J, Schober A, et al. TGF-beta and the regulation of neuron survival and death. J Physiol Paris. 2002;96:25–30. doi: 10.1016/s0928-4257(01)00077-8. [DOI] [PubMed] [Google Scholar]
- [16].Chao CC, Hu S, Frey WH, 2nd, et al. Transforming growth factor beta in Alzheimer's disease. Clin Diagn Lab Immunol. 1994;1:109–110. doi: 10.1128/cdli.1.1.109-110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].De Servi B, La Porta CA, Bontempelli M, et al. Decrease of TGF-beta1 plasma levels and increase of nitric oxide synthase activity in leukocytes as potential biomarkers of Alzheimer's disease. Exp Gerontol. 2002;37:813–821. doi: 10.1016/s0531-5565(02)00018-9. [DOI] [PubMed] [Google Scholar]
- [18].Wyss-Coray T, Lin C, Yan F, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- [19].Tichauer JE, von Bernhardi R. Transforming growth factor-beta stimulates beta amyloid uptake by microglia through Smad3-dependent mechanisms. J Neurosci Res. 2012;90:1970–1980. doi: 10.1002/jnr.23082. [DOI] [PubMed] [Google Scholar]
- [20].Caraci F, Battaglia G, Busceti C, et al. TGF-beta 1 protects against Abeta-neurotoxicity via the phosphatidy-linositol-3-kinase pathway. Neurobiol Dis. 2008;30:234–242. doi: 10.1016/j.nbd.2008.01.007. [DOI] [PubMed] [Google Scholar]
- [21].Caraci F, Battaglia G, Bruno V, et al. TGF-beta1 pathway as a new target for neuroprotection in Alzheimer's disease. CNS Neurosci Ther. 2011;17:237–249. doi: 10.1111/j.1755-5949.2009.00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tesseur I, Zou K, Esposito L, et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest. 2006;116:3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee HG, Ueda M, Zhu X, et al. Ectopic expression of phospho-Smad2 in Alzheimer's disease: uncoupling of the transforming growth factor-beta pathway? J Neurosci Res. 2006;84:1856–1861. doi: 10.1002/jnr.21072. [DOI] [PubMed] [Google Scholar]
- [24].Das P, Golde T. Dysfunction of TGF-beta signaling in Alzheimer's disease. J Clin Invest. 2006;116:2855–2857. doi: 10.1172/JCI30284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Caraci F, Spampinato S, Sortino MA, et al. Dysfunction of TGF-beta1 signaling in Alzheimer's disease: perspectives for neuroprotection. Cell Tissue Res. 2012;347:291–301. doi: 10.1007/s00441-011-1230-6. [DOI] [PubMed] [Google Scholar]
- [26].Wang H, Liu J, Zong Y, et al. miR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer's disease targets TGF-beta type II receptor. Brain Res. 2010;1357:166–174. doi: 10.1016/j.brainres.2010.08.023. [DOI] [PubMed] [Google Scholar]
- [27].Penheiter SG, Singh RD, Repellin CE, et al. Type II transforming growth factor-beta receptor recycling is dependent upon the clathrin adaptor protein Dab2. Mol Biol Cell. 2010;21:4009–4019. doi: 10.1091/mbc.E09-12-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bao H. Ann Arbor: Proquest, Umi Dissertation Publishing; 2008. RNA Profiling in an Alzheimer's Disease Mouse Model. [Google Scholar]
- [29].Low K, Aebischer P. Use of viral vectors to create animal models for Parkinson's disease. Neurobiol Dis. 2012;48:189–201. doi: 10.1016/j.nbd.2011.12.038. [DOI] [PubMed] [Google Scholar]
- [30].Lentz TB, Gray SJ, Samulski RJ. Viral vectors for gene delivery to the central nervous system. Neurobiol Dis. 2012;48:179–188. doi: 10.1016/j.nbd.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Thorne BA, Takeya RK, Peluso RW. Manufacturing recombinant adeno-associated viral vectors from producer cell clones. Hum Gene Ther. 2009;20:707–714. doi: 10.1089/hum.2009.070. [DOI] [PubMed] [Google Scholar]
- [32].Foust KD, Nurre E, Montgomery CL, et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dayton RD, Wang DB, Klein RL. The advent of AAV9 expands applications for brain and spinal cord gene delivery. Expert Opin Biol Ther. 2012;12:757–766. doi: 10.1517/14712598.2012.681463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Carty N, Lee D, Dickey C, et al. Convection-enhanced delivery and systemic mannitol increase gene product distribution of AAV vectors 5, 8, and 9 and increase gene product in the adult mouse brain. J Neurosci Methods. 2010;194:144–153. doi: 10.1016/j.jneumeth.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Doetschman T, Barnett JV, Runyan RB, et al. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2012;347:203–223. doi: 10.1007/s00441-011-1241-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kovacic P, Somanathan R. Cell signaling and receptors in toxicity of advanced glycation end products (AGEs): alpha-dicarbonyls, radicals, oxidative stress and antioxidants. J Recept Signal Transduct Res. 2011;31:332–339. doi: 10.3109/10799893.2011.607171. [DOI] [PubMed] [Google Scholar]
- [37].Konkel JE, Chen W. Balancing acts: the role of TGF-beta in the mucosal immune system. Trends Mol Med. 2011;17:668–676. doi: 10.1016/j.molmed.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang F, Chen YG. Regulation of TGF-beta receptor activity. Cell Biosci. 2012;2:9. doi: 10.1186/2045-3701-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xu P, Liu J, Derynck R. Post-translational regulation of TGF-beta receptor and Smad signaling. FEBS Lett. 2012;586:1871–1884. doi: 10.1016/j.febslet.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mitchell H, Choudhury A, Pagano RE, et al. Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell. 2004;15:4166–4178. doi: 10.1091/mbc.E04-03-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cavallucci V, D’Amelio M, Cecconi F. Abeta toxicity in Alzheimer's disease. Mol Neurobiol. 2012;45:366–378. doi: 10.1007/s12035-012-8251-3. [DOI] [PubMed] [Google Scholar]
- [42].Pithadia AS, Lim MH. Metal-associated amyloid-beta species in Alzheimer's disease. Curr Opin Chem Biol. 2012;16:67–73. doi: 10.1016/j.cbpa.2012.01.016. [DOI] [PubMed] [Google Scholar]
- [43].Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- [44].Dobolyi A, Vincze C, Pal G, et al. The neuroprotective functions of transforming growth factor Beta proteins. Int J Mol Sci. 2012;13:8219–8258. doi: 10.3390/ijms13078219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Copani A, Condorelli F, Caruso A, et al. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999;13:2225–2234. [PubMed] [Google Scholar]
- [46].Copani A, Sortino MA, Caricasole A, et al. Erratic expression of DNA polymerases by beta-amyloid causes neuronal death. FASEB J. 2002;16:2006–2008. doi: 10.1096/fj.02-0422fje. [DOI] [PubMed] [Google Scholar]
- [47].Herrup K, Neve R, Ackerman SL, et al. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vorhees CV, Williams MT. Morris water maze: pro-cedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].D’Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- [50].Sardi F, Fassina L, Venturini L, et al. Alzheimer's disease, autoimmunity and inflammation. The good, the bad and the ugly. Autoimmun Rev. 2011;11:149–153. doi: 10.1016/j.autrev.2011.09.005. [DOI] [PubMed] [Google Scholar]
- [51].Barber R. Inflammatory signaling in Alzheimer disease and depression. Cleve Clin J Med. 2011;78(Suppl 1):S47–49. doi: 10.3949/ccjm.78.s1.08. [DOI] [PubMed] [Google Scholar]
- [52].Johnston H, Boutin H, Allan SM. Assessing the con-tribution of inflammation in models of Alzheimer's disease. Biochem Soc Trans. 2011;39:886–890. doi: 10.1042/BST0390886. [DOI] [PubMed] [Google Scholar]
- [53].Makwana M, Jones LL, Cuthill D, et al. Endogenous transforming growth factor beta 1 suppresses inflammation and promotes survival in adult CNS. J Neurosci. 2007;27:11201–11213. doi: 10.1523/JNEUROSCI.2255-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Boche D, Cunningham C, Docagne F, et al. TGFbeta1 regulates the inflammatory response during chronic neurodegeneration. Neurobiol Dis. 2006;22:638–650. doi: 10.1016/j.nbd.2006.01.004. [DOI] [PubMed] [Google Scholar]
- [55].Tone AA, Virtanen C, Shaw P, et al. Prolonged postovulatory proinflammatory signaling in the fallopian tube epithelium may be mediated through a BRCA1/DAB2 axis. Clin Cancer Res. 2012;18:4334–4344. doi: 10.1158/1078-0432.CCR-12-0199. [DOI] [PubMed] [Google Scholar]
- [56].Kiyota T, Okuyama S, Swan RJ, et al. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer's disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010;24:3093–3102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].El-Amouri SS, Zhu H, Yu J, et al. Neprilysin: an enzyme candidate to slow the progression of Alzheimer's disease. Am J Pathol. 2008;172:1342–1354. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Donkin JJ, Stukas S, Hirsch-Reinshagen V, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yang Z, Bian C, Zhou H, et al. MicroRNA hsa-miR-138 inhibits adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells through adenovirus EID-1. Stem Cells Dev. 2011;20:259–267. doi: 10.1089/scd.2010.0072. [DOI] [PubMed] [Google Scholar]
- [60].Zhao YN, Wang F, Fan YX, et al. Activated microglia are implicated in cognitive deficits, neuronal death, and successful recovery following intermittent ethanol exposure. Behav Brain Res. 2013;236:270–282. doi: 10.1016/j.bbr.2012.08.052. [DOI] [PubMed] [Google Scholar]