

We are all familiar with the idea of cellular function being driven by spatiotemporal changes in the concentration of intracellular signalling ions, such as Ca2+. The reviews by Swietach et al. (2014) and Zhang et al. (2014), in the this issue of The Journal of Physiology, address the localised control of H+ and nitric oxide (NO) levels, respectively, within the cardiac myocyte, and their coupling to intracellular Ca2+. This raises the possibility of local H+, NO and Ca2+ signals sometimes behaving as a functional unit, given that their spatial spread is constrained by diffusion–reaction processes, and that the individual components exhibit cross-talk.
Low intracellular Ca2+ mobility encourages local microdomains
Ca2+ signalling drives a multiplicity of cellular events, including excitation–contraction coupling, exocytosis, and gene transcription and translation. Ca2+ signalling in the cardiac myocyte is shaped partly by the kinetics of cytoplasmic Ca2+ diffusion, operating in combination with buffering by proteins (e.g. calmodulin and troponin-C), and membrane transport at the sarcoplasmic reticulum (SR) and sarcolemma (Bers, 2002). Buffering and transport reactions greatly slow passive cytoplasmic Ca2+ mobility. Under some circumstances, this results in spatial Ca2+ non-uniformity (i.e. the formation of a cytoplasmic Ca2+ microdomain). A classic example of this is the Ca2+ spark (Cheng & Lederer, 2008), a transient spot of elevated cytoplasmic Ca2+, caused by localised SR release, which is the unitary building block of the larger cytoplasmic Ca2+ transient (CaT) that drives cellular contraction.
Low intracellular H+ mobility also encourages local microdomains
The mobility of other intracellular signalling ions and small molecules can be similarly constrained by diffusion–reaction phenomena. The H+ ion is a universal end product of metabolism, of such potent chemical reactivity that it must be kept at a low ‘baseline’ level in cytoplasm, similar to that for Ca2+, i.e. ∼60 nm (pHi 7.20). And, like Ca2+, the intracellular H+ level can fluctuate physiologically or pathophysiologically by tens to hundreds of nanomolar, albeit on a much slower timescale (minutes rather than milliseconds, Vaughan-Jones et al. 2009). Because of powerful buffering by intracellular protein, cytoplasmic H+ mobility is also low. Indeed, in cardiac myocytes, H+ ions can only move passively in significant quantities by attaching reversibly to small, diffusible buffer molecules, such as the histidyl dipeptides (HDPs) carnosine and homocarnosine, which are collectively present at 10–15 mm. Even so, H+ mobility remains low, which can lead to spatial cytoplasmic H+ gradients of tens of nanomolar in amplitude (local H+ microdomains), for example during stimulation of sarcolemmal H+ extrusion. An intriguing discovery, reviewed here by Swietach et al. (2014), is that HDPs competitively bind and shuttle both H+ and Ca2+ ions. Thus HDPs represent a new class of small intracellular molecule with common Ca2+/H+ binding. This HDP mechanism contributes to a well-known phenomenon in cardiac myocytes, whereby a rise of cytoplasmic [H+] results in a parallel rise of baseline [Ca2+]i, consistent with competitive ion binding. More surprising is the finding that this interaction can become highly localised, due to the ability of HDPs to perform spatial Ca2+/H+ exchange transport within the cytoplasmic compartment. As a result, Ca2+ levels map spatially onto cytoplasmic H+ microdomains. This novel form of Ca2+ control may serve to compensate for local inhibitory effects of H+ ions on proteins that are targeted by Ca2+ signalling. The local control occurs because the cytoplasmic mobility of Ca2+ and H+ ions shares a common diffusion–reaction mechanism within the myocyte, mediated via mobile HDPs.
Restricted intracellular NO dispersion may encourage local microdomains
NO is another example of a diffusible but chemically constrained solute. It is a widely distributed signalling molecule. In cardiac myocytes, it exerts indirect inhibitory effects on the L-type Ca2+ current, an effect that contributes to modulation of the CaT. Zhang et al. (2014) review how the NO molecule is generated locally within the cell (from l-arginine), but with an effective mobility that is limited by chemical oxidation and scavenging reactions. Its production is catalysed by NO synthase (NOS), while the scavenging is via haem groups on myoglobin molecules. This will retard the spatial dispersion of NO, even though the molecule is readily diffusible. Thus a diffusion–reaction scheme again provides the potential for the generation of local signalling microdomains, in this case for NO. The problem here is that, although such microdomains may be predicted theoretically, in practice they have yet to be definitively measured, and so remain highly contentious. Zhang et al. (2014) discuss the emerging role of the nNOS isoform as a major catalytic source for local NO generation within the healthy or diseased myocardium (isoform defined as nNOS because its expression was originally observed neuronally). In healthy myocytes, the enzyme is associated particularly with SR and mitochondrial membranes. In contrast, the eNOS isoform (named after endothelial-derived NOS) is associated with caveolae in the surface sarcolemma and transverse tubules. Sources of NO generation are thus sited in specific regions within the cell. This affords a local control of other signalling events such as those involving Ca2+. For example, nNOS-derived NO modulates SR Ca2+ reuptake by SERCA (sarcoplasmic endoplasmic reticulum Ca2+-ATPase), while eNOS-derived NO has been proposed to modulate mechanical stress-induced SR Ca2+ release. The differential expression of NOS isoforms (and their observed redistribution in diseased states, like maladaptive hypertrophy and heart failure), coupled with the restricted spatial mobility of NO, emphasises the potential importance of NOS as a local controller of signalling (Paton et al. 2002).
Can these microdomains be linked?
Ca2+, H+ and NO signals may display considerable cross-talk. For example, H+ ions modulate Ca2+ by a variety of routes (Vaughan-Jones et al. 2009) including, as discussed here, HDPs. There is evidence, however, that H+ ions may also directly or indirectly modulate NOS activity (e.g. Boedtkjer et al. 2011), and thus the sourcing of NO. In addition, as discussed above and by Zhang et al. (2014), NO influences Ca2+ signalling, while reverse modulation (i.e. Ca2+ affecting NOS activity and hence NO) may occur through interactions involving calmodulin. Finally, cytoplasmic H+ levels may, themselves, be modulated by NO, through the molecule's targeting of Na+/H+ membrane transporters (Ito et al. 1997), and by Ca2+ that, under the right circumstances, may liberate H+ from HDPs (Swietach et al. 2014). There is thus the possibility of interaction among all three signalling systems. As each may be constrained spatially by diffusion–reaction processes, this raises the intriguing possibility that the three signals will sometimes interact as a local unit (Fig. 1) to control cell function. For full integration within a myocyte, the signals would need to interact reversibly and possess common spatial domains. HDPs, being small, mobile Ca2+/H+ carriers, should readily access dyadic, sub-membranous and bulk cytoplasmic regions. The molecules are thus likely to come into close proximity with membrane-located and any cytoplasmically located NOS. A tripartite collaboration among Ca2+, H+ and NO therefore seems feasible. It remains to be seen, however, if local cross-talk is physiologically important. It would certainly provide another layer of complexity to the intracellular influence of the three signals. Cross-talk emphasises that the functional effects of H+ and NO on Ca2+ signalling, as reviewed individually by Swietach et al. (2014) and Zhang et al. (2014), cannot be evaluated fully in isolation. An integrated approach will ultimately be necessary.
Figure 1.
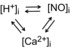
Reversible cross-talk (arrows) among three intracellular signalling domains
Additional information
Competing interests
None declared.
References
- Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Boedtkjer E, Praetorius J, Matchkov VV, Stankevicius E, Mogensen S, Füchtbauer AC, Simonsen U, Füchtbauer EM, Aalkjaer C. Disruption of Na+-HCO3− cotransporter NBCn1 (slc4a7) inhibits NO-mediated vasorelaxation, smooth muscle Ca2+ sensitivity, and hypertension development in mice. Circulation. 2011;124:1819–1829. doi: 10.1161/CIRCULATIONAHA.110.015974. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- Ito N, Bartunek J, Spitzer KW, Lorell BH. Effects of the nitric oxide donor sodium nitroprusside on intracellular pH and contraction in hypertrophied myocytes. Circulation. 1997;95:2303–2311. doi: 10.1161/01.cir.95.9.2303. [DOI] [PubMed] [Google Scholar]
- Paton JRF, Kasparov S, Paterson DJ. Nitric oxide and autonomic control of heart rate: a question of specificity. Trends in Neuroscience. 2002;25:626–631. doi: 10.1016/s0166-2236(02)02261-0. [DOI] [PubMed] [Google Scholar]
- Swietach P, Leem C-H, Spitzer KW, Vaughan-Jones RD. Pumping Ca2+ ions up H+ gradients: a cytoplasmic Ca2+/H+ exchanger without a membrane. J Physiol. 2014;592:3179–3188. doi: 10.1113/jphysiol.2013.265959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol. 2009;46:318–331. doi: 10.1016/j.yjmcc.2008.10.024. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Jin CZ, Jang JH, Wang Y. Molecular mechanisms of neuronal nitric oxide synthase in cardiac function and pathophysiology. J Physiol. 2014;592:3189–3200. doi: 10.1113/jphysiol.2013.270306. [DOI] [PMC free article] [PubMed] [Google Scholar]