

Abstract
There is much interest in α7 nicotinic acetylcholine receptors (nAChRs) in CNS function since they are found throughout peripheral tissues as well as being highly expressed in brain regions implicated in attention, learning and memory. As such, the role of these receptors in many aspects of CNS function and disease is being actively investigated. To date, only one null mouse model (A7KO) is available which is non-conditional and constitutive. Since α7 nAChRs are present on neurons and glia (including astrocytes), as well as being developmentally regulated, there is an unmet need for the technical capability to control α7 nAChR gene expression. Therefore we have generated mice in which the fourth exon of the α7 nAChR gene (Chrna7) is flanked by loxP sites (B6-Chrna7LBDEx4007Ehs) which we refer to as floxed α7 nAChR conditional knockout or α7nAChRflox. We validated the chosen approach by mating α7nAChRflox with mice expressing Cre recombinase driven by the glial acidic fibrillary protein (GFAP)-Cre promoter (GFAP-A7KO) to test whether α7nAChRflox, GFAP-A7KO and appropriate littermate controls performed equally in our standard Rodent In Vivo Assessment Core battery to assess general health, locomotion, emotional and cognitive behaviours. Neither α7nAChRflox nor GFAP-A7KO exhibited significant differences from littermate controls in any of the baseline behavioural assessments we conducted, similar to the ‘first generation’ non-conditional A7KO mice. We also determined that α7 nAChR binding sites were absent on GFAP-positive astrocytes in hippocampal slices obtained from GFAP-A7KO offspring from α7nAChRflox and GFAP-Cre crosses. Finally, we validated that Cre recombinase (Cre)-mediated excision led to functional, cell- and tissue-specific loss of α7 nAChRs by demonstrating that choline-induced α7 nAChR currents were present in Cre-negative, but not synapsin promoter-driven Cre-positive, CA1 pyramidal neurons. Additionally, electrophysiological characterization of α7 nAChR-mediated current traces was similar in terms of amplitude and time constants of decay (during desensitization) for the α7nAChRflox and wild-type (WT) mice. Thus, we have in vivo and in vitro evidence that the Chrna7 exon 4 targeting strategy does not alter behavioural, cognitive, or electrophysiological properties compared to WT and that Cre-mediated excision is an effective approach to delete α7 nAChR expression in a cell-specific manner.
Key points
Currently, no animal model exists in which selective deletion of α7 nAChRs from a specific cell type or tissue is possible through genetic manipulation.
We have generated mice in which the fourth exon of the α7 nAChR gene (Chrna7) is flanked by loxP sites (B6-Chrna7LBDEx4007Ehs) which we refer to as floxed α7 nAChR conditional knockout or α7nAChRflox.
In the brain, α7 nAChRs are expressed both on neurons and astrocytes.
Proper Cre recombinase excision of the targeted gene was verified with a combination of in vitro and in vivo approaches including demonstrating that α7 nAChR binding sites were absent on glial fibrillary acidic protein (GFAP)-positive astrocytes in hippocampal slices obtained from offspring of α7nAChRflox and GFAP-Cre mating.
This study validates the chosen approach for deletion of α7 nAChR gene expression from any tissue for which appropriate Cre constructs are available, thus providing the nicotinic receptor research field with new technical capabilities.
Introduction
Alpha7 (α7) nicotinic acetylcholine receptors (nAChRs) are widely distributed throughout the peripheral and central nervous systems as well as the immune system and a variety of peripheral tissues (Seguela et al. 1993; Dominguez del Toro et al. 1994; Rubboli et al. 1994a,b1994b; Drago et al. 2003). As such, α7 nAChRs have been implicated in a variety of biological functions and are considered a prominent therapeutic target in several disease states (Gallowitsch-Puerta & Tracey, 2005; Zoheir et al. 2012; Wallace & Bertrand, 2013; Young & Geyer, 2013). Therefore, the ability to control α7 nAChR expression in developmental and cell-specific manners is crucial to understanding the various roles α7 nAChRs play. Currently available α7 nAChR knock-out mouse models exhibit gene deletion in all tissues de novo (Orr-Urtreger et al. 1995, 1997). To fill this technological gap, we generated conditional A7KO (α7nAChRflox) mice using Cre-loxP methodology followed by in vivo and in vitro validation of the approach to provide the research community with a new tool to investigate α7 nAChR biology.
The α7nAChRflox mice have a Chrna7 gene in which the fourth exon is flanked by loxP sites (B6-Chrna7LBDEx4007Ehs). Based upon our and others’ recent discoveries of functional α7 nAChRs on astrocytes (Sharma & Vijayaraghavan, 2001; Shen & Yakel, 2012; Lee et al. 2014) and their role in glutamate gliotransmission (Pirttimaki et al. 2013; Talantova et al. 2013), these α7nAChRflox were mated to glial fibrillary acidic protein (GFAP)-Cre recombinase (Cre) hemizygous transgenic mice to generate a mouse strain in which α7 nAChR expression was expected to be selectively deleted from astrocytes, referred to as GFAP-A7KO. Genotypes were confirmed by PCR to detect the floxed and excised Chrna7 alleles as well as the presence of the Cre recombinase (Cre) transgene. Utilizing our standard Rodent In Vivo Assessment Core battery to assess general health, locomotion, emotional and cognitive behaviours, α7nAChRflox and GFAP-A7KO lines were found to be indistinguishable from their respective wild-type (WT) and heterozygous littermates. These nominal findings are consistent with the phenotype determined for the original A7KO line subjected to similar assessments (Paylor et al. 1998); however, since then, a cadre of more subtle cognitive and sensory deficiencies have been reported utilizing specialized protocols (Franceschini et al. 2000; Morley & Rodriguez-Sierra, 2004; Young et al. 2004; Keller et al. 2005; Levin et al. 2009; Brown et al. 2010; Young et al. 2011).
Additionally, we confirmed Cre-mediated excision of the loxP-targeted Chrna7 gene by demonstrating that hippocampal slice cultures infected with an AAV containing a Cre-tdTomato fusion cDNA driven by the neuron-specific synapsin promoter lacked choline-induced α7 nAChR currents in Cre-positive, but not Cre-negative, neurons through whole cell patch clamp recordings. Furthermore in murine hippocampal primary cultures, α7 nAChR-mediated currents were of similar profile and the magnitude of the current amplitude and time constants of decay (during desensitization) between the α7nAChRflox and WT mice were not significantly different. Thus, we have in vivo and in vitro evidence that the Chrna7 exon 4 targeting strategy does not introduce artifactual effects on baseline health, behaviour, cognition, or electrophysiological properties. Furthermore, in the presence of the Cre recombinase, it is an effective approach to delete α7 nAChR expression in a cell-specific manner. This validation study provides the nAChR research field with new technical capabilities to delete α7 nAChR gene expression from any tissue for which appropriate Cre constructs are available.
Methods
Chrna7 gene targeting strategy
We chose to flox exon 4 within the murine Chrna7 gene that encodes a portion of the extracellular ligand-binding domain. It is larger than the first three exons and its deletion was predicted to introduce a frame shift and many stop codons downstream. Although the resulting transcript was capable of being translated into truncated protein, the mRNA was expected to be unstable and subjected to nonsense-mediated mRNA decay.
Generation of conditional knockout targeting vectors
Figure 1 depicts schema of the Chrna7 exon 4 targeting strategy and the different Chrna7 alleles to obtain Cre recombinase-driven deletion of exon 4 (Fig. 1E). A 7 kb PacI-EcoRI genomic DNA fragment containing exon 4 was subcloned from a Chrna7 BAC (RP24-336F13) obtained from BACPAC CHORI into a plasmid vector. A loxP was inserted ∼290 bp upstream from exon 4; a second loxP and a flippase recognition-targeted (Frt) PGKneobpA cassette were inserted ∼380 bp downstream from exon 4 (Fig. 1B). The PGKneobpA was flanked by a 3.2 kb of genomic DNA on the 5′ and 3.8 kb on the 3′ side. Correctly targeted embryonic stem (ES) cell clones were identified by Southern blotting using BamHI digestion and 5′ flanking probe or 3′ flanking probe (wild-type, 20 kb Fig. 1A; targeted, 7.7 kb with 5′ probe Fig. 1C or 14.4 kb with 3′ probe Fig. 1C).
Figure 1.

Exon 4 targeting strategy
Schematic representation showing the position of the 5′-flanking probe (5′) and 3′-flanking probe (3′) to be used for Southern blot; the internal probe was to the ‘neo’ cassette. BamHI genomic DNA digestion yielded the indicated band sizes by Southern blot to identify the following: WT (murine) Chrna7 allele (A); Frt-loxP targeting vector (B); neo-positive targeted Chrna7 allele (C); floxed allele following FLP recombinase (FLPe, D); and the deleted allele (E). Vertical arrows indicate which recombinase was used to obtain the designated alleles. N47R and N47F indicate primers used for PCR genotyping.
Generation of Chrna7 mutants carrying floxed alleles
The Chrna7 exon 4 knockout targeting construct was delivered to Taconic (Hudson, NY, USA) in a linearized form and electroporated into B6-3 ES cells (origin = C57BL/6T). Colonies resistant to the antibiotic Geneticin® (G418) were identified by Southern blot screening (as described above). Genotypes and deletion of the neo cassette in these clones was confirmed by PCR using primers: N47_F (GTGCCAGACCACATGTGCATTGG) and N47_R (GGTCACTGAGCAGTGGTGGACAG). Two subclones confirmed for flippase (FLP)-Frt recombination were injected into albino C57BL/6 (C57BL/6J-Tyrc-2J/J) blastocysts. Highly pigmented chimeras were crossed with albino C57BL/6 mice to obtain heterozygous mutants (neo/+). Genotypes were confirmed by PCR as described above.
Animals
Animals were bred in the UTMB and NIEHS/NIH animal care facilities. Mice were housed, n ≤ 5 per cage, with food and water ad libitum. UTMB and NIEHS/NIH operate in compliance with the USDA Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, and UTMB Institutional Animal Care and Use Committee approved protocols. Genotypes were determined and validated from tail clip biopsies obtained at weaning and after the animals were killed. All genotyping services were outsourced (Transnetyx – Taconic, Cordova, TN, USA).
Hemizygous female Tg(GFAP-Cre)77.6Mvs/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) (stock no. 012887) and mated to homozygous α7nAChRflox males (Fig. 2). Mice homozygous for the α7nAChRflox mutation and hemizygous for the GFAP-Cre transgene are given the nomenclature GFAP-A7KO (Fig. 2). α7nAChRflox and GFAP-A7KO groups with appropriate control WT and/or heterozygous littermates were analysed.
Figure 2.
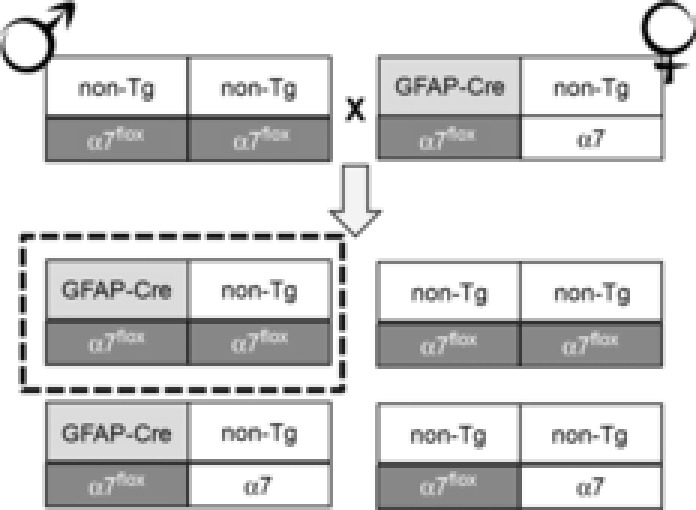
Breeding strategy for generating GFAP-α7nAChR mutant mice
Homozygous α7nAChRflox (α7flox) mice are crossed to hemizygous GFAP-Cre to generate a mouse that is both heterozygous for the floxed α7 allele and hemizygous for the GFAP-Cre transgene. These genotypes are interbred as indicated. After 5 generations, the parental α7nAChRflox and the transgenic Cre genotypes are backcrossed into WT C57BL/6J (Jackson Laboratories stock no. 000664) to avoid genetic drift. Tg = transgenic, α7 = Chrna7 allele.
Behavioural analyses
Behavioural experiments were performed during the lights-on phase (06.00–18.00 h) in the UTMB Rodent In Vivo Assessment Core (directed by K. T. Dineley) within the UTMB Center for Addiction Research (directed by Dr Kathryn Cunningham). Based upon power analyses of previous data, 10 (WT) to 15 (transgenic) mice per group (male and female) were subjected to baseline health, locomotor and cognitive testing. Specific numbers are as follows: 20, 23, 15 for WT, heterozygous and homozygous α7nAChRflox, respectively, and 10, 17, 15 for WT, heterozygous and homozygous GFAP-A7KO, respectively. All experimental groups were balanced to include approximately equivalent numbers of each sex.
Initially, naive animals (not previously handled) were first subjected to the elevated plus maze as this is a test for anxiety-like behaviour and the objective was to detect inherent anxiety-like tendencies in our novel genetic models. Next, the animals were handled daily for 3 consecutive days followed by general health and neurological reflex assessment. General health assessment was determined by evaluating individual animals’ fur and whisker appearance and noting any deviations from typical (Crawley & Paylor, 1997) followed by recording weight and temperature. Baseline neurological tests included gently approaching the eye, ear and whisker with a cotton-tipped swab to determine twitch response. In addition, tail suspension, righting reflex, and general home cage behaviours were noted. Tail suspension test checked that all four mouse appendages were extended outward as though bracing for a fall. Righting reflex was tested by manually placing the rodent on its back and noting the ability to right itself within a few seconds. Home cage activity observations evaluated whether running, jumping, freezing, sniffing, licking, grooming, defaecation and urination were duly observed within <5 min observation period.
Plantar and tail flick tests
Next, plantar (Stoelting, Wood Dale, IL, USA) and tail flick (Columbus Instruments, Columbus, OH, USA) tests were performed to determine analgesic threshold by measuring latency to withdraw from an infra-red heat generator that is placed below a glass pane upon which the operator places the rodent. The infra-red source is positioned under the rear paw or tail. The heat source and a timer start simultaneously. When the mouse withdraws its paw or tail, the infra-red heat source and the timer automatically turn off, determining the withdrawal latency.
Open field
Each mouse was tested for normal exploratory behaviour and locomotion in an open field. Each mouse was allowed to explore a 30 cm × 30 cm open field box for a 15 min test session. Using TopScan software (CleverSys Inc., Reston, VA, USA), videos were subsequently analysed to determine the distance travelled and the number of centre and peripheral zone entries. Parameters analysed include total distance travelled during the test session and the ratio of time spent in the peripheral 4/5ths of the gridded arena and the centre 1/5th.
Elevated plus maze
Using a standard elevated plus maze (two open arms perpendicular to two arms enclosed by high walls placed approximately 50 cm above the floor), each mouse was assessed for exploration in the open arms during a 5 min session using CleverSys automated video capture and TopScan software. Parameters analysed include time spent in the open arms of the maze and number of entries into the open arms.
Fear conditioning
Using our standard two-pairings fear conditioning (FC) training protocol (Dineley et al. 2002; Taglialatela et al. 2009; Rodriguez-Rivera et al. 2011; Denner et al. 2012), mice were assessed for contextual and cued FC. Twenty-four hours after training, hippocampus-dependent contextual learning was assessed by quantifying freezing behaviour when the animal was placed back into the training chamber. Four hours later the cued test was performed in a completely novel context. Freezing was quantified using FreezeFrame automated video capture and software analysis (Coulbourn Instruments, Whitehall, PA, USA) and evaluated as percentage freezing in 30 s (training) or 60 s (contextual, cued) bins.
Statistics for behaviour
Data were reported as mean ± SEM. α7nAChRflox and GFAP-A7KO groups were analysed separately by one-way ANOVA followed by Dunnett's post hoc multiple comparisons test.
Immunohistochemistry
Mice were anaesthetized with avertin (0.2%) and transcardially perfused with PBS, pH 7.4, followed by 4% (w/v) paraformaldehyde fixative in PBS, pH 7.4. Whole brains were extracted, post-fixed and cryoprotected by immersion in 30% (w/v) sucrose in PBS at 4°C. Frozen sagittal sections, 25 μm thick, were collected and stored in cryoprotectant at −20°C until ready for use. Prior to blocking, all sections underwent heated antigen retrieval with 0.01 m sodium citrate (pH 6.0), and autofluorescence clearance with 0.1% sodium borohydride in PBS. After thorough rinsing, sections were incubated for 10 min at room temperature in blocking buffer (BB) diluent prepared in PBS: 1% (w/v) bovine serum albumin, 0.1% (v/v) cold fish gelatn, 0.1% (v/v) Triton X-100 and 0.05% (v/v) Tween 20. Sections were blocked for 1 h at room temperature in BB containing 10% (v/v) normal goat serum (NGS), then incubated overnight in AlexaFluor488 (AF488)-conjugated α-bungarotoxin (2.5 nm; no. B13422, Life Technologies, Grand Island, NY, USA) diluted in BB with 1% (v/v) NGS at 4°C. Subsequently, sections were washed 4 × 60 min in BB at 4°C, then incubated overnight in anti-glial fibrillary acidic protein (GFAP; 1:100; no. A2182, Life Technologies) in BB containing 1% NGS at 4°C. Anti-GFAP was detected using the ABC method (Universal ABC kit; Vector Laboratories, Burlingame, CA, USA) and streptavidin–cyanine 3 (Cy3; 1:400; no. 016-160-084; Jackson ImmunoResearch, West Grove, PA, USA). Sections were counterstained using Vectashield mounting media (Vector Laboratories) with 4′,6-diamidino-2-phenylindole (DAPI). To assess for gross morphological differences between genotypes, a subset of representative sections (every 150 μm) were stained with 0.5% cresyl violet (Watson et al. 2012) to assist with identifying brain structures and neuroanatomical landmarks. Images were acquired with a Axioplan 2 (Zeiss, Thornwood, NY, USA) epifluorescence microscope equipped with an Axiocam camera to image GFAP (red, Cy3), α-bungarotoxin (green, AF488) and nuclear DNA (blue, DAPI) using three different fluorescent filters.
Hippocampal neuron primary cultures
Primary cultures were prepared at embryonic day 18 using a modified version from Quitsch et al. (2005). Briefly hippocampi were mechanically dissociated after treatment for 30 min at 37°C with 0.05% Trypsin EDTA (Life Technologies). Cells were plated in Neurobasal media (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Hyclone Laboratories, GE Healthcare Life Sciences, Logan, UT, USA) and 1%, Glutamax™ (Life Technologies) at 3,500 mm–2 on poly-d-lysine (Sigma-Aldrich, St. Louis, MO, USA)-coated coverslips and grown in a humidified atmosphere of 5% CO2 at 37°C. Half the medium was replaced 24 h after plating and every 72 h thereafter with Neurobasal media in which FBS was substituted with 2% B27 (Life Technologies).
Measurement of acetylcholine (ACh)-evoked currents
Hippocampal neurons were transferred at DIV 14–15 to a chamber containing (in mm): 165 NaCl, 5 KCl, 2 CaCl2, 10 glucose, 5 Hepes, 0.001 atropine, and 0.0003 tetrodotoxin (TTX); pH was adjusted to 7.3 with NaOH. Bath solution was perfused continuously through the chamber (1 ml volume) at 2 ml min−1 throughout the experiments. Neurons were visualized using a Eclipse TE300 (Nikon Instruments, Melville, NY, USA) microscope. Borosilicate patch pipettes (3–6 MΩ) were filled with (in mm): 120 CsCl, 2 MgCl2, 10 EGTA, 10 Hepes, and ATP-regenerating compounds 5 ATP and 20 phosphocreatine; pH was adjusted to 7.3 with CsOH. Currents were recorded at −70 mV. Experiments were performed at room temperature (22°C). Whole cell recordings were done using an Axopatch-200A amplifier connected to a Digidata 1322A and software (pCLAMP v. 10.1) from Molecular Devices (Sunnyvale, CA, USA). Currents were filtered at 1 kHz and digitized at 10 kHz with an output gain of one. One-second ACh (1 mm) was applied using a synthetic quartz perfusion tube (0.7 mm i.d.) operated by a computer-controlled valve (AutoMate Scientific, Berkeley, CA, USA).
Hippocampal slice cultures
Slice cultures were prepared as described previously (Dineley et al. 2001; Bell et al. 2004; Gu et al. 2012), which was adapted from (Stoppini et al. 2000). Brain slices of 300 μm were cut with a Leica VT1000S vibratome (Leica Microsystems, Buffalo Grove, IL, USA). The detachable parts of the vibratome and surgery instruments for dissecting brains were all autoclave sterilized. Briefly, mice (6–8 days old) were deeply anaesthetized with isoflurane, dipped in 70% ethanol, briefly rinsed with ice-cold cutting medium, and decapitated. Brains were quickly removed and placed into ice-cold cutting medium (MEM (pH 7.2; Life Technologies) supplemented with (in mm): 25 Hepes, 10 Tris base, 10 glucose, and 3 MgCl2). Horizontal hippocampal slices were cut in cutting medium with a vibratome; the hippocampi were then dissected out from the slices and placed onto Transwell® membrane inserts (Corning, Tewksbury, MA, USA) sitting in 6-well culture plates prefilled with 1.2 ml culture medium (pH 7.2), which was prepared as a 2:1 mixture of Eagle's basal medium (Sigma) and Earle's balanced salts solution (Sigma), and supplemented with (in mm) 20 NaCl, 5 NaHCO3, 0.2 CaCl2, 1.7 MgSO4, 48 glucose, 26.7 Hepes, 10 ml l−1 penicillin–streptomycin (Life Technologies), insulin (1.32 mg l−1) (Sigma), and 5% horse serum (Life Technologies). The slices were stored in a 5% CO2 incubator at 34°C and fed twice a week with a half change of media.
Virus infection
Cre cDNA was subcloned into an AAV vector with a neuron-specific synapsin promoter fused with tdTomato, kindly provided by Bernd Gloss (NIEHS) for neuronal expression. The viruses were packaged with serotype 9 helper at the Viral Vector Core at NIEHS. The AAV vector (Addgene no. 26972) was provided by Karl Deisseroth (Stanford University) and the AAV serotype 9 helper plasmid was from James Wilson (University of Pennsylvania). After 24 h in vitro, desired areas were injected with viruses (5 nl) using a Nanoject (Drummond Scientific, Broomall, PA, USA) microinjecting pipette. Experiments were done 10 days after virus infection to ensure adequate protein expression in the hippocampus.
Choline-induced α7 nAChR currents
α7 nAChR-mediated currents were recorded via whole cell patch clamp induced by pressure application (50 ms, 10 p.s.i.) of 10 mm choline (Sigma) (Fayuk & Yakel, 2004) (to selectively activate the α7 nAChRs) at 2 min intervals through a glass pipette located in the stratum radiatum area (about 150 μm away from the soma of the neurons under recording). Pressure was applied through a PPM-2 pneumatic pump (Harvard Apparatus, Holliston, MA, USA). TTX (1 μm) to the ACSF perfusate and choline solution.
Whole cell patch clamp was performed under the guidance of infrared differential interference contrast (IR-DIC) optics using an Axopatch 200B patch amplifier (Axon Instruments) with a glass pipette filled with an internal solution containing (in mm): 120 potassium gluconate, 2 NaCl, 5 MgATP, 0.3 Na2GTP, 20 KCl, 10 Hepes, and 1 EGTA, with pH ∼7.2–7.3 and osmolarity of ∼270–280 mosmol l–1. Data were digitized with the Digidata 1322A, collected with Clampex (Molecular Devices) and analysed with Clampfit (Molecular Devices). Choline-induced α7 nAChR currents were recorded under voltage clamp at −60 mV. Cre-expressing neurons were visualized with 543 nm laser light that was controlled by a Zeiss LSM 510 NLO META system. Choline-induced α7 nAChR currents were recorded in neurons located in the hippocampal CA1 pyramidal layer. α7 nAChR currents were selectively blocked using 10 nm methyllycaconitine (MLA, Sigma).
Statistics for electrophysiology
The amplitude and kinetics of ACh and of choline-induced α7 nAChR currents were analysed with Clampfit 10. Values were presented as mean ± SEM. Two-tailed Student's t tests were performed using Prism 5 software (GraphPad, La Jolla, CA, USA).
Results
The purpose of this study was to verify that the chosen exon 4 Chrna7 gene targeting strategy was accomplished at the molecular level and did not result in artifactual effects on baseline α7 nAChR currents as well as in vivo baseline health, behaviour, or cognitive status. We also confirmed that functional α7 nAChRs were absent from neurons expressing the Cre recombinase through viral infection of synapsin promoter-driven Cre recombinase and that offspring resulting from α7nAChRflox mated to glial fibrillary acidic protein (GFAP)-Cre hemizygous transgenic mice to generate a mouse strain in which α7 nAChR expression was selectively deleted from astrocytes (GFAP-A7KO).
Confirmation of targeted ES cells, neo deletion from targeted sensitive clones, and generation of α7nAChRflox mutant mice
From 288 G418-resistant colonies, eight potentially targeted clones were identified in the primary Southern blot screening using BamHI digestion of genomic DNA and the 5′-flanking probe to detect a 7.7 kb targeted allele (Fig. 3A). All eight were expanded and frozen. The confirmation of the potentially targeted clones was conducted by Southern blot using BamHI digestion, and 5′, 3′ and internal (neo) probes. As shown in Fig. 3B, five clones were properly targeted on both 5′ and 3′ ends (B4, D4, B11, D8 and G11). However, only one clone (G11) was devoid of random insertions of the targeting vector (judged by multiple bands on the Southern blot, Fig. 3) and was chosen for FLP-mediated deletion of the PGKneobpA cassette. The clone G11 was electroporated with FLP-expressing vector, 192 colonies were picked, and 18 G418-sensitive clones were identified. Subsequently, seven G418-sensitive clones were expanded and deletion of the neo cassette in these clones was confirmed by PCR (Fig. 3C). Following blastocyst transfer, heterozygous mutants were confirmed by PCR (Fig. 3D).
Figure 3.
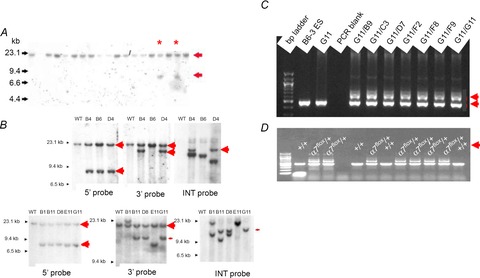
Screening and confirmation of ES cells injected with targeting vector and subsequent clones
A, BamHI-digested genomic DNA was agarose gel electrophoresed, then hybridized with 5′-flanking probe. The probe detected both a 20 kb wild-type (WT or +/+) and 7.7 kb targeted bands (arrows). Asterisks indicate correctly targeted heterozygote (+/−) genotypes. B, Southern blot confirmation of properly targeted ES cells following BamHI DNA digestion. Probe used for Southern blot is indicated on each gel frame. 5′ probe: wild-type = 20 kb; targeted allele = 7.7 kb. 3′ probe: wild-type = 20 kb; targeted allele = 14.4 kb. C, PCR confirmation of neo deletion in G418-sensitive ES clones from the original G11 subjected to neo deletion. WT (∼0.45 kb) and neo-deleted (∼0.6 kb) alleles are indicated by red arrows to the right of each figure. D, PCR confirmation of α7nAChRflox heterozygous (α7flox/+) mice.
Behavioural analyses
Both the α7nAChRflox and GFAP-A7KO cohorts were assessed for general health, neurological reflexes, baseline locomotor and anxiety-like behaviour, as well as cued and contextual fear conditioning. This battery of tests determined whether the floxed Chrna7 allele or deletion of Chrna7 from GFAP-positive astrocytes exhibited any overall detrimental effect resulting from the novel gene-targeting approach used to generate the animal models in this study.
Neurological reflex tests included eye blink, ear twitch and whisker twitch in response to a cotton-tipped swab gently approaching the sensory organ. Additional non-quantitative neurological tests that were performed included tail suspension, righting reflex and general home cage behaviours. All animals and groups exhibited normal behavioural responses and were not significantly different from each other by genotype (data not shown).
The quantitative physiological and baseline behavioural measures that were collected on the α7nAChRflox and GFAP-A7KO are shown in Fig. 4A. We found no significant differences when comparing either the α7nAChRflox with its control groups (WT and heterozygous α7nAChRflox) or the GFAP-A7KO with its control groups (WT and heterozygous GFAP-A7KO) in body weight, grip strength, tail flick and plantar tests, as well as in locomotor activity using the open field and elevated plus maze protocols. Furthermore, after completing a 2-pairings fear conditioning training protocol, α7nAChRflox and GFAP-A7KO cohorts did not exhibit significantly different freezing behaviours during testing for contextual and cued memory (Fig. 4B). These data suggest that acquisition and consolidation of hippocampus-dependent contextual and hippocampus-independent cued fear conditioning was equivalent within the two cohorts. One-way ANOVA test results for the data depicted in Fig. 4 are shown in Table 1. In all cases, neither Bartlett's test nor the Brown–Forsythe test for significantly different standard deviations detected significance (data not shown), suggesting that group numbers were sufficiently balanced. In conclusion, similar to ‘first generation’ non-conditional A7KO mice (Paylor et al. 1998), we found no deficiencies in any of these baseline general health, neurological, locomotor, or cognitive parameters.
Figure 4.

Baseline behaviour and cognitive testing of α7nAChRflox and GFAP-A7KO groups
Six genotypes within two statistical groups were monitored for general health and neurological reflexes (i.e. weight, grip strength, nociception, locomotor and anxiety-like behaviour; A) and cognition (top, training; middle, contextual; bottom: cued, B) for α7nAChRflox (first three bars in the histograms labelled WT, heterozygous, homozygous) or GFAP-A7KO (last three bars in the histograms labelled WT, heterozygous, homozygous). No significant differences were detected by one-way ANOVA within either group.
Table 1.
One-way ANOVA statistics for behavioural analysis of α7nAChRflox and GFAP-A7KO cohorts
| α7nAChRflox | GFAP-A7KO | ||||
|---|---|---|---|---|---|
| Assessment | F(2,47) | P value | F(2,42) | P value | |
| General health | Body weight | 0.79 | 0.46 | 1.2 | 0.30 |
| Neurological | |||||
| Analgesic threshold (nociception) | Tail flick | 1.6 | 0.23 | 1.1 | 0.35 |
| Plantar | 0.03 | 1.0 | 2.6 | 0.10 | |
| Exploratory behaviour | Open field (crossings) | 1.2 | 0.30 | 2.2 | 0.13 |
| Open field (total distance) | 0.69 | 0.51 | 0.67 | 0.52 | |
| EPM (time in open) | 0.77 | 0.47 | 0.47 | 0.63 | |
| EPM (open entries) | 2.3 | 0.11 | 0.89 | 0.42 | |
| Cognition (fear conditioning) | Training | 0.68 | 0.51 | 0.09 | 0.92 |
| Contextual | 3.7 | 0.11 | 2.6 | 0.18 | |
| Cued | 0.09 | 0.92 | 0.04 | 1.0 | |
F (including degrees of freedom) and P values obtained from one-way ANOVA statistical analyses of behavioural assessments for each test group, α7nAChRflox and GFAP-A7KO (Fig. 4A and B). These values demonstrate no significant differences between genotypes for either cohort. EPM, elevated plus maze.
α-Bungarotoxin binding in hippocampus of floxed α7nAChRflox and GFAP-A7KO mice
Since α7 nAChR are expressed by both neurons and glia (Sharma & Vijayaraghavan, 2001; Shytle et al. 2004; Shen & Yakel, 2012), we employed immunohistochemical methods to localize α7nAChR α-bungarotoxin (BTX) binding site distribution in the hippocampus of GFAP-A7KO and WT littermates. Although multiple regions were assessed (data not shown), we report findings in the hippocampus (CA1 and dentate gyrus (DG)) to complement our electrophysiological studies (Fig. 5). α7 nAChR binding sites were targeted with AlexaFluor-488 conjugated BTX and glia with Cy3-labelled anti-GFAP. DAPI counterstaining was utilized to identify nuclei under lower magnification. Both WT and GFAP-A7KO demonstrated similar BTX principal cell hippocampal expression, as previously described using 125I-BTX (Hernandez & Terry, 2005; Hernandez et al. 2006). Immunohistochemical analysis of BTX and GFAP demonstrates the absence of BTX binding sites on GFAP-positive astrocytes (Fig. 5H and P) in GFAP-A7KO hippocampus whereas we detected overlapping immunoreactivity on GFAP-positive astrocytes in hippocampus of WT littermates (Fig. 5D and L). Additionally, no gross hippocampal morphological differences were observed in cresyl violet-stained sections of α7nAChRflox or GFAP-A7KO (Fig. 6). These results suggest that α7 nAChRs were selectively deleted from astrocytes through the genetic cross of α7nAChRflox mice with GFAP-Cre mice. Furthermore, astrocytic deletion of α7 nAChRs does not induce any gross brain morphological abnormalities and is consistent with the behavioural testing performed thus far.
Figure 5.

Loss of α7nAChR expression on GFAP-positive cells in GFAP-A7KO hippocampus
Immunohistochemical localization of glia using anti-GFAP and α7 nAChR α-bungarotoxin (BTX) binding sites in CA1 (A–H) and dentate gyrus (DG) (I–P). Sections were counterstained with DAPI to localize nuclei: WT CA1 (A–D) and GFAP-A7KO CA1 (E–H); WT DG (I–L), GFAP-A7KO DG (M–P). Scale bar, 100 μm (A,E,I,M); all others, 10 μm.
Figure 6.

No gross morphological abnormalities in GFAP-A7KO hippocampus
Cresyl violet staining of representative α7nAChRflox (A) and GFAP-A7KO (B) sagittal brain sections indicates normal hippocampal anatomy and morphology. Scale bar, 300 μm.
Electrophysiological analyses
Next, we performed a set of electrophysiological experiments to determine that the functional properties of α7 nAChRs in α7nAChRflox mice were unaltered compared to WT. Representative ACh-evoked responses (Fig. 7) from hippocampal neurons at DIV 14–15 from WT (Fig. 7A) and α7nAChRflox (Fig. 7B) mice demonstrate that current amplitude (Fig. 7C) and desensitization kinetics (Fig. 7D) were not significantly different and matched previously published values for α7 nAChRs expressed in hippocampal pyramidal neurons (Colón-Sáez & Yakel, 2011). Therefore these basic functional properties of the α7 nAChRs were similar between WT and α7nAChRflox mice.
Figure 7.
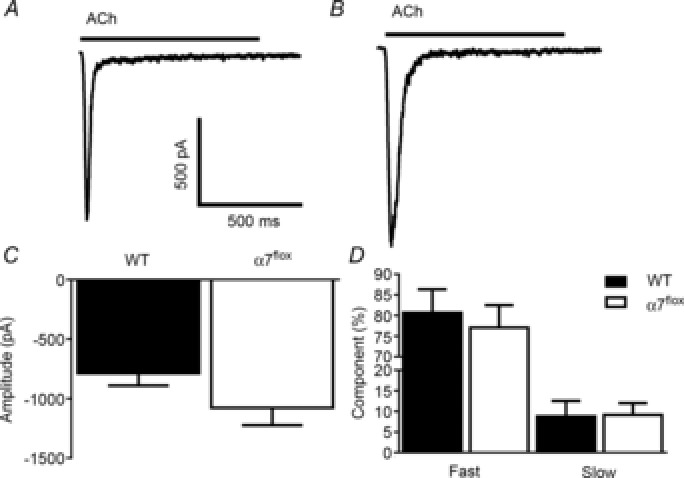
ACh-induced currents in WT and α7nAChRflox hippocampus exhibit similar amplitude and desensitization kinetics
Representative traces of ACh-evoked (1 mm) responses from cultured hippocampal neurons from WT (n = 7 neurons, A) and α7nAChRflox (α7flox; n = 6 neurons, B) at DIV 14–15. Current amplitudes (C) and the ratio of the fast and slow time constants of decay during desensitization (D) were not significantly different. The amplitude values as well as the percentage of fast and slow time constants of decay for WT were 787 ± 103 pA, 81 ± 6% and 9 ± 4%, respectively. For the α7flox mice, these values were 1078 ± 147 pA, 77 ± 5% and 9 ± 3%, respectively.
Next, we verified the effectiveness of Cre recombinase to control the functional expression of α7 nAChRs in α7nAChRflox mice by monitoring α7 nAChR-mediated currents in Cre-positive and -negative hippocampal pyramidal neurons in cultured slices (Fig. 8). α7 nAChR-mediated currents were recorded with whole cell patch clamp, and induced by pressure applications of choline. We found comparable choline-induced currents from α7nAChRflox and WT in the absence of Cre expression. The choline-induced currents in WT neurons were blocked by MLA, a specific α7 nAChR antagonist, indicating they were mediated by α7 nAChRs. The α7 nAChR-mediated currents were absent in Cre-expressing neurons from the α7nAChRflox mice, while normal α7 nAChR-mediated currents were recorded in Cre-expressing neurons from WT mice. These results indicate normal α7 nAChR function in α7nAChRflox mice, and the effective Cre-driven removal of functional α7 nAChRs in neurons expressing the recombinase.
Figure 8.
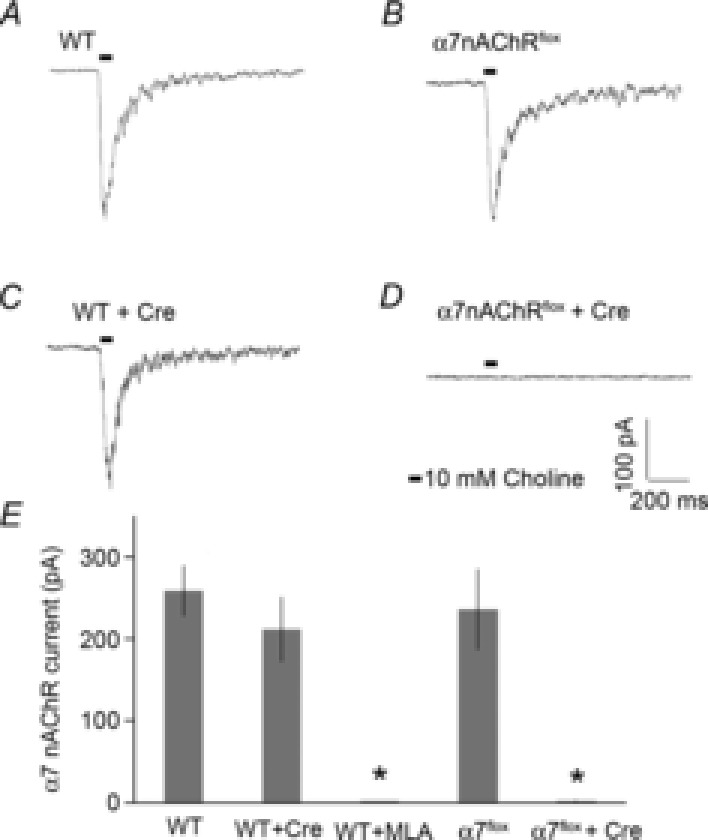
Choline-induced α7 nAChR currents are absent in Cre-infected neurons in organotypic hippocampal slice cultures from α7nAChRflox mice
A, typical traces of choline-induced currents from whole cell patch clamp recording of WT CA1 pyramidal neurons (258 ± 31 pA, n = 10 neurons). The currents were induced by pressure application (10 p.s.i. for 50 ms) of 10 mm choline to the CA1 stratum radiatum. B, Cre expression did not change α7 nAChR current properties recorded from WT neurons (212 ± 39 pA, n = 6 neurons). C, choline-induced currents recorded from α7nAChRflox hippocampal neurons were similar to those obtained from WT in the absence of Cre expression (236 ± 49 pA, n = 8 neurons). D, Cre-expression completely eliminated the choline-induced current in α7nAChRflox neurons (2.5 ± 1 pA, n = 6 neurons). E, bar graph depicting α7 nAChR CA1 pyramidal neuron current properties from WT or α7nAChRflox slices in the absence and presence of Cre expression. WT currents were completely blocked by the specific α7 nAChR antagonist methyllycacotinine (MLA) (2.6 ± 1 pA, n = 6 neurons). *P < 0.001 significant from WT, Student's t test.
These results demonstrate that the Chrna7 exon 4 targeting strategy did not result in unexpected gain-of-function properties and GFAP-Cre did not cause any abnormalities since the parent floxed animal and the resultant constitutive tissue-specific GFAP-A7KO line are viable, fertile, and do not possess any baseline behavioural or hippocampus-dependent cognitive abnormalities, as determined for the original A7KO mice (Orr-Urtreger et al. 1997; Paylor et al. 1998). We also provide evidence that α7 nAChR expression is lost in the presence of Cre recombinase.
Discussion
To date, a single α7 nAChR null mouse model is available in which expression is lost from all tissues constitutively (Orr-Urtreger et al. 1997). There is much interest in α7 nAChRs in peripheral and central disease states since these receptors are expressed on peripheral immune cells (Wang et al. 2003) and neurons (Si & Lee, 2002) as well as in brain regions that underlie learning and memory (Leiser et al. 2009; Levin, 2012) and several CNS disease states including nicotine reward and withdrawal, schizophrenia and Alzheimer's disease (AD) (Hernandez & Dineley, 2012; Leslie et al. 2013; Wallace & Bertrand, 2013). Furthermore, these receptors are developmentally regulated (Liu et al. 2007); therefore, there is an unmet need for the technical capability to control α7 nAChR gene expression to specific cell types or tissues. For example, this receptor has long been considered a viable AD therapeutic target due to its well-established interaction with amyloid-β (Aβ) peptide (Dineley, 2007; Parri & Dineley, 2010) and its broad distribution within the septo-hippocampal cholinergic system, which is a particularly vulnerable brain network in early AD (Hernandez & Dineley, 2012). Interestingly, α7 nAChRs are expressed on astrocytes (Sharma & Vijayaraghavan, 2001; Shen & Yakel, 2012). We and others recently showed that a key mediator of astrocyte α7 nAChR function is Aβ, a ubiquitously expressed signalling peptide and putative toxic trigger for AD (Parri & Dineley, 2010; Pirttimaki et al. 2013; Talantova et al. 2013; Lee et al. 2014). Astrocytes play a critical role in synaptic transmission, plasticity and memory formation through the release of chemical transmitters (gliotransmitters) such as glutamate (Pirttimaki et al. 2011; Hill et al. 2012; Pirttimaki & Parri, 2012). Thus, astrocytes are probably a significant component of the molecular, synaptic and cognitive consequences of aberrant Aβ accumulation in AD pathogenesis that have largely been overlooked (Teaktong et al. 2003); these proof-of-concept studies position us to specifically address their role in AD initiation and progression in preclinical models as well as several other cell-type-specific roles (e.g. neurogenesis and silent synapse development; Campbell et al. 2010; Wang et al. 2013).
A7KO mice were originally developed in the laboratories of Dr James Patrick and Dr Arthur Beaudet (Orr-Urtreger et al. 1997). The deletion strategy targeted exons 8–10; initial studies were performed on mice maintained on a mixed 129/SvEv and C57BL/6J background and were shown to lack α-bungarotoxin-binding sites and α7 nAChR-mediated currents in the hippocampus. This mutant mouse strain was then back-crossed onto the C57BL/6J background followed by behavioural analysis. An extensive analysis of general health, baseline behaviour and cognition found no apparent deficits (Paylor et al. 1998); however, subsequent studies have found altered baroreflex responses (Franceschini et al. 2000), asynchronous oestrous cycles (Morley & Rodriguez-Sierra, 2004), enhanced impulsivity (Reynolds & Hoidal, 2005), and attention deficits using prolonged sessions (Young et al. 2004; Guillem et al. 2011). Since the current work focused on an extensive validation of the α7nAChRflox mouse model as a research tool, future work will be necessary to determine if more recently discovered subtleties in the original A7KO model's phenotype are purely due to the receptor's absence at maturity or if they play a developmental role.
Using Cre-loxP technology, we developed a mouse line in which α7 nAChR expression can be deleted through a genetic cross with Cre-expressing mice or infection with a Cre recombinase-containing virus. We demonstrated that the parent floxed α7nAChRflox line appeared in good general health and exhibited normal baseline behaviour and cognition as did a line crossed with a GFAP-Cre transgenic mouse line using our standard neurological and behavioural battery of tests for new animal models (UTMB Rodent In Vivo Assessment Core directed by K.T.D.). We also demonstrated that α7 nAChRs were selectively deleted from astrocytes by showing an absence of BTX binding on GFAP-positive cells in GFAP-A7KO. Therefore, similar to the ‘first generation’ non-conditional A7KO mice, we found no deficiencies in any of these baseline physical and behavioural parameters. In addition, electrophysiological recordings in both neuronal and organotypic slice cultures prepared from hippocampus showed that the α7 nAChR-mediated currents were similar in the α7nAChRflox mice indicating that our loxP-targeting strategy did not alter normal channel function. Furthermore, neurons in cultured slices expressing the Cre recombinase driven by the neuron-specific synapsin promoter, α7 nAChR-mediated currents were completely absent. Thus, we have in vivo and in vitro evidence that the Chrna7 exon 4 targeting strategy is an effective approach to delete α7 nAChR expression in a cell-specific manner. The mouse we have engineered functions as intended, and therefore will be a valuable tool to take the α7 nAChR field to new levels.
Acknowledgments
None declared.
Glossary
- AD
Alzheimer's disease
- BTX
α-bungarotoxin
- Cre
Cre recombinase
- ES cell
embryonic stem cell
- FLP
flippase
- GFAP
glial fibrillary acidid protein
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- WT
wild-type
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
Experiments were performed in the laboratories of J.L.Y. and K.T.D. Conception of the experiments: J.L.Y. and K.T.D. Design of the experiments: C.M.H., M.W., J.L.Y. and K.T.D. Collection, analysis and interpretation of data: C.M.H., I.C., Z.G., J.O.C.-S., P.W.L., M.W., J.Y.L., K.T.D. Drafting the article or revising it critically for important intellectual content: C.M.H., I.C., Z.G., J.O.C.-S., P.W.L., M.W., J.L.Y. and K.T.D. All authors approved the final version of the manuscript. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This work was supported by the Intramural Research Program, NIEHS/NIH to J.L.Y., a kind gift to K.T.D. from J. & W. Mohn, and an Independent Investigator Research Grant from the Alzheimer's Association to K.T.D.
References
- Bell KA, O'Riordan KJ, Sweatt JD, Dineley KT. MAPK recruitment by β-amyloid in organotypic hippocampal slice cultures depends on physical state and exposure time. J Neurochem. 2004;91:349–361. doi: 10.1111/j.1471-4159.2004.02722.x. [DOI] [PubMed] [Google Scholar]
- Brown KL, Comalli DM, De Biasi M, Woodruff-Pak DS. Trace eyeblink conditioning is impaired in α7 but not in β2 nicotinic acetylcholine receptor knockout mice. Front Behav Neurosci. 2010;4:166. doi: 10.3389/fnbeh.2010.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW, Berg DK. Endogenous signaling through α7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci. 2010;30:8734–8744. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colón-Sáez JO, Yakel JL. The α7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J Physiol. 2011;589:3163–3174. doi: 10.1113/jphysiol.2011.209494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- Denner LA, Rodriguez-Rivera J, Haidacher SJ, Jahrling JB, Carmical JR, Hernandez CM, Zhao Y, Sadygov RG, Starkey JM, Spratt H, Luxon B, Wood TG, Dineley KT. Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. J Neurosci. 2012;32:16725–16735. doi: 10.1523/JNEUROSCI.2153-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT. Beta-amyloid peptide–nicotinic acetylcholine receptor interaction: the two faces of health and disease. Front Biosci. 2007;12:5030–5038. doi: 10.2741/2445. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits and up-regulation of α7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- Dominguez del Toro E, Juiz JM, Peng X, Lindstrom J, Criado M. Immunocytochemical localization of the alpha 7 subunit of the nicotinic acetylcholine receptor in the rat central nervous system. J Comp Neurol. 1994;349:325–342. doi: 10.1002/cne.903490302. [DOI] [PubMed] [Google Scholar]
- Drago J, McColl CD, Horne MK, Finkelstein DI, Ross SA. Neuronal nicotinic receptors: insights gained from gene knockout and knockin mutant mice. Cell Mol Life Sci. 2003;60:1267–1280. doi: 10.1007/s00018-003-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayuk D, Yakel JL. Regulation of nicotinic acetylcholine receptor channel function by acetylcholinesterase inhibitors in rat hippocampal CA1 interneurons. Mol Pharmacol. 2004;66:658–666. doi: 10.1124/mol.104.000042. [DOI] [PubMed] [Google Scholar]
- Franceschini D, Orr-Urtreger A, Yu W, Mackey LY, Bond RA, Armstrong D, Patrick JW, Beaudet AL, De Biasi M. Altered baroreflex responses in α7 deficient mice. Behav Brain Res. 2000;113:3–10. doi: 10.1016/s0166-4328(00)00195-9. [DOI] [PubMed] [Google Scholar]
- Gallowitsch-Puerta M, Tracey KJ. Immunologic role of the cholinergic anti-inflammatory pathway and the nicotinic acetylcholine α7 receptor. Ann N Y Acad Sci. 2005;1062:209–219. doi: 10.1196/annals.1358.024. [DOI] [PubMed] [Google Scholar]
- Gu Z, Lamb PW, Yakel JL. Cholinergic coordination of presynaptic and postsynaptic activity induces timing-dependent hippocampal synaptic plasticity. J Neurosci. 2012;32:12337–12348. doi: 10.1523/JNEUROSCI.2129-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillem K, Bloem B, Poorthuis RB, Loos M, Smit AB, Maskos U, Spijker S, Mansvelder HD. Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science. 2011;333:888–891. doi: 10.1126/science.1207079. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Dineley KT. α7 nicotinic acetylcholine receptors in Alzheimer's disease: neuroprotective, neurotrophic or both. Curr Drug Targets. 2012;13:613–622. doi: 10.2174/138945012800398973. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Gearhart DA, Parikh V, Hohnadel EJ, Davis LW, Middlemore ML, Warsi SP, Waller JL, Terry AV. Comparison of galantamine and donepezil for effects on nerve growth factor, cholinergic markers, and memory performance in aged rats. J Pharmacol Exp Ther. 2006;316:679–694. doi: 10.1124/jpet.105.093047. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Terry AV. Repeated nicotine exposure in rats: effects on memory function, cholinergic markers and nerve growth factor. Neuroscience. 2005;130:997–1012. doi: 10.1016/j.neuroscience.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Hill EJ, Jiménez-González C, Tarczyluk M, Nagel DA, Coleman MD, Parri HR. NT2 derived neuronal and astrocytic network signalling. PloS One. 2012;7:e36098. doi: 10.1371/journal.pone.0036098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JJ, Keller AB, Bowers BJ, Wehner JM. Performance of α7 nicotinic receptor null mutants is impaired in appetitive learning measured in a signalled nose poke task. Behav Brain Res. 2005;162:143–152. doi: 10.1016/j.bbr.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Lee L, Kosuri P, Arancio O. Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients. J Alzheimers Dis. 2014;38:49–62. doi: 10.3233/JAD-130740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser SC, Bowlby MR, Comery TA, Dunlop J. A cog in cognition: how the α7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol Ther. 2009;122:302–311. doi: 10.1016/j.pharmthera.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Leslie FM, Mojica CY, Reynaga DD. Nicotinic receptors in addiction pathways. Mol Pharmacol. 2013;83:753–758. doi: 10.1124/mol.112.083659. [DOI] [PubMed] [Google Scholar]
- Levin ED. α7-Nicotinic receptors and cognition. Curr Drug Targets. 2012;13:602–606. doi: 10.2174/138945012800398937. [DOI] [PubMed] [Google Scholar]
- Levin ED, Petro A, Rezvani AH, Pollard N, Christopher NC, Strauss M, Avery J, Nicholson J, Rose JE. Nicotinic α7- or β2-containing receptor knockout: effects on radial-arm maze learning and long-term nicotine consumption in mice. Behav Brain Res. 2009;196:207–213. doi: 10.1016/j.bbr.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhang J, Berg DK. Role of endogenous nicotinic signaling in guiding neuronal development. Biochem Pharmacol. 2007;74:1112–1119. doi: 10.1016/j.bcp.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley BJ, Rodriguez-Sierra JF. A phenotype for the α7 nicotinic acetylcholine receptor null mutant. Brain Res. 2004;1023:41–47. doi: 10.1016/j.brainres.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack α-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Seldin MF, Baldini A, Beaudet AL. Cloning and mapping of the mouse α7-neuronal nicotinic acetylcholine receptor. Genomics. 1995;26:399–402. doi: 10.1016/0888-7543(95)80228-e. [DOI] [PubMed] [Google Scholar]
- Parri HR, Dineley KT. Nicotinic acetylcholine receptor interaction with β-amyloid: molecular, cellular, and physiological consequences. Curr Alzheimer Res. 2010;7:27–39. doi: 10.2174/156720510790274464. [DOI] [PubMed] [Google Scholar]
- Paylor R, Nguyen M, Crawley JN, Patrick J, Beaudet A, Orr-Urtreger A. α7 nicotinic receptor subunits are not necessary for hippocampal-dependent learning or sensorimotor gating: a behavioral characterization of Acra7-deficient mice. Learn Mem. 1998;5:302–316. [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Codadu NK, Awni A, Pratik P, Nagel DA, Hill EJ, Dineley KT, Parri HR. α7 nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer's mouse model. PloS One. 2013;8:e81828. doi: 10.1371/journal.pone.0081828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Hall SD, Parri HR. Sustained neuronal activity generated by glial plasticity. J Neurosci. 2011;31:7637–7647. doi: 10.1523/JNEUROSCI.5783-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Parri HR. Glutamatergic input-output properties of thalamic astrocytes. Neuroscience. 2012;205:18–28. doi: 10.1016/j.neuroscience.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitsch A, Berhorster K, Liew CW, Richter D, Kreienkamp HJ. Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein Densin-180. J Neurosci. 2005;25:479–487. doi: 10.1523/JNEUROSCI.2699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds PR, Hoidal JR. Temporal-spatial expression and transcriptional regulation of α7 nicotinic acetylcholine receptor by thyroid transcription factor-1 and early growth response factor-1 during murine lung development. J Biol Chem. 2005;280:32548–32554. doi: 10.1074/jbc.M502231200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rivera J, Denner L, Dineley KT. Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behav Brain Res. 2011;216:255–261. doi: 10.1016/j.bbr.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubboli F, Court JA, Sala C, Morris C, Chini B, Perry E, Clementi F. Distribution of nicotinic receptors in the human hippocampus and thalamus. Eur J Neurosci. 1994a;6:1596–1604. doi: 10.1111/j.1460-9568.1994.tb00550.x. [DOI] [PubMed] [Google Scholar]
- Rubboli F, Court JA, Sala C, Morris C, Perry E, Clementi F. Distribution of neuronal nicotinic receptor subunits in human brain. Neurochem Int. 1994b;25:69–71. doi: 10.1016/0197-0186(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc Natl Acad Sci U S A. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JX, Yakel JL. Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci. 2012;48:14–21. doi: 10.1007/s12031-012-9719-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shytle RD, Mori T, Townsend K, Vendrame M, Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR, Tan J. Cholinergic modulation of microglial activation by α7 nicotinic receptors. J Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- Si ML, Lee TJ. α7-nicotinic acetylcholine receptors on cerebral perivascular sympathetic nerves mediate choline-induced nitrergic neurogenic vasodilation. Circ Res. 2002;91:62–69. doi: 10.1161/01.res.0000024417.79275.23. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Brun R, Muller D, Duport S, Parisi L, Seebeck T. Infection of organotypic slice cultures from rat central nervous tissue with Trypanosoma brucei brucei. Int J Med Microbiol. 2000;290:105–113. doi: 10.1016/S1438-4221(00)80113-7. [DOI] [PubMed] [Google Scholar]
- Taglialatela G, Hogan D, Zhang WR, Dineley KT. Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav Brain Res. 2009;200:95–99. doi: 10.1016/j.bbr.2008.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang YJ, Tu S, Molokanova E, McKercher SR, Hires SA, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, Powers ET, Kelly JW, Roberts A, Tong G, Fang-Newmeyer T, Parker J, Holland EA, Zhang D, Nakanishi N, Chen HS, Wolosker H, Wang Y, Parsons LH, Ambasudhan R, Masliah E, Heinemann SF, Pina-Crespo JC, Lipton SA. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–E2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, Hall R, Perry E. Alzheimer's disease is associated with a selective increase in α7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia. 2003;41:207–211. doi: 10.1002/glia.10132. [DOI] [PubMed] [Google Scholar]
- Wallace TL, Bertrand D. Alpha7 neuronal nicotinic receptors as a drug target in schizophrenia. Expert Opin Ther Targets. 2013;17:139–155. doi: 10.1517/14728222.2013.736498. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Wang X, Lippi G, Carlson DM, Berg DK. Activation of α7-containing nicotinic receptors on astrocytes triggers AMPA receptor recruitment to glutamatergic synapses. J Neurochem. 2013;127:632–643. doi: 10.1111/jnc.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Paxinos G, Puelles L. The Mouse Nervous System. London: Academic Press; 2012. [Google Scholar]
- Young JW, Finlayson K, Spratt C, Marston HM, Crawford N, Kelly JS, Sharkey J. Nicotine improves sustained attention in mice: evidence for involvement of the α7 nicotinic acetylcholine receptor. Neuropsychopharmacology. 2004;29:891–900. doi: 10.1038/sj.npp.1300393. [DOI] [PubMed] [Google Scholar]
- Young JW, Geyer MA. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatment of schizophrenia. Biochem Pharmacol. 2013;86:1122–1132. doi: 10.1016/j.bcp.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Meves JM, Tarantino IS, Caldwell S, Geyer MA. Delayed procedural learning in α7-nicotinic acetylcholine receptor knockout mice. Genes Brain Behav. 2011;10:720–733. doi: 10.1111/j.1601-183X.2011.00711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoheir N, Lappin DF, Nile CJ. Acetylcholine and the alpha 7 nicotinic receptor: a potential therapeutic target for the treatment of periodontal disease. Inflamm Res. 2012;61:915–926. doi: 10.1007/s00011-012-0513-z. [DOI] [PMC free article] [PubMed] [Google Scholar]