

Abstract
Dystrophin is a large, submembrane cytoskeletal protein, absence of which causes Duchenne muscular dystrophy. Utrophin is a dystrophin homologue found in both muscle and brain whose physiological function is unknown. Recordings of single-channel activity were made from membrane patches on skeletal muscle from mdx, mdx/utrn+/– heterozygotes and mdx/utrn–/– double knockout mice to investigate the role of these cytoskeletal proteins in mechanosensitive (MS) channel gating. We find complex, gene dose-dependent effects of utrophin depletion in dystrophin-deficient mdx muscle: (1) increased MS channel open probability, (2) a shift of MS channel gating to larger pressures, (3) appearance of modal gating of MS channels and small conductance channels and (4) expression of large conductance MS channels. We suggest a physical model in which utrophin acts as a scaffolding protein that stabilizes lipid microdomains and clusters MS channel subunits. Depletion of utrophin disrupts domain composition in a manner that favours open channel area expansion, as well as allowing diffusion and aggregation of additional MS channel subunits.
Key points
Loss of the cytoskeletal protein dystrophin leads to muscle degeneration in Duchenne muscular dystrophy. The mdx mouse lacks dystrophin but muscle disease is mild, in part because upregulation of utrophin compensates for dystrophin. Evidence suggests that Ca2+ entry through mechanosensitive (MS) ion channels contributes to disease pathogenesis, but the role of utrophin in regulating Ca2+ entry has not been studied.
Depletion of utrophin in mdx mice produces a gene dose-dependent increase in MS channel open probability, single-channel conductance in some patches, and an increase in the pressure required to MS channels. The overall effect of loss of utrophin is increased Ca2+ entry.
The results suggest utrophin organizes local membrane microdomains containing MS channels. Disruption of these domains causes changes in membrane composition, which favour channel open states and subunit aggregation. Utrophin-replacement therapies may act by correcting MS channel gating, and specific MS channel inhibitors have the potential to prevent disease progression.
Introduction
Skeletal muscle possesses a highly structured cytoskeletal lattice designed to prevent membrane damage by contraction-induced stresses (Lynch, 2004). A main component of this protein assembly is dystrophin, a 427 kDa cytoskeletal protein, absence of which causes Duchenne muscular dystrophy (DMD) in humans. Dystrophin is held tightly to the membrane by a glycoprotein complex composed of six sarcoglycans (α, β, δ, γ, ε, ω) and two dystroglycans (α and β), syntrophin and dystrobrevin (reviewed by Ervasti, 2007). The glycoprotein complex binds to laminin in the extracellular basement membrane and the entire complex links the extracellular matrix to the intracellular actin cytoskeletal network. While much is known about the organization of the dystrophin–glycoprotein complex, few studies have addressed the precise physiological function of this complex cystoskeletal–membrane protein interactome.
The mdx mouse lacks full-length dystrophin and has been used extensively as a model for human DMD (Dangain & Vrbova, 1984; Tanabe et al. 1986; Carnwath & Shotton, 1987). A body of evidence shows an increase in intracellular calcium concentration ([Ca2+]i) is an early stage of disease pathogenesis. We and others have reported that mechanosensitive (MS) ion channels in muscle from mdx mice show prolonged openings that probably contribute to increased resting Ca2+ fluxes (Franco & Lansman, 1990b; Franco-Obregon & Lansman, 1994; De Backer et al. 2002; Suchyna & Sachs, 2007; reviewed by Lansman & Franco-Obregon, 2006; Lansman, 2007). In addition, pharmacological blockers of MS channels block an early rise in [Ca2+]i in mdx skeletal muscle fibres and prevent contraction-induced membrane damage (Yeung et al. 2005; Whitehead et al. 2006).
We recently reported evidence that transient receptor potential vanilloid 4 (TRPV4) contributes to the MS channel in skeletal muscle (Ho et al. 2012). TRPV4 is expressed in mouse skeletal muscle where it contributes to resting Ca2+ influx (Pritschow et al. 2011). Other studies, however, suggest that transient receptor potential channel 1 (TRPC1) contributes to Ca2+ entry in normal and mdx muscle (Vandebrouck et al. 2002; Millay et al. 2009; Zhang et al. 2012). In addition, TRPV2 contributes to Ca2+ entry and contraction-induced injury in mdx muscle (Iwata et al. 2003; Zanou et al. 2009). Given the diversity of TRP channel proteins in skeletal muscle (reviewed by Gailly, 2012), the relative contributions of various TRP channel proteins to Ca2+ entry in normal and dystrophic muscle remain of debate.
While the mdx mouse has been a useful model for studying the pathogenesis of muscular dystrophy, it is limited by the fact that disease progression is much less severe than in human DMD. Although there is some degeneration of muscle in mdx mice, overall muscle performance and life expectancy are mostly normal. The mild disease phenotype of the mdx mouse may result from compensation for the loss of dystrophin by an increased expression of utrophin, an autosomal gene product with significant homology to dystrophin (Love et al. 1989; Tinsley et al. 1992; Deconinick et al. 1997). Utrophin is highly expressed in the membrane of fetal and regenerating muscle, but its expression is markedly reduced at birth and, subsequently, it is found localized mainly at the myotendinous and neuromuscular junctions of adult muscle (Gramolini & Jasmin, 1999; Weir et al. 2002, 2004; reviewed by Blake et al. 2002). In the mdx mouse, utrophin binds to several dystrophin-associated proteins (Matsumura et al. 1992). Utrophin is also expressed in the brain in choroid plexus, blood vessels and neuronal soma, although the precise function is not understood (reviewed by Perronnet & Vaillend, 2010).
Although knockout of utrophin produces phenotypically normal mice with only minor structural abnormalities at the neuromuscular junction (Grady et al. 1997), mice lacking both dystrophin and utrophin have severe muscle disease that more closely resembles DMD in humans (Deconinck et al. 1997; Grady et al. 1997). The double knockout mice weigh less, show marked deformity and impaired mobility, have difficulty breathing, and die within 8–10 weeks. Increased transgenic expression of utrophin reduces muscle pathology in the mdx mouse (Squire et al. 2002) and efforts to discover a treatment for DMD have focused on genetic approaches to replace or increase expression of utrophin (reviewed by Fairclough et al. 2013). Despite more than 15 years of research on utrophin, its physiological function is unknown.
In this paper, we examine the effects of genetic depletion of utrophin on the behaviour of single MS channels in skeletal muscle from mdx, mdx/utrn+/– heterozyotes and mdx/utrn–/– double knockout (DKO) mice. Patch recordings provide high spatial resolution and information on single molecule dynamics of MS channels reflecting its coupling to the dystrophin-associated glycoprotein network. Our first goal was to test whether an absence of utrophin in mdx muscle influences ion fluxes through MS channels to provide support for a role in the pathogenesis of muscular dystrophy. We find, however, much more complex effects of genetic depletion of utrophin in mdx muscle. Loss of only a single utrophin allele in mdx muscle modifies MS channel gating behaviour so that channels enter a prolonged opening mode. We also find an increased single-channel conductance of MS channels in some patches. In addition, depletion of utrophin leads to changes in pressure-dependence of channel gating and the probability of occupancy of subconductance states during single activations. The results suggest a model in which utrophin stabilizes membrane microdomains containing MS channels. Genetic depletion of utrophin may cause breakdown of domain structure, allowing bilayer expansion which favours both MS channel opening, as well as recruitment and clustering of individually gating channel subunits.
Methods
Experimental animals
mdx (C57BL/10Scn-mdx) mice were obtained from the Jackson Laboratory (Bar Harbour, ME, USA). Utrophin-deficient mice (strain B6.129-Utrntm1Jrs) were obtained from the Mutant Mouse Regional Resource Center (MMRRC) at the University of California, Davis, and were originally donated by Dr Joshua Sanes. DKO mice were obtained using the following breeding scheme: female mdx mice were mated with male utrophin-deficient mice. Male F1 offspring (XmdxY/utrn+/–) were mated with mdx females (XmdxXmdx/utrn+/+) to produce an F2 generation consisting of mdx/utrn+/+ and mdx/utrn+/– mice of both sexes. Mating male and female F2 mdx/utrn+/– mice produced progeny that were 25% double knockout mdx/utrn–/–; remaining progeny were either mdx/utrn+/– (50%) or mdx/utrn+/+ (25%). Genotyping was performed by Transnetyx Corp. (Memphis, TN, USA) from tail samples using PCR to identify the dystrophin mutation, the utrophin wild-type gene and the neomycin insert used to produce the utrophin transgenic knockout. DKO mice could be distinguished at about 2–3 weeks of age by their small size compared with normal sized mdx littermates. All experiments were performed on mice between 2 and 4 weeks of age. Mice were killed after isoflurane anaesthesia according to a protocol approved by the UCSF Committee on Animal Research. Methods for isolating single skeletal muscle fibres and general electrophysiological methods are identical to those described in previous studies (Franco-Obregón & Lansman, 1994, 2002: Vasquez et al. 2012). Dissociated single fibres were incubated in oxygenated culture media at 34°C. After each experiment, we rinsed the experimental chamber with normal saline and added a fresh aliquot of cells that were stored in incubation media.
Electrophysiological methods
Patch electrodes were pulled in two steps from borosilicate capillaries (Custom 8520, Warner Instruments, Hamden, CT, USA), the shanks coated with Sylgard (Dow Corning, Midland, MI, USA) close to the tips, and the tips heat polished to give a resistance of 3–5 MΩ when filed with a physiological saline solution and immersed in an isotonic K+-aspartate bathing solution. The electrode solution contained (mm): 150 NaCl, 5 KCl, 1 MgCl2, 10 EGTA, 10 Hepes and 17 glucose. The pH was adjusted to 7.4 by adding tetraethylammonium hydroxide (TEA-OH). The isotonic K+ bathing solution contained (mm): 150 potassium aspartate, 5 MgCl2, 10 EGTA, 10 Hepes and 10 glucose. The pH was adjusted to 7.4 with TEA-OH. The high K+ bathing solution was used to zero the cell's membrane potential so the patch potential would be the same as the voltage command. Excision of the membrane patch from the cell at the end of an experiment indicated a voltage error <5 mV. Experiments in which the electrode potential drifted by more than 5 mV at the end of an experiment were not used for analysis. We also excluded patches in which the uncorrected leakage current exceeded 5 pA.
Single-channel currents were recorded with an EPC-9 patch clamp amplifier (HEKA Instruments, Southboro, MA, USA) using Pulse (HEKA) software for stimulus generation and data acquisition. Pressure was applied to the patch electrode using a pressure clamp device (HSPC-1, ALA Scientific, Farmingdale, NY, USA). During an experiment, the tip of the electrode was gently placed on the surface of an isolated muscle fibre and negative pressure of −3 to −5 mmHg applied to the electrode via the pressure clamp. Gigaohm seals formed within ∼30 s after the pressure was returned to 0 mmHg.
Data analysis
Patch currents were digitized at either 2.5 or 50 kHz, filtered at 1 or 10 kHz, and stored directly on the hard disk of a Power Mac G4. Digitized currents were exported into Igor Pro (Wavemetrics, Portland, OR, USA) or TAC (Bruxton Corp., Seattle, WA, USA) for analysis. Subconductance transitions were analysed by compiling amplitude histograms of the open channel current. Cursors were set by visual inspection to mark the period during which the channel was open and to exclude transitions to the fully closed level. Data points were accumulated from a large number of openings in an experiment. The accuracy of this procedure depends critically on subtraction of linear leakage current. We corrected records of single-channel activity by fitting a line through neighbouring records without channel activity and subtracting the fitted line from the record. The baseline correction was updated throughout the recording and was checked by forming an amplitude histogram of closed channel noise and fitting it with a single Gaussian to ensure the peak corresponded to zero current (±0.05 pA). Amplitude histograms of the open channel current were fit with the sum of Gaussian components. The number of components was determined with use of a maximum-likelihood routine which fitted all points in the distribution of current amplitudes (see Vasquez et al. 2012). An additional component was fitted to the distribution if it maximized likelihood values by 5 units or more. The starting parameters were changed several times for each fit to ensure we obtained the best fit as judged by convergence and maximization of likelihood values.
Open times and burst durations were measured with TAC event detection software using a threshold detection algorithm for event idealization. We set the threshold for event detection at 60–70% of the fully open current. While this approach misses short events, it excluded small openings that fail to reach the fully open state (see Vasquez et al. 2012). Setting a high threshold also excluded most false events triggered by baseline noise fluctuations. Because MS channels fluctuate between subconductance levels, threshold detection captures only transitions between the fully closed and fully open states and larger subconductance levels. However, the threshold detection procedure captures longer closings with sufficient accuracy to identify bursts of openings. In this study, we did not analyse closed times because of patch to patch variability in brief spike-like openings, which may arise from acetylcholine receptor channels or other cation channels. We defined a burst as a group of openings separated by a closed period greater than 10 ms. Channel closings shorter than this were included as part of the burst duration. Short bursts with a duration less than 5 ms were excluded from the histograms. Burst duration histograms were well fitted with a single exponential in recordings from mdx fibres. In recordings from fibres isolated from mdx/utrn+/– heterozygotes and DKO muscle, an additional exponential component was needed to fit the burst duration histogram. Values of channel open probabilities and kinetic parameters were averaged from different experiments and a Wilcoxon signed rank test was used to determine significance between mean values.
Results
Figure 1 shows three separate recordings from cell-attached patches on skeletal muscle fibres isolated from wild-type (top), mdx (middle), and mdx/utrn–/– DKO mice at a low time resolution. In recordings from wild-type flexor digitorum brevis (FDB) fibres with normal saline in the patch electrode and the pressure set at 0 mmHg, single-channel activity appeared as brief bursts of openings (Fig. 1, top traces). A recording from an mdx fibre under similar conditions (Fig. 1, middle traces) shows an increase in the duration of individual bursts of openings, as described previously (Vasquez et al. 2012). Recordings from muscle fibres isolated from mdx/utrn–/–DKO mice, however, often showed a qualitatively different pattern of activity (Fig. 1, bottom traces). Channel openings appeared as short bursts, like those in recordings from wild-type and mdx fibres. Subsequently, channels entered a prolonged open state characterized by rapid, unresolved transitions. Entry into the prolonged open state is characteristic of a modal gating mechanism in which the channel shows abrupt shifts in kinetic behaviour.
Figure 1.
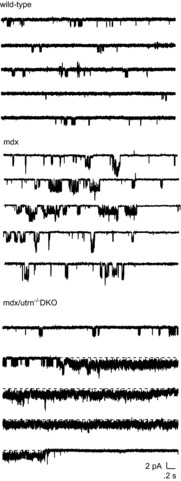
Examples of single-channel activity recorded from cell-attached patches on FDB fibres isolated from mdx (top), mdx/utrophin heterozygous (middle) and mdx/utrophin DKO mice (bottom)
The patch holding potential was −60 mV and the pressure in the electrode was set to 0 mmHg. Currents were sampled at 2.5 Hz and filtered at 1 kHz.
Modal gating of MS channels in DKO muscle differed from modal gating of MS channels previously described in cultured mdx myotubes (Franco & Lansman, 1990a; Franco-Obregon & Lansman, 2002): (1) the high open probability gating mode was not maintained, but was interspersed with periods of low open probability gating; and (2) it was not terminated by a pressure stimulus (data not shown). Subsequent experiments examined modal gating of MS channels in FDB fibres from mdx mice and from mdx mice lacking either a single (mdx/utrn+/– heterozygotes) or both utrophin alleles (mdx/utrn–/– DKO).
Figure 2 shows the combined results of recordings from experiments on muscle fibres isolated from mdx, mdx/utrophin heterozygotes (het) and mdx/utrophin DKO mice. Figure 2A shows the distribution of single-channel conductances measured for channel activity recorded in different experiments on fibres isolated from each of the three mouse genotypes. In mdx muscle, the predominant single-channel activity was from MS channels with a single-channel conductance of ∼22 pS. Smaller channels with conductances of ∼10 and ∼15–17 pS were also detected (Vasquez et al. 2012). Surprisingly, the distribution of single-channel conductances differed in muscle from mdx/utrophin heterozygotes and DKOs. Notably, single channels with conductances exceeding ∼30 pS were observed in utrophin heterozygous and DKO muscle fibres. Despite the limited sample size, the results suggest a gene dosage-dependent increase in the occurrence of both small and large conductance channels with depletion of utrophin.
Figure 2.
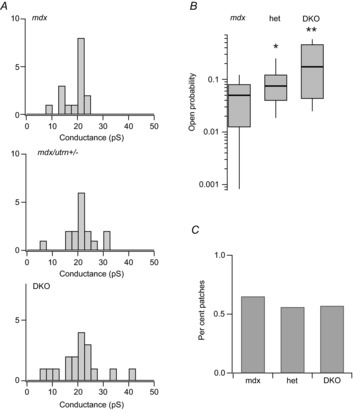
Conductance and open probability of single-channel activity recorded from muscle fibres isolated from mdx, mdx/utrophin heterozygotes and DKO mice
A, distribution of single-channel conductances determined by measurements of muscle fibres of the indicated genotype. B, box plots of channel open probability measured during the first 60 s of channel activity recorded from patches on mdx, mdx/utrophin heterozygotes and DKO muscle fibres. The values of mean open probability were 0.05 ± 0.04 (n = 13), 0.11 ± 0.1 (n = 18), and 0.25 ± 0.20 (n = 16) for experiments on mdx, utrophin heterozygotes and DKO fibres, respectively. A value of 0.4 was determined for wild-type fibres in an earlier study (Franco-Obregon & Lansman, 1994). Mean open probabilities are significantly different at α = 0.05 using a two-sample t test for mdx versus heterozygotes and heterozygotes versus DKOs, giving P values of 0.016 and 0.017. In this and all experiments, each recording was from a different fibre that was kept incubated in dissociation media at 37°C. C, percentage of patches showing single-channel activity in recordings from patches on muscle fibres of the indicated genotype (0.65, mdx; 0.56, mdx/utrophin heterozygotes; and 0.57, DKO).
In addition to differences in single-channel conductance in the different genotypes, the number of utrophin alleles expressed in mdx muscle strongly influenced channel open probability. Figure 2B shows MS channel open probability measured in recordings from muscle of each genotype. There was considerable variability in resting MS channel activity in all three genotypes. For example, several patches from DKO fibres showed low levels of channel activity. By contrast, none of the patches on mdx fibres showed high levels of MS channel activity seen in heterozygotes and DKOs. The mean MS channel open probability in mdx fibers measured in this study was 0.05, close to the value of 0.07 measured in an earlier study (Franco-Obregon & Lansman, 1994). Despite the patch to patch variability, many recordings from heterozygous and DKO fibres showed long lasting periods during which the channel was open continuously (see Fig. 1, bottom records). The combined results from many recordings show that MS channel open probability increased with genetic depletion of utrophin. Although there were clear genotype-dependent differences in single-channel conductance and open probability, Fig. 2C shows MS channel density, as measured by the fraction of recordings showing channel activity, was roughly the same in each genotype (∼60% of patches). Evidently, MS channel functional expression remained unchanged despite the loss of utrophin.
In an earlier study, high open probability gating was not detected in recordings from mdx FDB fibres (Franco-Obregon & Lansman, 1994). Here, we observed two patches on mdx fibres which showed reversible entry into a high open probability gating mode. Figure 3A shows an example of a recording from a patch on an mdx fibre that contained a single MS channel that opened to either a small (∼17 pS, left records) or large (∼21 pS, right records) conductance level. We previously showed that openings to different conductance levels reflects gating of a single-channel as determined by a statistical test for serial, non-random gating (Vasquez et al. 2012). This behaviour was interpreted as a change in the number of independently gating MS channel subunits that open during a single activation. The mechanism which controls the number of open subunits during a single activation is not known, although this question is considered later. In this example, short bursts of openings were followed by periods in which the channel entered a high open probability gating mode. We refer to the short bursts of openings as mode I and the longer openings as mode II, a simple convention first adopted for gating of L-type Ca2+ channels in heart muscle (Hess et al. 1984)
Figure 3.
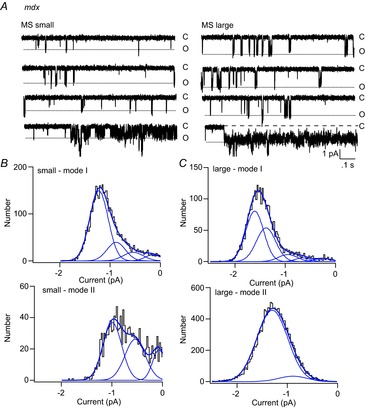
Modal gating of an MS channel showing two conductance states in a recording from an mdx muscle fibre
A, records of single-channel activity showing small (left) and large (right) openings of a single MS channel in an mdx fibre. Channel activity occurs as short bursts of openings and occasional periods in which the channel enters a high probability open state. B, amplitude distribution of the open channel current for mode I (short) and mode II (long) openings to the small conductance state. The amplitude distribution of the open channel current was fit as the sum of four Gaussian components for mode I openings and 3 Gaussian components for mode II openings. C, amplitude distribution of the open channel current for mode I and mode II openings to the large conductance state. The amplitude distribution was fit as the sum of five Gaussian components for mode I openings and one major Gaussian component for mode II openings.
Figure 3B and C shows the amplitude distributions of the open channel currents for both conventional mode I gating as well as for mode II gating observed in this recording. The gating transitions within a mode II burst were too fast to be measured as opening and closings to discrete levels. Figure 3B (top) shows that the amplitude distribution of the open channel current for small mode I events is well fit with the sum of four Gaussian components with the channel spending most of the time in the fully open state. Figure 3C (top) shows the amplitude distribution of large mode I events fit as the sum of five Gaussian components, again with the channel spending most of the time in the fully open state. Compared with the smaller events, large mode I events have a larger contribution from a sub-state smaller than the fully open one. The amplitude distributions of small and large mode I events are consistent with the opening of either four or five independently gating subunits. By contrast, the amplitude distribution of the open channel current for mode II gating was best fit with three Gaussian components for small events and only a single major Gaussian component for the larger events. The smaller mode II events probably reflect rapid transitions between three conductance states. By contrast, large conductance mode II events represent rapid, partially unresolved fluctuations between the two largest conductance states. Consequently, large mode II events appear as prolonged openings with an increase in the open channel noise. Qualitatively, the results show two types of mode II gating for MS channels in mdx muscle: one involving smaller, rapid opening and closing transitions and the other involving a shift to a larger open state.
Recordings from fibres from mdx/utrophin heterozygotes and DKO mice showed much more frequent mode II gating than recordings from mdx muscle. We found ∼40% of patches on fibres from both heterozygotes and DKO mice have MS channels showing prolonged openings characteristic of mode II gating. In addition, mode II gating of MS channels in DKO muscle lasted much longer than in utrophin-containing mdx muscle. Nonetheless, the fraction of patches showing mode II gating was the same whether muscle contained either one or no utrophin alleles. By contrast, mode II charge transfer was greater in DKO muscle than in muscle from utrophin heterozygotes, as described below.
Figure 4 shows examples of modal gating of MS channels in recordings from the utrophin deletion mutants. Figure 4A shows records of single-channel activity recorded from an mdx/utrophin heterozygous fibre (left records) and from a DKO fibre (right records). The patch potential was –60 mV and the pressure in the electrode was set to 0 mmHg. In both patches, short bursts of channel openings were followed by a prolonged period of mode II activity in which the channel was open continuously, but subsequently closed and returned to mode I low open probability gating. Compared with mode I bursts in the heterozygote (Fig. 4A, left records), mode I bursts in DKO muscle were short and infrequent (Fig. 4A, left records). However, the duration of mode I bursts was highly variable from patch to patch. Figure 4B and D shows plots of channel open probability during the entire recording period. The open probability plots show ∼5 min of continuous MS channel activity at a fixed holding potential.
Figure 4.
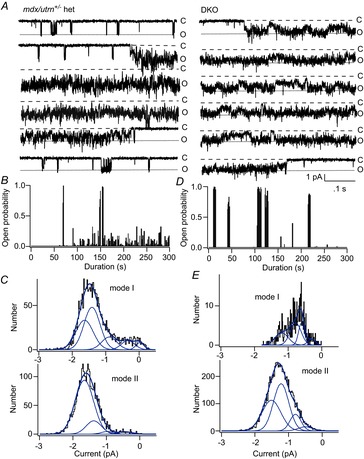
Modal gating of MS channels in utrophin heterozygotes and DKO mice
A, records of continuous single-channel activity recorded from membrane patches on utrophin heterozygous (left) and DKO (right) fibres. The patch potential was −60 mV and the electrode pressure was set to 0 mmHg. Closed and open states are indicated as ‘C’ and ‘O’, respectively. Note periods in which channel openings appeared as short bursts of openings (mode I) interspersed with closed periods and periods in which the channel entered a prolonged open state (mode II). b, plot of MS channel open probability versus time during the recording from the utrophin heterozygote. The plot shows approximately 5 min of channel activity. In this particular experiment, channel activity during the first ∼50 s is not shown as the patch potential was varied to measure the single-channel I–V relationship. C, amplitude distribution of the open channel current for mode I (top) and mode II (openings). The amplitude distribution for mode I openings was fit with the sum of five Gaussian components, while the distribution for mode II openings was fit with four Gaussian components, although the Gaussian component with the largest amplitude dominated the fit to mode II openings. D, plot of channel open probability versus time during the recording. The plot shows approximately 5 min of channel activity from a DKO fibre with the patch potential held at –60 mV. Note that in this recording mode I openings are very brief and so the probability plot shows very little channel activity except for the long clusters of mode II activity. E, amplitude distribution of the open channel current for mode I (top) and mode II (openings) in the recording from the DKO fibre. The amplitude distributions for mode I and mode II openings were fit with the sum of four Gaussian components. Because mode I openings were relatively infrequent and brief in this recording, the amplitude distribution of the open channel current is not completely resolved.
Figure 4B shows that in the utrophin heterozygote, episodes of mode II gating were relatively infrequent and most episodes of channel activity occurred as low open probability mode I bursts. Figure 4D shows, by contrast, that high open probability mode II gating in the recording from DKO muscle was much more frequent and persisted for a greater length of time. Figure 4C and E shows the amplitude distribution of the open channel current for mode I (low open probability) and mode II (high open probability) gating for single-channel activity recorded from the utrophin heterozygote (Fig. 4C) and the DKO (Fig. 4E). As shown previously, mode I activity shows discrete transitions between five conductance levels (Vasquez et al. 2012 and Fig. 3). By contrast, the distribution of current amplitudes for mode II openings is generally dominated by higher conductance levels. However, the noise in the open channel during mode II gating makes it difficult to distinguish discrete subconductance states and therefore the Gaussian fits to mode II activity are complicate by fast, unresolved transitions between multiple subconductance levels. Nonetheless, the results suggest depletion of utrophin produces a gene dose-dependent increase in high open probability mode II gating. To investigate this in more detail, we analysed the single-channel kinetics in muscle fibres of each genotype.
Figure 5 shows the analysis of the kinetics of MS channel gating in muscle fibres of all three genotypes. We restricted the analysis to channel open times and burst durations. Analysis of closed times was subject to errors from the presence of brief (<2 ms) channel openings that varied from patch to patch. These brief openings may arise from unliganded acetylcholine receptors or other cation channels present in skeletal muscle. Analysis of channel open times and burst durations, however, provides information on MS channel charge transfer in the different cytoskeletal mutants.
Figure 5.
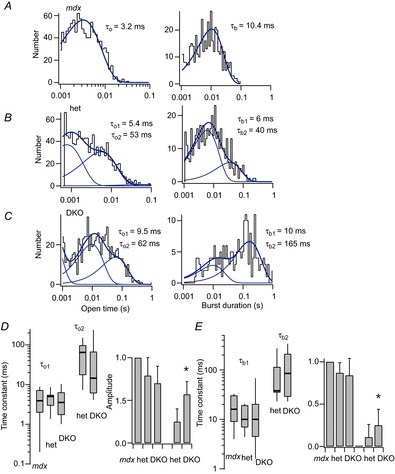
Kinetic analysis of MS channel activity recorded from cell-attached patches on mdx, mdx/utrophin heterozygous and mdx/utrophin DKO mice
Recordings were made at a patch holding potential of −60 mV and with the electrode pressure set to 0 mmHg. A, histograms of channel open times (left) and burst durations (right) obtained from a recording on an mdx fibre. A single exponential was fit to each distribution with the time constant indicated to the right of the histogram. B, histograms of channel open times (left) and burst durations (right) obtained from a recording on an mdx/utrophin heterozygous fibre. The histograms of open times and burst durations were each fit as the sum of two exponential components with the time constants indicated. C, histograms of channel open times (left) and burst durations (right) obtained from a recording of a DKO fibre. The histograms of open times and burst durations were each fit as the sum of two exponential components with the indicated time constants. D, compiled values of the open time constants obtained from the exponential fits to the open time histograms plotted as box plots (right) and the amplitudes of each component plotted as a simple bar graph (left). The values for τo1 were 4.3 ± 2.8 (n = 7), 4.7 ± 2.3 (n = 11) and 4.4 ± 3.2 (n = 14) with amplitudes, a1, of 1, 0.87 ± 0.12 (n = 11) and 0.83 ± 0.2 (n = 14) for mdx, utrophin heterozygous and DKO muscle, respectively. The values for τo2 were 63.8 ± 50 (n = 10) and 43.8 ± 77 (n = 10) with amplitudes, a2, of 0.11 ± 0.15 (n = 10) and 0.25 ± 0.2 (n = 10) for utrophin heterozygotes and DKOs, respectively. The relative amplitudes of τo2 for heterozygotes and DKOs were significantly different at α = 0.05 with a P value of 0.03. E, compiled values of channel burst durations obtained from the exponential fits to the burst durations histograms plotted as box plots (right) and the amplitudes of each component plotted as a simple bar graph (left). The values for τb1 were 17.6 ± 11 (n = 7), 12.0 ± 5.8 (n = 11) and 18.4 ± 26 (n = 14) with amplitudes, ab1, of 1, 0.79 ± 0.29 (n = 11) and 0.80 ± 0.28 (n = 14) for mdx, utrophin heterozygous and DKO muscle, respectively. The values for τb2 were 83.3 ± 79 (n = 10) and 121 ± 121 (n = 7) with amplitudes, ab2, 0.25 ± 0.20 (n = 9) and 0.57 ± 0.20 (n = 7) for utrophin heterozygotes and DKOs, respectively. The relative amplitudes ab2 of τo2 for heterozygotes and DKOs were significantly different at α = 0.05 with a P value of 0.015.
The distributions of channel open times and burst durations are shown in Fig. 5A–C for channels in mdx, mdx/utrophin heterozygotes and DKO muscle, respectively. Measurement of open times provides only an approximate fit to the single-channel currents because of the presence of subconductance fluctuations in the open channel (see Methods for details). Figure 5A shows the distribution of open times and burst durations obtained from a recording on an mdx fibre was well fit with a single exponential component. By contrast, at least two exponential components were required to fit the distributions of open times and burst durations in recordings from utrophin heterozygotes and DKOs (Fig. 5B, C). This presence of a large slow component to the histograms of open times and burst durations reflects the higher frequency of mode II gating in the utrophin mutants.
Figure 5D and E shows the combined kinetic measurements obtained from many recordings from each genotype shown as box plots (see figure legend for details). The longer open time constant for channels in utrophin heterozygotes and DKO muscle varied over a wide range and there was no statistical difference between the two (Fig. 5D). By contrast, the amplitude of the slow component of the open time histogram was greater in recordings from DKO muscle than from utrophin heterozygotes. This was also the case with the distribution of channel burst durations: while the slower component of the fit to the burst duration distribution was similar in heterozygotes and DKOs, the amplitude of the slow component was greater in DKOs than in the utrophin heterozygotes (Fig. 5E). The combined results show that genetic depletion of utrophin causes a gene dose-dependent increase in the number, rather than the duration, of long openings, as well as an increase in the number of mode II bursts. Evidently, loss of utrophin results in increased charge transfer and Ca2+ entry through MS channels as a result of an increase in the number of long MS channel openings and bursts.
Although MS channel activity was detected in ∼60% of patches on muscle from all three genotypes, we found considerable patch to patch variability in the response to pressure stimuli. Pressure steps applied to some patches failed to increase single-channel activity, despite the presence of bursts of activity in the absence of a pressure stimulus. In other patches, pressure steps only increased channel activity at pressures exceeding ∼−30 to −40 mmHg and patches often ruptured with large pressure steps. An increased threshold for MS gating, in addition to an apparent increase in the fragility of membrane patches on DKO fibres, prevented a detailed analysis of pressure-dependent channel gating. However, in a few experiments we were able to obtain a complete set of current records in response to step increases in patch electrode pressure. Figure 6 shows an example of one such experiment recorded from a DKO fibre.
Figure 6.
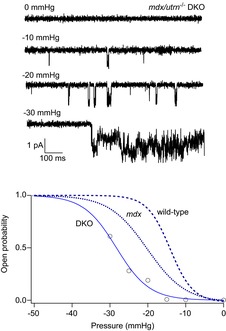
Pressure-dependent gating of the 22 pS MS channel recorded from a cell-attached patch on a muscle fibre isolated from a DKO mouse
An example of an experiment in which single MS channel activity was recorded from a cell-attached patch at −60 mV with the pressure set to the indicated values. The data points represent channel open probability as a function of pressure (mmHg) on the x-axis. The data points were fitted with a Boltzmann relation of the form 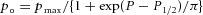 where po is the channel open probability, pmax is the maximum channel open probability, P is the pressure applied to the electrode (mmHg), P1/2 is the pressure required for po = 0.5, and π is the steepness of the relation (mmHg). The fit to the DKO data gave P1/2 = −26 mm Hg and π = −4 mmHg. The dashed and dotted curves are from MS channels in wild-type and mdx muscle, which were drawn with P1/2 = −14 and −20 mmHg and π = −3 and −5 mmHg, respectively (Franco-Obregon & Lansman, 1994).
where po is the channel open probability, pmax is the maximum channel open probability, P is the pressure applied to the electrode (mmHg), P1/2 is the pressure required for po = 0.5, and π is the steepness of the relation (mmHg). The fit to the DKO data gave P1/2 = −26 mm Hg and π = −4 mmHg. The dashed and dotted curves are from MS channels in wild-type and mdx muscle, which were drawn with P1/2 = −14 and −20 mmHg and π = −3 and −5 mmHg, respectively (Franco-Obregon & Lansman, 1994).
The records in Fig. 6 show the MS channel current recorded from a DKO fibre in response to pressure steps to −10, −20 and −30 mmHg. Note that the pressure steps to −10 and −20 mmHg evoked little channel activity. During the pressure step to −30 mmHg, there was a large, but delayed increase in MS channel activity. Mean MS channel open probability during a pressure step was measured and plotted as a function of the electrode pressure (Fig. 6, bottom graph). The dashed and dotted lines are the pressure–open probability relationships measured for MS channels in wild-type and mdx FDB fibres, respectively, in an earlier study (Franco-Obregon & Lansman, 1994, see figure legend for details). Comparing the gating curves shows that a pressure step to −20 mmHg fully activates MS channels in wild-type muscle, but only ∼50% of MS channels in mdx muscle and less than 20% of channels in DKO muscle. The relationship between pressure and channel open probability in the DKO fibre was shifted to larger negative pressures compared with wild-type and mdx fibres. The results indicate complete depletion of utrophin in DKO muscle alters MS gating by shifting gating to higher pressures. Possible explanations for this shift are considered in the Discussion.
In a recent study, we showed that MS channels in wild-type muscle show well-resolved transitions between distinct subconductance states (Vasquez et al. 2012). Furthermore, an absence of dystrophin in muscle from mdx mice was associated with reduced occupancy of the fully open state, with MS channels spending a greater fraction of the time at well-defined, smaller subconductance levels. These findings suggested that MS channels are composed of multiple subunits that gate independently. Sukharev et al. (1999) described subconductance transitions in the large conductance bacterial MS channel but concluded only the transition from the closed state to the smallest subconductance state involved a channel conformational change. By contrast, the analysis of Gil et al. (2001) for MS channels in oocytes suggested a model in which subunit transitions are not coupled but may occur independent of one another.
As a first approximation, we considered the MS channel in muscle to be composed of five subunits, each with an individual probability P of opening. Assuming a binomial distribution of sub-state occupancy, a fit to the sub-state occupancy data gave the binomial parameter P = 0.8 and 0.5 for MS channels in wild-type and mdx fibres, respectively. Thus, the absence of dystrophin in mdx muscle reduces subunit opening probability. This might occur if dystrophin contributes to force transmission to individual subunits. If this were the case, depletion of utrophin would be expected to produce further changes in opening of individual MS channel subunits. In subsequent experiments, we measured sub-state occupancy of the open channel current in the utrophin heterozygous and DKO muscle to test whether (1) utrophin participates in individual MS channel subunit gating and (2) the shift in the pressure dependence of MS channel gating in DKO muscle reflects a reduced probability of individual subunit gating.
Figure 7 shows the results of experiments investigating subconductance transitions within individual activations of the 22 pS MS channel. Figure 7 (top records) shows the single channel activity recorded from mdx (top), mdx/utrophin heterozygous (middle) and DKO (bottom) muscle fibres at high time resolution. The amplitude distribution of the open channel current is shown to the left of each record. The amplitude histograms were measured from many channel activations in a single patch, excluding all transitions to the fully closed level. As can be seen, the amplitude distributions are highly skewed, indicating the contribution of multiple Gaussian components. The amplitude distributions were fit with the sum of five Gaussian components, which included the fully open state and four subconductance levels. These are indicated by dashed lines in the records. A maximum likelihood fitting method was used as described previously (Vasquez et al. 2012). The fits show that MS channels in mdx muscle spend approximately equal amounts of time in the fully open state and the next smaller subconductance level. This contrasts with MS channels in wild-type muscle, in which channels occupied the fully open state most of the time (Vasquez et al. 2012). By contrast, Fig. 7 shows that genetic depletion of dystrophin in heterozygotes and DKOs reduced occupancy of the fully open state even further, so that channel activations occurred primarily to the largest subconductance level S1.
Figure 7.
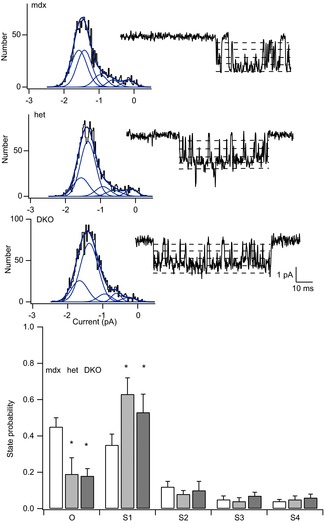
Amplitude distribution analysis of subconductance states of MS channels in mdx, mdx/utrophin heterozygotes and mdx/utrophin DKO mice
A, amplitude distribution of the open channel current measured in a recording from an mdx fibre. All amplitude distributions were fit with the sum of five Gaussian components which included the fully open state and four subconductance levels. B, amplitude distribution of the open channel current measured in recording from an mdx/utrophin heterozygous fibre. C, amplitude distribution of the open channel current measured in recording from a DKO fibre. D, occupancy probability of subconductance levels in MS channels from fibres isolated from mice of the indicated genotype. Each bar represents the fraction of time MS channels spent in the fully open state, O, and either of four subconductance levels, S1, S2, S3 and S4. Open bars are from mdx muscle; filled bars are from mdx/utrophin heterozygous and DKO fibres. Currents were measured during channel openings recorded at a constant holding potential of –60 mV with the pressure set to 0 mm Hg. Closed channel levels were excluded from the measurements. The single-channel amplitudes for O, S1, S2, S3 and S4 states were −1.63 ± 0.22, −1.36 ± 0.18, −1 ± 0.17, −0.6 ± 0.19 and −0.22 ± 0.10 pA (mean ± SD; n = 9) for MS channels in mdx muscle; −1.66 ± 0.20, −1.4 ± 0.20, −0.87 ± 0.10, −0.47 ± 0.13 and −0.13 ± 0.10 (n = 7) for MS channels in mdx/utrophin heterozygous muscle; and −1.9 ± 0.30, −1.6 ± 0.30, −1.2 ± 0.20, −0.73 ± 0.18 and −0.24 ± 0.10 (n = 6) for MS channels in mdx/utrophin DKO muscle. The normalized single-channel amplitudes were 1.0, 0.83, 0.61, 0.37 and 0.1; 1, 0.86, 0.53, 0.29 and 0.1; and 1, 0.84, 0.63, 0.38 and 0.13 for channels in mdx, mdx/utrophin heterozygotes and mdx/utrophin DKO mice. The state probabilities for O, S1, S2, S3 and S4 were 0.45 ± 0.05, 0.35 ± 0.06, 0.12 ± 0.03, 0.05 ± 0.02 and 0.04 ± 0.01 (mean ± SD; n = 9) for MS channels mdx muscle; 0.19 ± 0.09, 0.63 ± 0.09, 0.08 ± 0.02, 0.04 ± 0.02 and 0.05 ± 0.02 (n = 7) for MS channels in mdx/utrophin heterozygous muscle; and 0.18 ± 0.04, 0.56 ± 0.12, 0.10 ± 0.05, 0.07 ± 0.02 and 0.06 ± 0.02 (n = 6), for MS channels in mdx/utrophin DKOs. Occupancy probabilities of the O and S1 levels are significantly different at α = 0.05 using a two-sample t test mdx versus heterozygotes and DKOs giving P values of 0.002 and 0.002.
Figure 7 (bottom) shows the combined results obtained from 7–10 patches. The graph shows the probability of occupying the fully open state (O) or subconductance states S1, S2, S3 or S4. The combined results show a striking reduction in occupancy of the fully open state, O, with an increase in occupancy of the largest subconductance state, S1. The fit to a binomial distribution gave a P value of ∼0.2, assuming each subunit gates independently. Surprisingly, the change in MS channel sub-state occupancy probability is roughly the same in recordings from muscle containing either one or no utrophin alleles. Evidently, even partial depletion of utrophin in the heterozygote is sufficient to reduce MS channel occupancy of the fully open state. Complete loss of utrophin in the DKO muscle, however, produces no further change in subconductance state occupancy. The results show partial genetic depletion of utrophin is sufficient to alter the probability of subunit gating and suggest that utrophin partially compensates for a reduction in the probability that a single subunit opens in dystrophin-deficient muscle.
Recordings from muscle from utrophin heterozygotes and DKO muscle showed the presence of novel, large conductance MS channels (∼30–42 pS). Large channels showed MS gating, although this was not studied in detail because large conductance channels were relatively rare and patches tended to rupture with pressure stimuli. Figure 8A shows an example of a high time resolution recording of the activity of a large conductance channel from a DKO muscle fibre. The amplitide of the single-channel current was ∼4 pA at −60 mV with a single channel conductance of ∼40 pS (Fig. 8B, C), significantly larger than MS channels in wild-type and mdx muscle (Franco & Lansman, 1990a; Franco-Obregon and Lansman, 1994; Vasquez et al. 2012).
Figure 8.
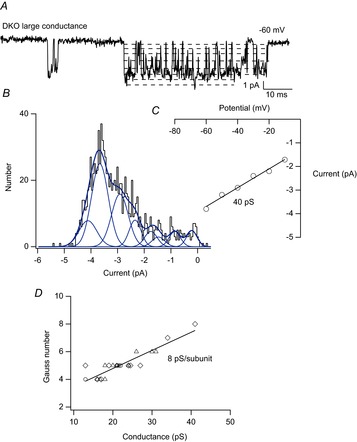
Subconductance states in a large conductance MS channel recorded from a DKO muscle fibre
A, single-channel activity of a large conductance MS channel recorded from a membrane patch on a DKO fibre at high time resolution. The dashed lines indicate subconductance levels. Well-resolved long-lasting transitions to the larger S4, S5, S6 and S7 levels can be seen in the record. The channel opens only briefly to the fully open state, O. B, amplitude distribution of the open channel current measured from many openings of the large conductance channel from the same experiment from which the record in A was taken. This amplitude distribution of the open channel current was fit as the sum of eight Gaussian components which included seven subconductance states (S1–S7) and the fully open state (O). The eight Gaussian fit was determined by maximizing the likelihood value of the fit using different numbers of Gaussian components. The inset shows the single-channel I–V relationship of the large conductance channel in the DKO fibre. The slope gave a conductance of ∼40 pS. C, plot of the number of subconductance states versus the single-channel conductance in recordings from muscle fibres of all three genotypes. Open circles, mdx; open triangles, mdx/utrophin heterozygotes; open diamond, DKO. The line was fit to all the data points using a least squares algorithm with correlation coefficient 0.9 and slope 8 pS.
Inspection of the current record in Fig. 8A shows multiple subconductance levels. In this record, at least seven subconductance levels can be seen, in addition to the fully open level, which the channel enters for only a very brief time. We hypothesized that large conductance MS channels may be produced when additional subunits in close proximity aggregate to form a functional channel. Figure 8B shows the amplitude distribution of the open channel current obtained from a large number of openings in this experiment. We fit the distribution as the sum of multiple Gaussian components. We began using four components and then increased the number of Gaussian components to maximize the log likelihood of the fit of the amplitude distribution. The best fit was obtained with eight Gaussian components, which included the fully open state and seven subconductance states. The fit was consistent with the number of subconductance levels that could be detected by visual inspection of the current records. Visual inspection of many records like that shown in Fig. 8A provided examples of well-resolved sojourns at each subconductance level that lasted at least 3–5 ms.
In a previous paper, we described small single-channel events that appeared to be produced by MS channels that failed to reach the fully open state (Vasquez et al. 2012). These ‘partial openings’ occurred more frequently in mdx than in wild-type skeletal muscle. An alternative explanation is that these small events are individual MS channel subunits that open independently and that variations in MS channel conductance depend on dynamic aggregation of individual subunits. This could occur, for example, by clustering of subunits within a membrane microdomain. Large conductance MS channels in DKO muscle would then represent MS channels composed of a larger number of subunits. In this view, each subunit contributes a fixed conductance to the multi-subunit channel.
To test the possibility that large conductance channels are composed of higher order subunit aggregates, we fitted the amplitude distribution of the open channel current for all single-channel recordings obtained from muscle of all three genotypes. The single-channel activity included the full range of MS channel conductances, which ranged from 15 to 40 pS. Figure 8C shows a plot of the number of Gaussian components versus the single-channel conductance (each symbol represents a different patch). The relationship was linear, consistent with linear summation of subunit conductance with a value of ∼8 pS per subunit.
Recordings from membrane patches on skeletal muscle fibres also showed the existence of a small conductance channel of ∼10 pS in a small fraction of patches (Fig. 2A). There is increased expression of the ∼10 pS channel in DKO muscle, although the small number of 10 pS channels makes it difficult to test this statistically. Zanou et al. (2010) showed a small conductance channel in skeletal muscle was absent in TRPC1 knockout mice. Zanou et al. (2010) also showed the small conductance channel was relatively insensitive to changing the pressure applied to the membrane patch, indicating it was not mechanosensitive. Although Zanou et al. (2010) used somewhat different ionic solutions in the patch electrode, it is likely that the small conductance channel detected in our electrophysiological recordings is TRPC1 based on their recordings from TRPC1 knockout mice.
Figure 9A shows the activity of the small conductance channel recorded from a cell-attached patch on an mdx (left) and a DKO (right) fibre. In the recording from an mdx fibre, channel events occurred as more or less discrete, relatively brief transitions between the closed state and a fully open state. Figure 9B shows the amplitude distribution of the open channel current, which confirms the existence of a single conducting open state, although there was a minor small component to the amplitude distribution. The single-channel current–voltage relationship gave a conductance of 11 pS in this experiment. Figure 9C shows the histogram of channel open times, which was well fitted with a single exponential giving a mean open time of ∼16 ms. By contrast, the small conductance channel showed a very different pattern of activity in recordings from DKO muscle. Channel activity appeared as very long openings and there was evidence of multiple channels in the patch as seen as superimposed openings (Fig. 9A, second record in right panel). Figure 9D shows that the amplitude of the open channel current was well fitted with a single Gaussian component and the slope of the I–V relationship gave a single-channel conductance of 10 pS. Figure 9E shows the histogram of channel open times obtained from the open durations measured from activity of the 10 pS channel in the DKO fibre. The open time distribution was well fitted with a single exponential with a time constant of 900 ms, more than 50 times longer than in mdx muscle. Evidently, openings of small conductance channels are greatly prolonged in muscle lacking both dystrophin and utrophin, suggesting a role of the small conductance channel in pathogenic Ca2+ entry in DKO muscle. This pattern was observed in three recordings from mdx muscle and five recordings from DKO muscle. In some patches on DKO fibres, the small conductance channel initially exhibited short openings, like those in mdx muscle. However, applying pressure to the electrode during the recording produced an irreversible shift to a gating mode with greatly prolonged openings. Mode II gating of the small conductance channel in DKO fibres was not altered by applying pressure to the electrode. However, the behaviour more closely resembled the irreversible, pressure-induced induction of mode II gating of MS channels in mdx myotubes (Franco-Obregon & Lansman, 2002).
Figure 9.
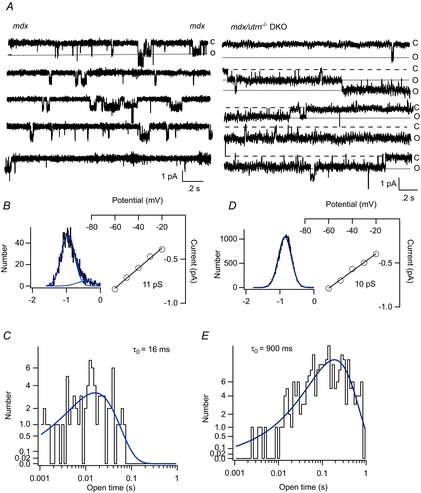
Gating of single TRPC1 channels in muscle fibres from mdx and DKO mice
A, single-channel activity recorded from membrane patches on an mdx fibre (left records) and a DKO fibre (right records). B, amplitude distribution of the open channel current for the single-channel activity measured from the mdx fibre. The amplitude distribution was fit as the sum of two Gaussian components, although the second component contributed to only a small area of the distribution. The patch potential was −60 mV. The inset shows the single-channel I–V relationship measured in the experiment in which the records in A were obtained. The slope of the I–V relationship gives a conductance of 11 pS. C, histogram of channel open times from the same recording on an mdx fibre. The open time distribution was fit with a single exponential with a time constant τo = 16 ms. D, amplitude distribution of the open channel current for the single-channel activity measured from the DKO fibre. The amplitude distribution was fit as a single Gaussian component. The patch potential was −60 mV. The inset shows the single-channel I–V relationship with a single-channel conductance of 10 pS. E, histogram of channel open times obtained from channel activity obtained from the DKO fibre. The open time distribution was fit with a single exponential with a time constant τo = 900 ms.
Discussion
This paper presents the first recordings of single MS channel activity from muscle lacking both dystrophin and utrophin. The possibility that utrophin compensates for dystrophin in mdx muscle has made it difficult to determine the extent to which dystrophin participates in mechanotransduction and changes in MS channel open probability. The data in this paper confirm a role for MS channels in pathogenic Ca2+ entry in dystrophic muscle. MS channel open probability increases with genetic depletion of utrophin (Fig. 3B), consistent with Ca2+ entry through MS channels contributing to disease severity.
Most studies have focused on differences in muscle function between mdx and DKO mice. DKO mice show greater muscle weakness, more extensive degeneration and fibrosis, and greater fatigue at 6–8 weeks than mdx mice (Connolly et al. 2001). More recently, comparison of mdx, mdx/utrophin heterozygotes and DKO mice has shown greater functional impairment in the utrophin heterozygotes than in mdx mice (van Putten, 2012). There is also increased expression of genes involved in regeneration, inflammation and fibrosis in heterozygotes compared with mdx mice. Apparently, loss of just a single utrophin allele is sufficient to worsen the pathology of the mdx mouse. Heterozygotes, however, do not die prematurely nor do they show the severe disease of DKO mice. Our finding of a utrophin gene dose-dependent effect on the behaviour of single MS channels suggests a close correlation between utrophin expression and changes in membrane ion permeability that are an early stage in the pathogenesis of muscular dystrophy.
Genetic depletion of utrophin in dystrophin-deficient mdx muscle produces complex effects on MS channel behaviour. These include: (1) an increase in channel open probability and mode II gating; (2) an increase in MS channel conductance in some patches; (3) an increase in the pressure required to activate MS channels; and (4) an increase in sub-state occupancy in utrophin mutants. Except for the change in sub-state occupancy, these effects are utrophin gene dose-dependent. The changes in MS channel gating showed considerable cell to cell variability within each genotype. For example, in many patches on muscle fibres from DKO mice, MS channel open probability was low and there was little mode II activity. In addition, there was a range of single-channel conductances in the utrophin mutants with only a small proportion of patches containing large conductance channels.
The variability of MS channel properties in the utrophin mutants would seem generally inconsistent with global changes in either the level of an intracellular second messenger or the loss of a regulatory protein. The results would be more consistent with utrophin deficiency causing discrete and more localized changes in the membrane cytoskeletal lattice, which influences local membrane structure or composition. For example, a reduced sensitivity to pressure and increased occupancy of smaller sub-states would be consistent with an increase in hydrophobic mismatch produced by accumulation of longer chain membrane lipids at the boundary of the channel protein–membrane interface (Markin & Sachs, 2004). Alternatively, there may be a loss of direct protein–protein interactions involved in force transmission in the utrophin mutants. The present data do not provide direct information on the precise mechanism for changes in MS gating in utrophin mutants. However, the structural organization of dystrophic muscle in the mutants provides some insight into possible mechanisms.
An absence of dystrophin in skeletal muscle from mdx mice disrupts the normal costameric organization of the β-spectrin cytoskeletal lattice. There are also corresponding changes in the sarcolemmal distribution of utrophin, vinculin and other proteins (Williams & Bloch, 1999). In addition, dystrophin deficiency in mdx muscle is associated with a loss of costameric actin (Rybakova et al. 2000), as well as disorganization of the microtubule lattice (Prins et al. 2009; Belanto et al. 2014). The variability in the disruption of the cytoskeletal lattice in mdx muscle observed in morphological studies may be related to differences in contractile activity and mechanical loading of individual fibres prior to isolation from the animal for study. It seems likely that the observed patch to patch variability in MS channel behaviour reflects the local variability of cytoskeletal disruption in the various mutants and the corresponding variations in local membrane lipid and protein composition.
As utrophin partially compensates for dystrophin, it would be expected that loss of utrophin would cause more extensive disruption of the submembrane cytoskeletal lattice in skeletal muscle. These cytoskeletal changes would be expected to be associated with marked changes in the surface morphology of the sarcolemma, as well as changes in the lipid/protein composition of local membrane microdomains. We suggest that the changes in MS channel behaviour described here reflect changes in the local structure and composition of membrane microdomains in which channels are embedded. The domain hypothesis provides a simple physical model that explains how loss of utrophin leads to increased MS channel open probability and mode II gating. For example, loss of local membrane domain structure may cause a change in the lipid composition of the membrane adjacent to MS channel proteins. Alternatively, loss of domain structure may alter adaptor or scaffolding protein availability. In either case, a change in domain compliance may provide a favourable lipid environment that stabilizes open channel conformational states. As we find only ∼40% of patches on utrophin mutants show mode II gating, MS channels may be more uniformly distributed on the sarcolemma and the effects of utrophin depletion on channel properties occur only at specific sites. It is possible that these sites coincide with costameres based on the known localization of dystrophin and utrophin in skeletal muscle, but we cannot rule out that utrophin-ordered domains have a wider sarcolemma distribution.
Anishkin and Chung (2013) recently suggested a similar model in which MS channels are embedded in stiffened, cholesterol-rich domains. Their model focused on cadherin and integrin stabilized domains consisting of cholesterol and sphingomylin, which produce a more ordered bilayer structure. MS channels embedded in stiff domains would suppress channel conformational fluctuations, for example, by preventing tilting of α-helical domains of the channel or other open channel structural rearrangements associated with an increase in protein area.
In skeletal muscle, cavelolin 3 organizes cholesterol-rich domains and interacts with β-dystroglycan (Sotgia et al. 2000). Dystrophin, through its interaction with β-dystroglycan, probably contributes to the stability of cholesterol-rich domains by binding actin. In endothelial cells, utrophin-associated protein complexes exist in both caveolar and non-caveolar lipid microdomains (Ramirez-Sánchez et al. 2012). Huang et al. (2013) recently reported localization of MS currents in caveolae in skeletal muscle myotubes. Huang et al. (2013) focused on macroscopic currents and did not investigate the single-channel properties of caveolar MS currents. Several MS channel proteins appear to be associated with caveolar domains, including TRPC1 and TRPV4 (Saliez et al. 2008; reviewed by Sabourin et al. 2009). Localization of MS channels to caveolar microdomains may be associated with a high local density of channel proteins. Clustering of channels may explain the appearance of large conductance MS channels in the utrophin mutants. Experimental and theoretical work on the bacterial MscL channel suggest closely clustered channels may lead to coupled gating in which conformational changes in one channel affect the gating of adjacent channels through elastic interactions in the bilayer (Ursell et al. 2007; Grage et al. 2011; Haselwandter & Phillips, 2013; Mika et al. 2013). Further studies of reconstituted mammalian MS channels are required to test the role of bilayer elastic interactions in coupled gating.
Alternatively, clustering may lead to increases in MS channel conductance via direct interactions between specific structural domains on channel-forming subunits. In acetylcholine and other ligand-gated channels, a conserved cytoplasmic MA helix in each subunit appears as a ‘hanging gondola’ beneath the channel pore and mutations in this region alter single-channel conductance (Unwin, 2005; Peters et al. 2010). GABAA receptor channels show changes in sub-state gating and single-channel conductance when clustered rather than when dispersed at low densities, presumably through MA domain interactions between adjacent channels (Tierney, 2011). MS channels in embryonic stem cells show transitions between three current levels both within single activations and also as isolated events (Soria et al. 2013). Soria et al. (2013) suggested a ‘gondola’ type model to explain sub-state transitions that arise from changes in ion permeation through protein–protein interactions.
It is possible that clustering of MS channel subunits in localized domains in skeletal muscle favours direct interactions involving a ‘gondola-like’ cytoplasmic helical domain that regulates single-channel conductance. In this view, depletion of utrophin would favour occupancy of smaller subconductance levels if it reduced domain interactions. On the other hand, depletion of utrophin might allow aggregation of channel subunits and coupled gating by direct interaction of MA-like domains in a less ordered domain if channels remain clustered. However, there is no evidence for the existence of MA-like domains in mammalian MS channels. Despite the precise mechanism, we find a clear linear correlation between the number of MS channel subconductance states and single-channel conductance. It is of interest that channel clustering has been shown to produce coupled gating associated with incremental increases in single-channel conductance in Na+ channels (Huang et al. 2012), K+ channels (Molina et al. 2006) and Ca2+ channels (Navedo et al. 2010).
The identity of the MS channel in skeletal muscle has been subject to considerable controversy. We previously presented evidence that MS channels in skeletal muscle are composed of TRPV4 subunits: MS channels are blocked by TRPV antagonists, insensitive to the TRPC and Orai antagonist 2-aminoethoxydiphenyl borate (2-APB), activated by hypotonic solutions and absent from skeletal muscle from TRPV4 knockout mice (Ho et al. 2012). However, the single-channel conductance of MS channels in skeletal muscle is substantially smaller than recombinant TRPV4 (∼22–24 vs. 83–95 pS, Ma et al. 2011). The present results suggest that MS channel conductance may be substantially larger depending on the state of cytoskeleton-organized microdomains and extent of subunit coupling.
Our results also show changes in a small (∼10 pS) channel in muscle from DKO mice which result in irreversible entry into a high open probability gating mode. The small conductance channel is likely to be TRPC1 based on its absence from TRPC1 knockout mice (Zanou et al. 2010). As shown in Fig. 3A, the small conductance channel was detected infrequently in muscle from mdx and mdx/utrophin heterozygous mice, but somewhat more frequently in muscle from DKO mice. Because small conductance channels were detected only infrequently, they could not be studied in any detail. The available data, however, showed that modal gating of these channels, which we consider to be TRPC1 based on the results of Zanou et al. (2010), differed in important ways from that of TRPV4 MS channels. First, entry into the high open probability gating mode was not reversible. Prolonged open times (Fig. 9, τo = 900 ms in DKO muscle vs. 16 ms in mdx muscle) persisted for the duration of the recording. Second, in some patches irreversible entry into high open probability mode II gating could be induced by applying a strong pressure pulse (data not shown). This is similar to the irreversible conversion of MS channels by strong pressure stimuli in mdx myotubes grown in tissue culture from precursor myoblasts (Franco-Obregon & Lansman, 2002). Third, we found no evidence of mode II gating of small TRPC1 channels in the heterozygotes, indicating that the changes in TRPC1 gating require complete loss of utrophin. These results lead to two conclusions: (1) TRPV4 and TRPC1 MS channels may be localized to different membrane domains and (2) TRPC1 may contribute to pathophysiological Ca2+ entry. Our findings suggest that therapeutic strategies to replace utrophin in patients with muscular dystrophy may act by stabilizing membrane domains and reducing Ca2+ entry through both TRPV4 and TRPC1 MS channels by preventing both open channel conformational transitions favoured by compliant lipid domains and aggregation of TRPV4 subunits. Further experiments are required to understand the precise localization of TRPV4 and TRPC1 channels in relation to dystrophin, utrophin and other costameric proteins in the various mouse models of muscular dystrophy.
Finally, our results suggest a general physical mechanism that would explain modal gating of other ion channel proteins. Modal gating has been detected in a wide range of ion channel proteins, including Na+, K+, Ca2+ and N-methyl-d-aspartate receptor channels. Modal gating in ion channels has generally been described in terms of discrete kinetic models with multiple open states and with stochastic transitions into a long-lived open state (e.g. Hess, Lansman, & Tsien, 1984). In these models, transitions between gating modes is a random process determined by a fixed set of rate constants. The data presented here suggest that stochastic models may be insufficient to explain modal gating of single ion channels. We suggest that modal gating arises from dynamic changes in membrane composition and/or structure in channel-containing microdomains such that open channel conformation states are thermodynamically favoured. Such changes would occur in response to mechanical forces in the membrane that lead to transient changes in domain structure. These changes may be produced by contractile proteins, adhesive interations of the membrane with the extracellular matrix and, perhaps, movements of adaptor proteins that act to stabilize domain structure. Future experiments need to focus of the structure and physiology of membrane microdomains.
Acknowledgments
We thank Drs Alfredo Franco-Obregon and Owen Hamill for comments on an earlier version of the manuscript.
Glossary
- DKO
mdx/utrn–/– double knockout
- DMD
Duchenne muscular dystrophy
- MS
mechanosensitive
- FDB
flexor digitorum brevis
- TRPC1
transient receptor potential channel 1
- TRPV4
transient receptor potential vanilloid 4
Additional information
Competing interests
The authors have no competing interests.
Author contributions
This work was done in the laboratory of J.B.L. in the Department of Cellular & Molecular Pharmacology, UCSF School of Medicine. N.T. contributed to the collection, analysis and interpretation of data, as well as preliminary experimental design. J.B.L. contributed to (1) conception and design of experiments; (2) collection, analysis and interpretation of data; and (3) writing and revising the article.
Funding
This work was supported by a grant from the Krasnow Foundation.
References
- Anishkin A, Kung C. Stiffened lipid platforms at molecular force loci. Proc Natl Acad Sci U S A. 2013;110:4886–4892. doi: 10.1073/pnas.1302018110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanto JJ, Mader TL, Eckhoff MD, Strandjord DM, Banks GB, Gardner MK, Lowe DA, Ervasti JM. Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci U S A. 2014;111:5723–5728. doi: 10.1073/pnas.1323842111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey S, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–321. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Carnwath JW, Shotton DM. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J Neurol Sci. 1987;80:39–54. doi: 10.1016/0022-510x(87)90219-x. [DOI] [PubMed] [Google Scholar]
- Connolly AM, Keeling RM, Mehta S, Pestronk A, Sanes JR. Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin α2-deficient mice. Neuromuscul Disord. 2001;11:703–712. doi: 10.1016/s0960-8966(01)00232-2. [DOI] [PubMed] [Google Scholar]
- Dangain J, Vrbova G. Muscle development in mdx mutant mice. Muscle Nerve. 1984;7:700–704. doi: 10.1002/mus.880070903. [DOI] [PubMed] [Google Scholar]
- De Backer F, DeBacker C, Gailly P, Gillis JM. Long-term study of Ca2+ homeostasis and of survival in collagenase-isolated muscle fibres from normal and mdx mice. J Physiol. 2002;542:855–865. doi: 10.1113/jphysiol.2002.020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck N, Tinsley J, De Backer F, Fisher R, Kahn D, Phelps S, Davies K, Gillis JM. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med. 1997;3:1216–1221. doi: 10.1038/nm1197-1216. [DOI] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- Ervasti JM. Dystophin, its interation with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772:1008–1117. doi: 10.1016/j.bbadis.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet. 2013;14:373–378. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- Franco A, Jr, Lansman JB. Stretch-sensitive channels in developing muscle cells from a mouse cell line. J Physiol. 1990a;427:361–80. doi: 10.1113/jphysiol.1990.sp018176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A, Jr, Lansman JB. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature. 1990b;344:670–673. doi: 10.1038/344670a0. [DOI] [PubMed] [Google Scholar]
- Franco-Obregón A, Lansman JB. Mechanosensitive ion channels in skeletal muscle from normal and dystrophic mice. J Physiol. 1994;481:299–309. doi: 10.1113/jphysiol.1994.sp020440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Obregón A, Lansman JB. Changes in mechanosensitive channel gating following mechanical stimulation in skeletal muscle myotubes from the mdx mouse. J Physiol. 2002;539:391–407. doi: 10.1113/jphysiol.2001.013043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailly P. TRP channels in normal and dystrophic skeletal muscle. Curr Opin Pharmacol. 2012;12:326–334. doi: 10.1016/j.coph.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Gil Z, Magleby KL, Silberberg SD. Two-dimensional kinetic analysis suggests nonsequential gating of mechanosensitive channels in Xenopus oocytes. Biophys J. 2001;81:2082–2099. doi: 10.1016/S0006-3495(01)75857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Merlie JP, Sanes JR. Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol. 1997;136:871–882. doi: 10.1083/jcb.136.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Gramolini A, Jasmin BJ. Expression of the utrophin gene during myogenic differentiation. Nucleic Acids Res. 1999;27:3603–3609. doi: 10.1093/nar/27.17.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grage SL, Keleshian AM, Turdzeladze T, Battle AR, Tay WC, May RP, Holt SA, Contera SA, Haertlein M, Moulin M, Pal P, Rohde PR, Forsyth VT, Watts A, Huang KC, Ulrich AS, Martinac B. Bilayer-mediated clustering and functional interaction of MscL channels. Biophys J. 2011;100:1252–1260. doi: 10.1016/j.bpj.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselwandter CA, Phillips R. Connection between oligomeric state and gating characteristics of mechanosensitive ion channels. PLoS Comput Biol. 2013;9:e1003055. doi: 10.1371/journal.pcbi.1003055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine agonists and antagonists. Nature. 1984;311:538–544. doi: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- Ho TC, Horn NA, Huynh T, Kelava L, Lansman JB. Evidence TRPV4 contributes to mechanosensitive ion channels in mouse skeletal muscle fibers. Channels (Austin) 2012;6:246–54. doi: 10.4161/chan.20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Volgushev M, Wolf F. A small fraction of strongly cooperative sodium channels boosts neuronal encoding of high frequencies. PLoS ONE. 2012;7:e37629. doi: 10.1371/journal.pone.0037629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Bae C, Sachs F, Suchyna TM. Caveolae regulation of mechanosensitive channel function in myotubes. PLoS ONE. 2013;8:e72894. doi: 10.1371/journal.pone.0072894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata Y, Katanosaka Y, Arai Y, Komamura K, Miyatake K, Shigekawa M. A novel mechanism of myocyte degeneration involving the Ca2+ permeable growth factor-regulated channel. J Cell Biol. 2003;161:957–967. doi: 10.1083/jcb.200301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansman JB, Franco-Obregón A. Mechanosensitive ion channels in skeletal muscle: a link in the membrane pathology of muscular dystrophy. Clin Exp Pharmacol Physiol. 2006;33:649–656. doi: 10.1111/j.1440-1681.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- Lansman JB. Mechanosensitive ion channels in dystrophic muscle. In: Hamill OP, editor. Current Topics in Membranes, Volume 59: Mechanosensitive Ion Channels, Part B. Amsterdam: Elsevier; 2007. pp. 467–484. [DOI] [PubMed] [Google Scholar]
- Love DR, Hill DF, Dickson G, Spurr NK, Byth BC, Marsden RF, Walsh FS, Edwards YH, Davies KE. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. 1989;339:55–58. doi: 10.1038/339055a0. [DOI] [PubMed] [Google Scholar]
- Lynch GS. Role of contraction-induced injury in the mechanisms of muscle damage in muscular dystrophy. Clin Exp Pharmacol Physiol. 2004;31:557–561. doi: 10.1111/j.1440-1681.2004.04026.x. [DOI] [PubMed] [Google Scholar]
- Ma X, Nilius B, Wong JW, Huang Y, Yao X. Electrophysiological properties of heteromeric TRPV4-C1 channels. Biochim Biophys Acta. 2011;1808:2789–2797. doi: 10.1016/j.bbamem.2011.07.049. [DOI] [PubMed] [Google Scholar]
- Markin VS, Sachs F. Thermodynamics of mechanosensitivity. Phys Biol. 2004;1:110–124. doi: 10.1088/1478-3967/1/2/007. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- Mika JT, Birkner JP, Poolman B, Koçer A. On the role of individual subunits in MscL gating: ‘all for one, one for all? FASEB J. 2013;27:882–892. doi: 10.1096/fj.12-214361. [DOI] [PubMed] [Google Scholar]
- Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci U S A. 2009;106:19023–19028. doi: 10.1073/pnas.0906591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina ML, Barrera FN, Fernández AM, Poveda JA, Renart ML, Encinar JA, Riquelme G, González-Ros JM. Clustering and coupled gating modulate the activity in KcsA, a potassium channel model. J Biol Chem. 2006;281:18837–18848. doi: 10.1074/jbc.M600342200. [DOI] [PubMed] [Google Scholar]
- Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010;106:748–56. doi: 10.1161/CIRCRESAHA.109.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perronnet C, Vaillend C. Dystrophins, utrophins, and associated scaffolding complexes: role in mammalian brain and implications for therapeutic strategies. J Biomed Biotech. 2010;2010:1–19. doi: 10.1155/2010/849426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JA, Cooper MA, Carland JE, Livesey MR, Hales TG, Lambert JJ. Novel structural determinants of single channel conductance and ion selectivity in 5-hydroxytryptamine type 3 and nicotinic acetylcholine receptors. J Physiol. 2010;588:587–596. doi: 10.1113/jphysiol.2009.183137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM. Dystrophin is a microtubule-associated protein. J Cell Biol. 2009;186(3):363–369. doi: 10.1083/jcb.200905048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritschow BW, Lange T, Kasch J, Kunert-Keil C, Liedtke W, Brinkmeier H. Functional TRPV4 channels are expressed in mouse skeletal muscle and can modulate resting Ca2+ influx and muscle fatigue. Pflugers Arch. 2011;461:115–122. doi: 10.1007/s00424-010-0883-4. [DOI] [PubMed] [Google Scholar]
- Ramírez-Sánchez I, Mendoza-Lorenzo P, Zentella-Dehesa A, Méndez-Bolaina E, Lara-Padilla E, Ceballos-Reyes G, Canto P, Palma-Flores C, Coral-Vázquez RM. Caveolae and non-caveolae lipid raft microdomains of human umbilical vein endothelial cells contain utrophin-associated protein complexes. Biochimie. 2012;94:1884–1890. doi: 10.1016/j.biochi.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000;150:1209–1214. doi: 10.1083/jcb.150.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin J, Cognard C, Constantin B. Regulation by scaffolding proteins of canonical transient receptor potential channels in striated muscle. J Muscle Res Cell Motil. 2009;30:289–97. doi: 10.1007/s10974-010-9206-9. [DOI] [PubMed] [Google Scholar]
- Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, Rezzani R, Rodella LF, Vriens J, Nilius B, Feron O, Balligand JL, Dessy C. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation. 2008;117:1065–1074. doi: 10.1161/CIRCULATIONAHA.107.731679. [DOI] [PubMed] [Google Scholar]
- Soria B, Navas S, Hmadcha A, Hamill OP. Single mechanosensitive and Ca2+-sensitive channel currents recorded from mouse and human embryonic stem cells. J Membr Biol. 2013;246:215–230. doi: 10.1007/s00232-012-9523-6. [DOI] [PubMed] [Google Scholar]
- Sotgia F, Lee JK, Das K, Bedford M, Petrucci TC, Macioce P, Sargiacomo M, Bricarelli FD, Minetti C, Sudol M, Lisanti MP. Caveolin-3 directly interacts with the C-terminal tail of β-dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem. 2000;275:38048–38058. doi: 10.1074/jbc.M005321200. [DOI] [PubMed] [Google Scholar]
- Squire S, Raymackers JM, Vandebrouck C, Potter A, Tinsley J, Fisher R, Gillis JM, Davies KE. Prevention of pathology in mdx mice by expression of utrophin: analysis using an inducible transgenic expression system. Hum Mol Genet. 2002;11:3333–3344. doi: 10.1093/hmg/11.26.3333. [DOI] [PubMed] [Google Scholar]
- Suchyna TM, Sachs F. Mechanosensitive channel properties and membrane mechanics in mouse dystrophic myotubes. J Physiol. 2007;581:369–387. doi: 10.1113/jphysiol.2006.125021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukharev SI, Sigurdson WJ, Kung C, Sachs F. Energetic and spatial parameters for gating of the bacterial large conductance mechanosensitive channel, MscL. J Gen Physiol. 1999;113:525–540. doi: 10.1085/jgp.113.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, Esaki K, Nomura T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol (Berl) 1986;69:91–95. doi: 10.1007/BF00687043. [DOI] [PubMed] [Google Scholar]
- Tierney ML. Insights into the biophysical properties of GABAA receptor channels: modulation of ion permeation by drugs and protein interactions. Biochim Biophys Acta. 2011;1808:667–673. doi: 10.1016/j.bbamem.2010.11.022. [DOI] [PubMed] [Google Scholar]
- Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick-Jones J, Suthers GK, Love DR, Edwards YH, Davies KE. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–89. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Ursell T, Huang KC, Peterson E, Phillips R. Cooperative gating and spatial organization of membrane proteins through elastic interactions. PLoS Comput Biol. 2007;3:e81. doi: 10.1371/journal.pcbi.0030081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandebrouck C, Martin D, Colson-Van Schoor M, Debaix H, Gailly P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol. 2002;158:1089–1096. doi: 10.1083/jcb.200203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Putten M, Kumar D, Hulsker M, Hoogaars WM, Plomp JJ, van Opstal A, van Iterson M, Admiraal P, van Ommen GJ, ‘t Hoen PA, Aartsma-Rus A. Comparison of skeletal muscle pathology and motor function of dystrophin and utrophin deficient mouse strains. Neuromuscul Disord. 2012;22:406–417. doi: 10.1016/j.nmd.2011.10.011. [DOI] [PubMed] [Google Scholar]
- Vasquez I, Tan N, Boonyasampant M, Koppitch KA, Lansman JB. Partial opening and subconductance gating of mechanosensitive ion channels in dystrophic muscle. J Physiol. 2012;590:6167–6185. doi: 10.1113/jphysiol.2012.240044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir AP, Burton EA, Harrod G, Davies KE. A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J Biol Chem. 2002;277:45285–45290. doi: 10.1074/jbc.M205177200. [DOI] [PubMed] [Google Scholar]
- Weir AP, Morgan JE, Davies KE. A-utrophin up-regulation in mdx skeletal muscle is independent of regeneration. Neuromusc Disord. 2004;14:19–23. doi: 10.1016/j.nmd.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Whitehead NP, Streamer M, Lusambili LI, Sachs F, Allen DG. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul Disord. 2006;33(7):845–854. doi: 10.1016/j.nmd.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Williams MW, Bloch RJ. Extensive but coordinated reorganization of the membrane skeleton in myofibers of dystrophic (mdx) mice. J Cell Biol. 1999;144:1259–1270. doi: 10.1083/jcb.144.6.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung EW, Whitehead NP, Suchyna TM, Gottlieb PA, Sachs F, Allen DG. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J Physiol. 2005;562:367–380. doi: 10.1113/jphysiol.2004.075275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanou N, Iwata Y, Schakman O, Lebacq J, Wakabayashi S, Gailly P. Essential role of TRPV2 ion channel in the sensitivity of dystrophic muscle to eccentric contractions. FEBS Lett. 2009;583:3600–3604. doi: 10.1016/j.febslet.2009.10.033. [DOI] [PubMed] [Google Scholar]
- Zanou N, Shapovalov G, Louis M, Tajeddine N, Gallo C, Van Schoor M, Anguish I, Cao ML, Schakman O, Dietrich A, Lebacq J, Ruegg U, Roulet E, Birnbaumer L, Gailly P. Role of TRPC1 channel in skeletal muscle function. Am J Physiol Cell Physiol. 2010;298:C149–162. doi: 10.1152/ajpcell.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BT, Whitehead NP, Gervasio OL, Reardon TF, Vale M, Fatkin D, Dietrich A, Yeung EW, Allen DG. Pathways of Ca2+ entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J Appl Physiol. 2012;112:2077–2086. doi: 10.1152/japplphysiol.00770.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]