

Abstract
Fine-tuning of cytokine-inducing pathways is essential for immune homeostasis. Consistently, a dysregulated increase or decrease in pattern recognition receptors (PRR)-induced signaling and cytokine secretion can lead to inflammatory bowel disease (IBD). Multiple gene loci are associated with IBD, but their functional effects are largely unknown. One such region in chromosome 2q12 (rs917997), also associated with other immune-mediated diseases, encompasses IL18RAP. We found that human monocyte-derived macrophages (MDM) from rs917997 AA risk carriers secrete significantly less cytokines than G carriers upon stimulation of multiple PRRs including NOD2. We identified that IL-18 signaling through IL-18RAP was critical in amplifying PRR-induced cytokine secretion in MDM. IL-18RAP responded to NOD2-initiated early, caspase-1-dependent autocrine IL-18, which dramatically enhanced MAPK, NF-κB, PI3K and calcium signaling. Reconstituting MAPK activation was sufficient to rescue decreased cytokines in NOD2-stimulated IL-18RAP-deficient MDM. Relative to GG carriers, MDM from rs917997 AA carriers had decreased expression of cell surface IL-18RAP protein, as well as of IL-18R1 and IL-1R1, genes also located in the IL18RAP region. Accordingly, these risk-carrier MDM show diminished PRR-, IL-18-, and IL-1-induced MAPK and NF-κB signaling. Taken together, our results demonstrate clear functional consequences of the rs917997 risk-polymorphism; this polymorphism leads to a loss-of-function through decreased IL-18RAP, IL-18R1 and IL-1R1 protein expression, which impairs autocrine IL-18 and IL-1 signaling, thereby leading to decreased cytokine secretion in MDM upon stimulation of a broad range of PRR.
Introduction
Immune-mediated diseases show dysregulated cytokine secretion, often via dysregulated host responses to microbes through pattern-recognition receptors (PRR)(1–3). PRR pathway perturbations resulting in either a loss-of-function (e.g. MyD88−/− or TLR5−/− mice)(4, 5) or gain-in-function (e.g. IRAK-M−/− or A20−/− mice)(6,7) can increase susceptibility to intestinal inflammation(2). Furthermore, polymorphisms affecting PRR and cytokine-inducing pathways can contribute to human immune-mediated diseases(1,8). For example, the highest genetic risk toward developing Crohn’s disease, one form of inflammatory bowel disease (IBD), are loss-of-function NOD2 polymorphisms(1). These polymorphisms decrease cytokine secretion following stimulation with muramyl dipeptide (MDP)(1,9–12), the minimal bacterial peptidoglycan component activating NOD2(13,14). Microbes activate multiple PRR, which is particularly important in tissues undergoing ongoing microbial exposure, such as the intestine. Therefore, identifying disease-associated polymorphisms regulating pathways common to multiple PRR may highlight mechanisms that affect global immune outcomes. Although multiple loci have now been associated with immune-mediated diseases, the functional consequences of the majority of these loci are unknown. Understanding these consequences is essential to ultimately design disease therapies.
IL18RAP region polymorphisms are associated with multiple immune-mediated diseases, including IBD(15), atopic dermatitis(16), leprosy(17), celiac disease(18) and Type I diabetes(19). IL-18RAP interactions with IL-18R1 mediate signal transduction initiated by IL-18(20). IL-18 signaling has been well established in mediating Th1 responses(20), but also contributes to diverse biological processes, such as responses to commensal microbiota and integrity of the intestinal epithelial barrier(21–23). Consequently, diminished IL-18 induction by the inflammasome, a multi-molecular complex required for caspase-1 activation and IL-1 and IL-18 processing(24), exacerbates experimental colitis and intestinal injury(21,22). Moreover, IL-18 mediates resolution of lung infection(25). Conversely, IL-18 administration can induce murine colitis(26) and lupus-like disease(27). Therefore, balancing IL-18 pathways is important, as IL-18 can have both protective and detrimental roles. Given the IL18RAP region polymorphisms associated with IBD(15), and the importance of host:microbe interactions to intestinal immune homeostasis, we examined IL-18RAP signaling and the outcomes of the IL18RAP region rs917997 polymorphism during exposure to microbial components in primary human monocyte-derived macrophages (MDM), cells mediating responses to microbiota.
To directly address physiological relevance to human immune responses, we utilized human myeloid cells from a large cohort of individuals. We found that less cytokines were induced upon stimulation of NOD2 and multiple TLR alone or in combination in rs917997 AA risk carriers. Consistently, knock-down of IL-18RAP expression, as well as IL-18RAP or IL-18 blockade, significantly attenuated NOD2- and PRR-induced cytokines, highlighting an important role for autocrine IL-18. This NOD2-induced autocrine IL-18 was initiated by rapid caspase-1-dependent cleavage of pre-existing pro-IL-18, and led to optimal MAPK, NF-κB, PI3K and calcium flux activation. Independently inducing MAPK activation was sufficient to rescue decreased NOD2-induced cytokines in IL-18RAP deficient cells. Finally, we investigated the mechanism through which rs917997 regulates PRR signaling. Rs917997 is in a gene cluster containing IL18RAP, IL18R1, IL1R1, IL1R2, IL1RL1 and IL1RL2, and we found that MDM from rs917997 AA carriers express decreased IL-18RAP, IL-18R1 and IL-1R1 surface protein, and have significantly reduced NOD2-, IL-1- and IL-18-induced ERK, p38 and NF-κB activation relative to GG carriers. Therefore, rs917997 affects both IL-18- and IL-1 pathways. Taken together, we identify functional consequences for the rs917997 polymorphism associated with multiple immune-mediated diseases, and elucidate that autocrine IL-18 dramatically enhances PRR-induced signaling and cytokine secretion in human MDM.
Materials and Methods
Patient recruitment and genotyping
Informed consent was obtained per protocol approved by the institutional review board at Yale University. Healthy participants with no personal or family history of autoimmune/inflammatory disease, including psoriasis, systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, type I diabetes mellitus, Crohn’s disease, and ulcerative colitis, or a HIV history were enrolled. Due to limitations in primary human cell numbers two separate cohorts were collected and stimulated in a differential manner. A cohort of 100 individuals was recruited for NOD2 and TLR2 dose-response studies in MDM (as shown in Fig. 1A&B). A second cohort of 98 individuals was collected for TLR response and NOD2/TLR synergy studies in monocyte-derived dendritic cells (MDDCs) (as shown in Fig. 1C&D). Genotyping was performed by TaqMan SNP genotyping (Applied Biosystems, Foster City, CA) or Sequenom platform (Sequenom Inc., San Diego, CA).
Figure 1. Primary human myeloid cells from disease-associated rs917997 AA carriers demonstrate decreased cytokine secretion upon PRR stimulation.

Human MDMs (n=100) were treated for 24h with (A) 1, 10 or 100 μg/ml MDP, or (B) 1, 10 or 100 μg/ml Pam3Cys. Human MDDCs (n=98) were treated for 24h with 1 μg/ml MDP (NOD2 ligand) and 1 μg/ml Pam3Cys (TLR2 ligand), 0.1 μg/ml polyI:C (TLR3 ligand), 0.01 μg/ml lipid A (TLR4 ligand), 0.5 ng/ml flagellin (TLR5 ligand), 0.1 μg/ml CL097 (TLR7 ligand) or 0.1 μg/ml CpG DNA (TLR9 ligand) for 24 h alone (C) or in combination (D). Fold TNF-α induction (log2 transformed) upon PRR stimulation stratified on the rs917997 genotype + SEM. *, p<0.05; **, p<0.01.
Primary myeloid cell culture
Mononuclear cells were isolated from human peripheral blood by Ficoll-Hypaque centrifugation (GE Healthcare, Piscataway, NJ). Monocytes were then purified by CD14+ selection (Miltenyi Biotec, Auburn, CA) and cultured with M-CSF (10 ng/ml) or IL-4 (40 ng/ml) and GM-CSF (40 ng/ml) (R&D Systems Inc. Minneapolis, MN) for 7 days for MDM and MDDC differentiation, respectively.
Myeloid cell stimulation
Cultured myeloid cells were treated with muramyl dipeptide (MDP) (Bachem, King of Prussia, PA), Pam3Cys-Ser-(Lys)4 (Calbiochem, La Jolla, CA), lipid A (Peptides International, Louisville, KY), flagellin, CL097, CpG, poly I:C (Invivogen, San Diego, CA) or IL-18 (R&D Systems Inc.). MDM were incubated with neutralizing anti-IL-18RAP or anti-IL-18 antibodies (R&D Systems Inc.) 1h prior to stimulation. Supernatants were assayed for TNF-α, IL-8, IL-6, IL-10 (BD Biosciences), or IL-18 and IL-1β (eBioscience, San Diego, CA) by ELISA.
Transfection of small interfering RNAs (siRNAs) and plasmids
300 nM scrambled or ON-TARGETplus SMARTpool small interfering RNA (siRNA) against IL-18RAP (Dharmacon, Lafayette, CO) (4 pooled siRNAs for each gene) or 5μg pMCL-MKK1 (R4F) (constitutively active ERK kinase)(28), pSRα-3HA-JNKK2-JNK1-WT (constitutively active JNK)(29) (generous gifts from Dr. Ben Turk), pCDNA3-Flag MKK6(glu) (constitutively active p38 kinase)(30) (Addgene plasmid 13518) or empty vector were transfected into myeloid cells using Amaxa nucleofector technology (Amaxa, San Diego, CA). Cells were cultured for an additional 48h and then treated as indicated, or in some cases stained with annexin V-FITC (eBiosciences) to ensure cell viability.
Phosphoprotein, calcium flux and surface protein detection
Phosphoprotein and calcium flux induction was determined by flow cytometry using Alexa Fluor 647, phycoerythrin- or Alexa Fluor 488-labeled antibodies to phospho-ERK, phospho-p38, phospho-JNK, phospho-Akt, phospho-p70-S6K and phospho-IκBα (Cell Signaling, Danvers, MA) or calcium green (eBioscience) along with MDP- or IL-18-treated isotype controls. For phosphoprotein staining, cells were fixed for 15 min using BD Cytofix/Cytoperm buffer, permeablilized for 30 min using Perm Buffer III (BD Biosciences), and then stained with antibodies suspended in Perm Buffer III for 1h. Surface protein expression was measured using phycoerythrin-labeled anti-IL-18RAP (BD Biosciences), fluorescein-labeled anti-IL-18R1 (Abcam, Cambridge, MA) and allophycocynanin-labeled anti-IL-1R1 (R&D Systems, Inc).
mRNA expression analysis
Following stimulation, total RNA was isolated using Trizol reagent (Life Technologies, Gaithersburg, MD), reverse transcribed by Superscript III reverse transcriptase (Invitrogen, San Diego, CA), and quantitative PCR performed using Maxima Sybr Green qPCR Master Mix (Thermo Fisher Scientific, Waltham, MA) on the ABI 7500 Real-Time PCR system (Applied Biosystems). Each sample was run in duplicate and normalized to GAPDH. Primers sequences are available upon request.
Protein expression analysis
Western blot was performed as in (12) with anti-caspase-1 (Cell Signaling Technology) or anti-IL-18 (Abcam) antibodies. GAPDH (Calbiochem) was assessed on separate blots as a loading control.
Statistical analysis
Significance was assessed using two-tailed t-test. p < 0.05 was considered significant.
Results
The rs917997 disease-risk polymorphism in the IL18RAP region dramatically decreases PRR-induced cytokine secretion in primary human myeloid cells
IBD is characterized by dysregulated responses to microbes and cytokine production(1). IL-18 is primarily produced by myeloid cells upon microbial exposure(20). We therefore asked if rs917997 in the IL18RAP region associated with IBD(15), modulates PRR-induced cytokines in primary human myeloid cells. We initially examined NOD2 stimulation, given the association of NOD2 polymorphisms to Crohn’s disease(1). We stimulated NOD2 with MDP, the minimal peptidoglycan component specifically activating NOD2 (10–14,31). TNF-α is critical for inflammation in Crohn’s disease(1), such that we measured TNF-α secretion following NOD2 stimulation of MDM from 100 healthy individuals. To analyze this large cohort, we normalized NOD2-induced cytokine induction to untreated cells and log2 transformed the data. MDM from rs917997 AA risk carriers secreted significantly lower TNF-α (Fig. 1A) upon NOD2 stimulation than GA or GG carriers. This effect was most dramatic at low MDP doses, but persisted at higher MDP doses. In addition to peptidoglycan, Gram-positive bacteria cell walls express the TLR2 ligand lipotechoic acid, and MDM from rs917997 AA carriers also demonstrated decreased TLR2-mediated cytokine secretion relative to GA and GG carriers (Fig. 1B). Dendritic cells (DC) also mediate PRR-induced cytokine secretion, and MDDC from rs917997 AA carriers similarly showed decreased NOD2 and TLR2-induced cytokines (Fig. 1C). Microbial products activate multiple additional receptors, and NOD2 can synergize with other PRR(8). We therefore investigated if the rs917997 polymorphism modulated cytokine secretion following stimulation of additional PRR alone or combined with NOD2. We utilized MDDC from a separate cohort of 98 people given limitations in primary cell numbers. Cytokine secretion under all these conditions was dramatically reduced in rs917997 AA carriers (Fig. 1C&D). Taken together, the celiac- and IBD-associated rs917997 risk IL18RAP region polymorphism dramatically reduces cytokine secretion by multiple PRR in primary human myeloid cells.
PRR stimulation induces IL-18RAP expression
We next asked if PRR stimulation modulates IL-18RAP expression. We detected surface IL-18RAP in untreated MDM (Fig 2A&B). This expression was further increased upon NOD2 stimulation with 100 μg/ml MDP, peaking at 6h (Fig. 2A&B). This MDP concentration approximates muramic acid levels in stool(32), and results in optimal NOD2-mediated cytokine secretion in MDM (Fig. 1A). We then asked if NOD2-induced IL-18RAP expression is regulated at the mRNA level. Three IL18RAP isoforms were previously identified in human tissues(33). We measured the total IL18RAP mRNA using primers that detect all three IL18RAP isoforms, as well as each isoform separately. NOD2 stimulation increased the expression of each IL-18RAP isoform with the optimal peak at 2h (Fig. 2C&D). Taken together, NOD2 stimulation enhances IL-18RAP expression in MDM.
Figure 2. IL-18RAP is expressed on human MDM and is upregulated upon NOD2 stimulation.

(A&B) MDM (n=8) were treated for the indicated times with 100 μg/ml MDP. (A) Representative flow cytometry plots with IL-18RAP MFI values. (B) Fold IL-18RAP protein induction normalized to untreated cells + SEM. (C&D) MDMs (n=8) were treated for the indicated time points with 100 μg/ml MDP. Fold IL18RAP mRNA induction using (C) primers detecting all three IL18RAP isoforms or (D) primers detecting each of the three distinct IL18RAP isoforms normalized to untreated cells (represented by the dotted line at 1) + SEM. Isoform sizes are indicated. *, p<0.05; **, p<0.01; ***, p<0.001; †, p<1×10−4.
Reduced IL-18RAP expression and signaling dramatically decreases NOD2-induced cytokine induction
We next examined the mechanism through which IL-18RAP contributes to PRR-induced cytokine secretion. We hypothesized that PRR stimulation induces autocrine IL-18 secretion which then signals through IL-18RAP to augment PRR-induced cytokines. We addressed this hypothesis through two independent approaches. First, we used anti-IL-18RAP neutralizing antibodies to block this proposed autocrine IL-18. IL-18RAP blockade dramatically downregulated NOD2-induced pro- and anti-inflammatory cytokine secretion (Fig. 3A). Second, we knocked down IL-18RAP expression by siRNA (Supplemental Fig. 1A). We ensured that cell viability was unaffected. Similar to IL-18RAP blockade, IL-18RAP expression knock-down significantly decreased NOD2-induced cytokine secretion (Fig. 3B). Anti-inflammatory cytokine secretion through dectin ligands was not affected, indicating that the cells could respond to other stimuli (Supplementary Fig. 1B). To establish that additional PRR require IL-18RAP for cytokine secretion, we knocked-down IL-18RAP and measured cytokine secretion following NOD1, TLR2, TLR3, TLR4, TLR5, TLR7 and TLR9 stimulation. IL-18RAP was critical for cytokine secretion upon stimulating all these receptors (Fig. 4). Taken together, IL-18RAP is required for optimal cytokine secretion following stimulation of NOD2 and multiple PRR.
Figure 3. IL-18RAP signaling is required for optimal NOD2-mediated cytokine secretion.
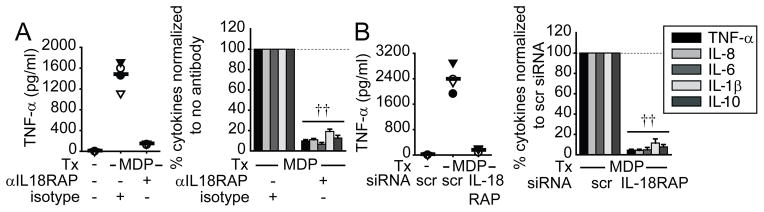
MDM were incubated with anti-IL-18RAP (300 ng/ml) blocking antibody or mIgG1 isotype control for 1h (A), or transfected with IL-18RAP siRNA for 48h (B), and then treated with 100 μg/ml MDP for 24h. (Left): TNF-α secretion in 4 representative individuals. (Right): Percent cytokine secretion (n=8) normalized to MDP-treated cells with isotype control or scrambled siRNA (dotted line at 100%)+SEM. Tx, treatment; scr, scrambled. ††, p<1×10−5.
Figure 4. IL-18RAP signaling is required for optimal PRR-mediated cytokine secretion.

MDM were transfected with scrambled or IL-18RAP siRNA for 48h, then treated with 100 μg/ml TriDAP, 10 μg/ml Pam3Cys, 100 μg/ml poly I:C, 0.1 μg/ml lipid A, 5 ng/ml flagellin, 1 μg/ml CL097 or 10 μg/ml CpG DNA for 24h. Top: TNF-α secretion for 4 representative individuals. Bottom: Percent cytokine secretion (n=8) normalized to cells transfected with scrambled siRNA (represented by the dotted line at 100%)+SEM. Tx, treatment; scr, scrambled. ††, p<1×10−5.
NOD2 stimulation induces autocrine IL-18 which dramatically augments NOD2-mediated cytokine secretion
We next sought to define how NOD2 regulates autocrine IL-18. IL-18 secretion peaked 4h following MDP treatment (Fig. 5A). As IL-18 secretion was low and decreased following 4h, we questioned if IL-18 was being rapidly consumed; we previously observed such consumption with IL-1β(10). We therefore blocked IL-18RAP to prevent early IL-18 consumption, and measured IL-18 secretion 15 min following MDP treatment. We observed previously undetectable secreted IL-18 at this early time (Fig. 5B), indicating that NOD2 stimulation results in rapid, but quickly consumed IL-18 secretion. This secretion occurred within a time frame (15 min) shorter than that required for IL-18 transcription and translation. Consistently, the transcriptional suppressor actinomycin did not affect early NOD2-induced IL-18 secretion (Fig. 5B). Therefore, we hypothesized that the early IL-18 secretion is due to a rapid caspase-1-mediated cleavage of pre-existing pro-IL-18 stores. Caspase-1 activation by MDP in primary human myeloid cells has been observed by us and others (34,35). We therefore knocked-down caspase-1 through siRNA (Fig. 5C), and ensured that the cells were viable (Fig. 5D). We then measured IL-18 secretion 15 min following MDP treatment under IL-18RAP blockade to prevent cytokine consumption. MDP-induced IL-18 was undetectable (Fig. 5E), indicating that caspase-1 is required for NOD2-induced early IL-18 secretion. We further confirmed mature IL-18 induction 15 min after NOD2 stimulation (Fig. 5F). Finally, to clearly confirm the role of autocrine IL-18 we neutralized IL-18; similar to IL-18RAP blockade results (Fig. 3A), MDP-induced secretion of additional cytokines was dramatically reduced (Fig. 5G). Therefore, NOD2 signaling activates rapid, caspase-1-dependent processing of pre-existing pro-IL-18, leading to autocrine IL-18 secretion which is then required for optimal MDP-initiated secretion of additional cytokines.
Figure 5. An early, caspase-1-dependent, IL-18 autocrine loop augments NOD2-initiated cytokine induction.

(A) MDM (n=8) were treated with 100 μg/ml MDP for the indicated time points and supernatants assessed for IL-18. (B) MDM (n=4) were left untreated or pretreated with actinomycin D (10 μg/ml), incubated with anti-IL-18RAP (300 ng/ml) blocking antibody or mIgG1 isotype control for 1h, then treated with 100 μg/ml MDP for 15 min. Supernatants were assessed for IL-18. (A&B) Similar results were observed in an additional n=4. (C) MDM were transfected with scrambled or caspase-1 siRNA for 48h. Pro-caspase-1 expression by Western blot from 2 of 4 representative donors. (D) MDM (n=4) were transfected with scrambled or caspase-1 siRNA for 48h, then stained with annexin V. Shown is percent annexin V+ cells as a measure of cell death. UV treatment at 50–100 J/m2 is shown as a positive control. (E) MDM (n=8) were transfected with caspase-1 siRNA for 48h, then incubated with anti-IL-18RAP (300 ng/ml) blocking antibody for 1h to prevent IL-18 consumption, and then treated with 100 μg/ml MDP for 15 min. Supernatants were assessed for IL-18. (F) MDMs were left untreated or treated with 100 μg/ml MDP for 15 min. Mature IL-18 expression by Western blot from 2 of 6 representative donors. (C, F) GAPDH from lysates was assessed on separate blots as a loading control. (G) MDM were incubated with an anti-IL-18 (100 ng/ml) neutralizing antibody or mIgG1 isotype control for 1h and then treated with 100 μg/ml MDP for 24h. Percent cytokine secretion (n=8) normalized to MDP-treated cells without antibody treatment (represented by the dotted line at 100%)+SEM. Tx, treatment; NS, not significant; casp, caspase; mIL18, mature IL-18. ***, p<0.001; ††, p<1×10−5.
IL-18 induces MAPK, NF-κB and PI3K signaling and calcium flux
We next investigated how IL-18 regulates cytokine-inducing signaling pathways that might cooperate with those initiated by NOD2. ERK, p38, JNK(20,36) and NF-κB(36) mediate IL-18-induced cytokine secretion in select cell subsets, and we asked if these pathways are activated specifically in IL-18-treated primary human MDM. IL-18 activated all three MAPK (Supplementary Fig. 2A) and the NF-κB pathway intermediate IκBα (Supplementary Fig. 2B). IL-18 also activated the PI3K pathway intermediate Akt (Supplementary Fig. 2C) which can contribute to IL-18-mediated cytokine secretion(36, 37). The PI3K intermediate mTOR contribution to IL-18-mediated cytokine production in human MDM has not been well investigated; we found that IL-18 also activated the mTOR pathway (Supplementary Fig. 2C). IL-18 can regulate calcium-related signaling in T cells(20,36). However, to our knowledge, whether IL-18 induces calcium flux has not been investigated in human MDM. We found that IL-18 also induces calcium flux in human MDM (Supplementary Fig. 2D). Taken together, IL-18 activates multiple cytokine-inducing pathways in primary human MDM.
IL-18RAP signaling is required for MDP-initiated MAPK, NF-κB, PI3K activation and calcium flux
We next questioned if the IL-18-activated signaling pathways we identified in Supplementary Fig. 2 are specifically enhanced by NOD2-induced autocrine IL-18/IL-18RAP signaling. Upon IL-18RAP knock-down, MAPK, NF-κB and PI3K activation and calcium flux were all significantly attenuated following MDP treatment (Fig. 6), indicating that autocrine IL-18 is required for optimal MDP-initiated activation of these cytokine-inducing pathways.
Figure 6. The IL-18 autocrine loop is required for optimal MDP-initiated MAPK, NF-κB, PI3K signaling and calcium flux in primary human MDM.

MDM (n=8) were transfected with scrambled or IL-18RAP siRNA. 48h later, cells were treated for 15 min with 100 μg/ml MDP. Left: Representative flow cytometry plots with MFI values as indicated for (A) phospho-ERK, phospho-p38, phospho-JNK, (B) phospho-IκBα, (C) phospho-Akt and phospho-p70S6K or (D) calcium green. Isotype controls are shown in grey. Right: Summarized data showing the fold phospho-protein or calcium flux induction normalized to untreated cells (dotted line at 1) + SEM. scr, scrambled. ***, p<0.001; †, p<1×10−4.
IL-18RAP signaling through MAPK is required for NOD2-induced cytokine secretion
Given that IL-18RAP signaling was required for both optimal MAPK signaling (Fig. 6) and cytokine secretion (Fig. 3) through NOD2, we questioned if rescuing MAPK during IL-18RAP knockdown is sufficient to restore diminished NOD2-induced cytokines. We therefore transfected plasmids constitutively activating ERK, p38 and JNK pathways into MDM knocked-down for IL-18RAP and treated with MDP, and confirmed restoration in activation of each respective MAPK to physiological levels (Supplementary Fig. 3). Importantly, transfection with each kinase, and particularly all three kinases combined, significantly restored cytokine secretion following NOD2 stimulation in MDM knocked-down for IL-18RAP (Fig. 7). Taken together, MAPK activation is sufficient to significantly rescue the impaired MDP-induced cytokine secretion in the absence of autocrine IL-18 signaling.
Figure 7. MAPK activation rescues the decreased cytokine secretion following NOD2 stimulation in the absence of IL-18RAP signaling.
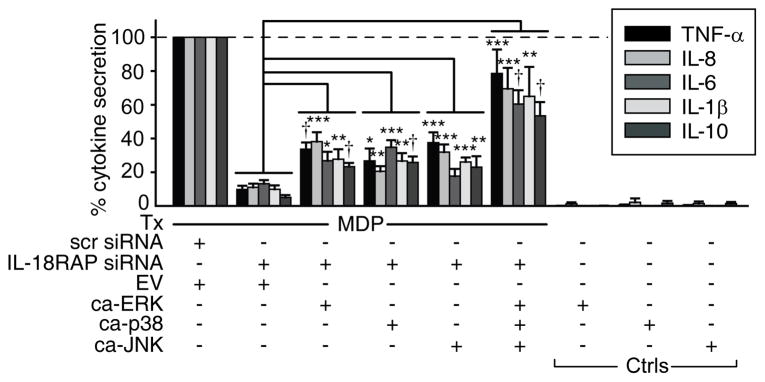
MDM (n=8) were transfected with IL-18RAP siRNA and/or 5 μg pMCL-MKK1 (R4F) (constitutively active ERK, ca-ERK), pCDNA3-Flag MKK6(glu) (constitutively active p38 kinase, ca-p38), pSRα-3HA-JNKK2-JNK1 WT (constitutively active JNK, ca-JNK), the three vectors in combination, or empty vector (EV) and left untreated or 48h later treated with 100μg/ml MDP for 24h. Cytokine secretion normalized to MDP treatment of EV and scrambled siRNA transfected cells (dotted line at 100%)+SEM. Selected significance comparisons are shown. *, p<0.05; **, p<0.01; ***, p<0.001; †, p<1×10−4. Tx, treatment; ctrls, controls.
rs917997 AA carrier cells demonstrate reduced IL-18RAP expression relative to GA and GG carriers
As rs917997 AA risk carriers showed dramatically reduced PRR-induced cytokine secretion relative to G allele carriers (Fig. 1), we next investigated the mechanism through which this polymorphism regulates cytokine secretion. Most IBD-associated polymorphisms are located in non-coding regions(1) and thus may modify risk-gene expression. As rs917997 is located approximately 1.5 kb downstream of the IL18RAP, we hypothesized that it would regulate IL18RAP expression. In untreated cells IL18RAP mRNA expression was highest in rs917997 GG carriers, intermediate in GA and lowest in AA carriers (Fig. 8A). A similar differential expression pattern was observed following MDP stimulation (Fig. 8A). Consistent with mRNA expression, AA carriers expressed significantly lower surface IL-18RAP protein than GG carriers. Interestingly, while IL-18RAP protein expression differed between GA and GG carriers in unstimulated MDM, this difference was lost upon NOD2 stimulation (Fig. 8B). This is consistent with Fig. 1 in which GA and GG carriers demonstrated similar NOD2-induced cytokines. Taken together, MDM from rs917997 AA carriers demonstrate reduced IL-18RAP mRNA and protein expression.
Figure 8. MDM from rs917997 AA risk carriers express decreased IL18RAP, IL18R1 and IL1R1 mRNA and surface protein relative to GA and GG carriers.

(A–B) MDM from rs917997 GG, GA or AA carriers (n=16, n=15 and n=9, respectively) were left untreated or treated with 100 μg/ml MDP for (A) 2h or (B) 6h. (A) IL18RAP mRNA expression (change in CT values normalized to GAPDH and represented as a linear scale) or (B, right) IL-18RAP surface protein expression, with average MFI values above bars. (B, left) Representative flow cytometry plots with MFI values. (C) Representation of the IL18RAP gene region on chromosome 2q12. (D–G) MDM from rs917997 GG, GA or AA carriers (n=16, n=15 and n=9, respectively) were left untreated or treated with 100 μg/ml MDP for (D,F) 2h or (E,G) 6h. (D,F) IL18R1 and IL1R1 mRNA expression (change in CT values normalized to GAPDH and represented as a linear scale) or (E,G right) protein expression, with average values indicated above bars. (E,G, left) Representative flow cytometry plots of IL-18R1 and IL-1R1 expression with MFI values. *, p<0.05; **, p<0.01; ***, p<0.001; †, p<1×10−4; ††, p<1×10−5.
rs917997 regulates the expression of IL-18R1 and IL-1R1
Interestingly, rs917997 is located in a region also containing IL18R1, IL1RL1, IL1RL2, IL1R1 and IL1R2 (Fig. 8C); some of these gene products bind and regulate additional inflammasome products. We therefore hypothesized that besides regulating IL18RAP, rs917997 would regulate the expression of a subset of these genes. We first asked if NOD2 stimulation upregulates the expression of these genes with similar kinetics to IL18RAP. IL18R1, IL1R1 and IL1RL1 mRNA was upregulated in MDM upon NOD2 stimulation with a peak at 2h (Supplementary Fig. 4A), paralleling the IL18RAP results in Fig. 2C. Importantly, MDM from rs917997 AA carriers had decreased IL18R1 and IL1R1 mRNA (Fig. 8D&F) and surface protein (Fig. 8E&G) expression compared to MDM from GA and GG individuals. Consistent with rs917997 effects on PRR-induced cytokines (Fig. 1) and IL-1R1 expression (Fig. 8F–G), we previously found that IL-1R autocrine signaling dramatically regulates PRR-induced cytokine secretion(10, 31). While IL1RL1 and IL1RL2, located on the chromosome between the above genes, were regulated in a genotype-dependent manner at the mRNA level (Supplementary Fig. 4B–C), surface protein expression of these genes was not significantly different between different genotypes (Supplementary Fig. 4E–F). IL1R2, which is the gene most distant from the rs917997 SNP, was regulated neither at the mRNA nor at the surface protein level in a genotype-dependent manner (Supplementary Fig. 4D&G). Of note, the expression of pro-caspase-1 and early secretion of IL-18 were not significantly modulated by rs917997 genotype (Supplementary Fig. 4H&I). This is consistent with the rs917997 genotype regulating the expression of the functional IL-18 receptor components, IL-18RAP and IL-18R1, and therefore later autocrine-dependent cytokine secretion, but not the initial PRR-induced signaling regulating early IL-18 secretion. We therefore establish that MDM rs917997 AA carriers have decreased IL18RAP, as well as decreased IL18R1 and IL1R1 mRNA and protein expression, relative to GA and GG carriers, indicating that this polymorphism impacts both IL-18- and IL-1-initiated signaling pathways.
rs917997 AA carrier MDM show diminished NOD2-, IL-18R- and IL-1R-induced signaling relative to GG carriers
We next questioned if rs917997 AA carriers demonstrate decreased NOD2-mediated signaling compared to GA and GG carrier MDM. MDM from AA carriers showed significantly decreased NOD2-induced MAPK (Fig. 9 & Supplementary Fig. 4J) and NF-κB (Supplementary Fig. 4K) pathway activation. Furthermore, consistent with the low IL-18RAP, IL-18R1 and IL-1 expression on rs917997 AA MDM, AA carriers showed significantly diminished MAPK (Fig. 9 and Supplementary Fig. 4J) and NF-κB (Supplementary Fig. 4K) signaling upon direct treatment with IL-18 or IL-1 relative to GA or GG carriers. Taken together, rs917997 AA risk allele carrier MDM show reduced IL-18R and IL-1R signaling relative to GA or GG carriers, which then affects multiple pathways requiring autocrine IL-18 and IL-1 for optimal responses.
Figure 9. rs917997 AA risk carrier MDM show decreased NOD2-, IL-18- and IL-1-mediated ERK activation relative to GA and GG carriers.

MDM from rs917997 GG, GA or AA carriers (n=10, n=9 and n=8, respectively) were treated for 15 min with 100 μg/ml MDP, 10 ng/ml IL-18 or 10 ng/ml IL-1β. Left: Representative flow cytometry plots of phospho-ERK with MFI values. Right: Summarized data of the fold phospho-ERK induction normalized to untreated cells + SEM. ***, p<0.001; †, p<1×10−4; ††, p<1×10−5.
Discussion
Functional perturbations through disease-associated polymorphisms can define pathophysiological mechanisms in fundamental primary human cell outcomes. Here we elucidate in a large cohort of individuals that the rs917997 IL18RAP region AA genotype, associated with IBD- and celiac disease risk(15,18), results in a loss-of-function with dramatically decreased IL-18RAP expression, signaling and cytokine secretion following stimulation across a broad range of PRR. Consistently, by three independent approaches, knock-down of IL-18RAP expression, and IL-18RAP and IL-18 neutralization in MDM, we show that decreased IL-18 signaling dramatically attenuates PRR-mediated cytokine secretion. We therefore establish that the IL-18 autocrine loop, acting through IL-18RAP, is critical in amplification of PRR-induced cytokine secretion in primary human MDM. Mechanistically, rapid, PRR-initiated, caspase-1-dependent cleavage of pre-existing pro-IL-18 leads to this autocrine IL-18, which in turn is critical for optimal PRR-induced MAPK, NF-κB, PI3K and calcium signaling and cytokine secretion. As such, reconstituting MAPK activation is sufficient to significantly rescue the decreased cytokines upon NOD2 stimulation in the absence of IL-18 signaling. Importantly, consistent with the cluster of cytokine receptors in the region, cells from rs917997 carriers show reduced surface protein expression of not only IL-18RAP, but also of IL-18R1, the other subunit of the IL-18R complex, as well as of IL-1R1, part of the receptor complex for IL-1. While rs917997 risk carrier MDM show only a relative defect in the NOD2-induced cytokines due to diminished IL-18RAP, IL-18R1 and IL-1R1 expression, complete elimination of IL-18RAP expression and signaling results in a dramatically decreased NOD2-induced cytokine secretion even at high doses of MDP treatment. Taken together, we now identify that autocrine IL-18 dramatically amplifies PRR-initiated signaling in human MDM, and the loss-of-function rs917997 disease-risk polymorphism in the IL18RAP region results in diminished PRR-initiated signaling and cytokine secretion via reduced IL-18 and IL-1 pathway signaling (Supplementary Fig. 4L).
IL18RAP locus polymorphisms are associated with inflammatory and infectious diseases(15–19). Either insufficient or excessive cytokine expression can contribute to the development of complex immune-mediated diseases and the regulation can be context- and tissue-dependent(1,4–7). Our findings that the IBD-associated IL18RAP/IL18R1/IL1R1 region polymorphism results in decreased signaling and cytokine expression following NOD2 stimulation parallel the outcomes of the loss-of-function Crohn’s disease-associated NOD2 Leu1007insC mutation(1, 10–12, 31). However, in addition to affecting NOD2-mediated cytokine secretion, rs917997 risk carriers show diminished cytokine secretion through multiple PRR, thus modulating global effects on immune responses. The increased disease risk conferred by the loss-of-function polymorphism in the IL18RAP/IL18R1/IL1R1 region may be associated with failure to mount proper cytokine secretion to intestinal microbial exposures, which could then lead to persistent microbial colonization and inflammation. Another possibility for the increased disease risk may be through IL-18-mediated protective and anti-inflammatory effects. For example, deficient IL-18 production in mice lacking NLRP6 exacerbates DSS-induced colitis through altered gut microbiota(21). In addition, IL-18-mediated downregulation of IL-22BP production in myeloid-derived cells allows for IL-22 availability, which in turn facilitates epithelial cell repair during episodes of intestinal injury(22).
We find that in addition to regulating the mRNA and protein expression of IL-18RAP, rs917997 disease-allele carriers also have reduced IL18R1, IL1RL1, IL1RL2 and IL1R1 transcripts. Therefore, this single polymorphism regulates transcript expression of multiple adjacent genes. However, in contrast to the genotype-dependent regulation of adjacent genes at the transcript level, neither basal nor NOD2-induced IL-1RL1 or IL-1RL2 surface protein expression was significantly different in MDM based on rs917997 genotype. This is consistent with the fact that the correlation between mRNA abundance and protein levels has been reported to be approximately 40%; other factors such as post-transcriptional and -translational regulation can account for discrepancies between mRNA and protein changes (38–40). This emphasizes the importance of assessing genotype-dependent modulation in expression of relevant genes at both the mRNA and protein levels. A recent study examining the large Framingham offspring cohort found that polymorphisms in IL1RL1, including rs917997, can regulate soluble IL-1RL1 plasma levels (41). We observed rs917997 genotype-dependent IL1RL1 regulation of mRNA, but not of transmembrane IL-1RL1 protein on MDM. This may indicate that the genotype-dependent regulation of IL1RL1 protein may be dependent on such factors as the cell subset or protein form (e.g. transmembrane vs soluble) examined. IL1R2, the gene furthest from rs917997 was regulated neither at the mRNA nor protein level in a genotype-dependent manner. Interestingly, in evaluating PRR-inducible expression of this gene, PRR stimulation did not upregulate IL1R2 mRNA, but upregulated IL-1R2 surface protein. This is consistent with the fact that IL-1R2 surface protein expression is regulated post-transcriptionally through cleavage of pre-existing IL-1R2 and subsequent transport to the cell surface (42).
Thus, the IL18RAP region polymorphism effects on multiple PRR-initiated pathways in MDM implicate the IL-18 pathway as a potentially important target of immune-mediated disease therapy. These findings suggest that one must use caution when designing inflammatory disease therapies targeting the IL-18 pathway, as maintaining or enhancing, rather than inhibiting IL-18 signaling, might be more efficacious for certain diseases.
Supplementary Material
Acknowledgments
This work was supported by The Broad Foundation, and NIH: DK099097, R01DK077905, DK062422, DK-P30-34989, U19-AI082713.
We gratefully acknowledge Dr. Ben Turk for reagents.
Abbreviations
- MDM
monocyte-derived macrophages
- MDP
muramyl dipeptide
- NOD
nucleotide-binding oligomerization domain
- PRR
pattern-recognition receptor
References
- 1.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140:1729–1737. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zenewicz LA, Abraham C, Flavell RA, Cho JH. Unraveling the genetics of autoimmunity. Cell. 2010;140:791–797. doi: 10.1016/j.cell.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, Gewirtz AT. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Biswas A, Wilmanski J, Forsman H, Hrncir T, Hao L, Tlaskalova-Hogenova H, Kobayashi KS. Negative regulation of Toll-like receptor signaling plays an essential role in homeostasis of the intestine. Eur J Immunol. 2011;41:182–194. doi: 10.1002/eji.201040479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, Barrera J, Huang EJ, Hou B, Malynn BA, Reizis B, DeFranco A, Criswell LA, Nakamura MC, Ma A. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12:1184–1193. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedl M, Abraham C. Negative regulation of human mononuclear phagocyte function. Mucosal Immunol. 2013;6:205–223. doi: 10.1038/mi.2012.139. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, Kitani A, Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hedl M, Abraham C. Distinct Roles for Nod2 Protein and Autocrine Interleukin-1{beta} in Muramyl Dipeptide-induced Mitogen-activated Protein Kinase Activation and Cytokine Secretion in Human Macrophages. J Biol Chem. 2011;286:26440–26449. doi: 10.1074/jbc.M111.237495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hedl M, Abraham C. Secretory mediators regulate Nod2-induced tolerance in human macrophages. Gastroenterology. 2011;140:231–241. doi: 10.1053/j.gastro.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedl M, Li J, Cho JH, Abraham C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci U S A. 2007;104:19440–19445. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girardin SE, I, Boneca G, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 14.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nunez G. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 15.Zhernakova A, Festen EM, Franke L, Trynka G, van Diemen CC, Monsuur AJ, Bevova M, Nijmeijer RM, van ‘t Slot R, Heijmans R, Boezen HM, van Heel DA, van Bodegraven AA, Stokkers PC, Wijmenga C, Crusius JB, Weersma RK. Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet. 2008;82:1202–1210. doi: 10.1016/j.ajhg.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Sakashita M, Yamada T, Fujieda S, Tanaka S, Doi S, Miyatake A, Enomoto T, Nishiyama C, Nakano N, Maeda K, Okumura K, Ogawa H, Ikeda S, Noguchi E, Sakamoto T, Hizawa N, Ebe K, Saeki H, Sasaki T, Ebihara T, Amagai M, Takeuchi S, Furue M, Nakamura Y, Tamari M. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat Genet. 2012;44:1222–1226. doi: 10.1038/ng.2438. [DOI] [PubMed] [Google Scholar]
- 17.Liu H, Irwanto A, Tian H, Fu X, Yu Y, Yu G, Low H, Chu T, Li Y, Shi B, Chen M, Sun Y, Yuan C, Lu N, You J, Bao F, Li J, Liu J, Liu H, Liu D, Yu X, Zhang L, Yang Q, Wang N, Niu G, Ma S, Zhou Y, Wang C, Chen S, Zhang X, Liu J, Zhang F. Identification of IL18RAP/IL18R1 and IL12B as leprosy risk genes demonstrates shared pathogenesis between inflammation and infectious diseases. Am J Hum Genet. 2012;91:935–941. doi: 10.1016/j.ajhg.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, Romanos J, Dinesen LC, Ryan AW, Panesar D, Gwilliam R, Takeuchi F, McLaren WM, Holmes GK, Howdle PD, Walters JR, Sanders DS, Playford RJ, Trynka G, Mulder CJ, Mearin ML, Verbeek WH, Trimble V, Stevens FM, O’Morain C, Kennedy NP, Kelleher D, Pennington DJ, Strachan DP, McArdle WL, Mein CA, Wapenaar MC, Deloukas P, McGinnis R, McManus R, Wijmenga C, van Heel DA. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, Howson JM, Stevens H, McManus R, Wijmenga C, Heap GA, Dubois PC, Clayton DG, Hunt KA, van Heel DA, Todd JA. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boraschi D, Dinarello CA. IL-18 in autoimmunity: review. Eur Cytokine Netw. 2006;17:224–252. [PubMed] [Google Scholar]
- 21.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O’Connor W, Jr, Murphy AJ, Valenzuela DM, Yancopoulos GD, Booth CJ, Cho JH, Ouyang W, Abraham C, Flavell RA. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawakami K, Qureshi MH, Zhang T, Okamura H, Kurimoto M, Saito A. IL-18 protects mice against pulmonary and disseminated infection with Cryptococcus neoformans by inducing IFN-gamma production. J Immunol. 1997;159:5528–5534. [PubMed] [Google Scholar]
- 26.Chikano S, Sawada K, Shimoyama T, Kashiwamura SI, Sugihara A, Sekikawa K, Terada N, Nakanishi K, Okamura H. IL-18 and IL-12 induce intestinal inflammation and fatty liver in mice in an IFN-gamma dependent manner. Gut. 2000;47:779–786. doi: 10.1136/gut.47.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esfandiari E, I, McInnes B, Lindop G, Huang FP, Field M, Komai-Koma M, Wei X, Liew FY. A proinflammatory role of IL-18 in the development of spontaneous autoimmune disease. J Immunol. 2001;167:5338–5347. doi: 10.4049/jimmunol.167.9.5338. [DOI] [PubMed] [Google Scholar]
- 28.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 29.Zheng C, Xiang J, Hunter T, Lin A. The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. J Biol Chem. 1999;274:28966–28971. doi: 10.1074/jbc.274.41.28966. [DOI] [PubMed] [Google Scholar]
- 30.Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hedl M, Abraham C. Nod2-induced autocrine interleukin-1 alters signaling by ERK and p38 to differentially regulate secretion of inflammatory cytokines. Gastroenterology. 2012;143:1530–1543. doi: 10.1053/j.gastro.2012.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vavricka SR, Musch MW, Chang JE, Nakagawa Y, Phanvijhitsiri K, Waypa TS, Merlin D, Schneewind O, Chang EB. hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology. 2004;127:1401–1409. doi: 10.1053/j.gastro.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 33.Fiszer D, Rozwadowska N, Rychlewski L, Kosicki W, Kurpisz M. Identification of IL-18RAP mRNA truncated splice variants in human testis and the other human tissues. Cytokine. 2007;39:178–183. doi: 10.1016/j.cyto.2007.07.186. [DOI] [PubMed] [Google Scholar]
- 34.Netea MG, Azam T, Ferwerda G, Girardin SE, Walsh M, Park JS, Abraham E, Kim JM, Yoon DY, Dinarello CA, Kim SH. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci U S A. 2005;102:16309–16314. doi: 10.1073/pnas.0508237102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hedl M, Abraham C. NLRP1 and NLRP3 inflammasomes are essential for distinct outcomes of decreased cytokines but enhanced bacterial killing upon chronic Nod2 stimulation. Am J Physiol Gastrointest Liver Physiol. 2013;304:G583–596. doi: 10.1152/ajpgi.00297.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fortin CF, Ear T, McDonald PP. Autocrine role of endogenous interleukin-18 on inflammatory cytokine generation by human neutrophils. Faseb J. 2009;23:194–203. doi: 10.1096/fj.08-110213. [DOI] [PubMed] [Google Scholar]
- 37.Yoo JK, Kwon H, Khil LY, Zhang L, Jun HS, Yoon JW. IL-18 induces monocyte chemotactic protein-1 production in macrophages through the phosphatidylinositol 3-kinase/Akt and MEK/ERK1/2 pathways. J Immunol. 2005;175:8280–8286. doi: 10.4049/jimmunol.175.12.8280. [DOI] [PubMed] [Google Scholar]
- 38.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan Z, Ford MJ, Cusanovich DA, Mitrano A, Pritchard JK, Gilad Y. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science. 2013;342:1100–1104. doi: 10.1126/science.1242379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H, Snyder M. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ho JE, Chen WY, Chen MH, Larson MG, McCabe EL, Cheng S, Ghorbani A, Coglianese E, Emilsson V, Johnson AD, Walter S, Franceschini N, O’Donnell CJ, Dehghan A, Lu C, Levy D, Newton-Cheh C, Lin H, Felix JF, Schreiter ER, Vasan RS, Januzzi JL, Lee RT, Wang TJ. Common genetic variation at the IL1RL1 locus regulates IL-33/ST2 signaling. J Clin Invest. 2013;123:4208–4218. doi: 10.1172/JCI67119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuhn PH, Marjaux E, Imhof A, De Strooper B, Haass C, Lichtenthaler SF. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J Biol Chem. 2007;282:11982–11995. doi: 10.1074/jbc.M700356200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.