

Abstract
A great challenge in orthopedic tumor operation faced by orthopedic implants is the high recurrence and metastasis of bone tumor as well as the bacterial infection associated with the implants. Thus ideal titanium (Ti)-based bone implants should be able to not only inhibit cancer cell adhesion and proliferation, promote cancer cell apoptosis, but also resist bacterial infections. Towards this end, we developed a new approach to modify the surface of Ti-based bone implants so that they can restrain functions of osteoclastoma (Giant cell tumor of bone) cancer cells (GCTs) and inhibit the adhesion of bacteria. First, the surface of pristine Ti substrates was functionalized with dopamine (DA) to form DA-Ti substrates. Then nanoparticles electrostatically assembled from poly-lysine (PLL) and heparin (Hep) were chemically immobilized onto the DA-Ti substrates to form PLL/Hep-Ti substrates. Chitosan (CH) and methotrexate (MTX) were then electrostatically immobilized onto the PLL/Hep-Ti substrates to generate CH-MTX-Ti substrates. The successful functionalization of the Ti substrates was confirmed by X-ray photoelectron spectroscopy. GCTs cultured on differently functionalized Ti substrates were investigated in terms of cell adhesion, cytoskeleton, proliferation, cytotoxicity and apoptosis. The growth of Staphylococcus aureus bacteria in the presence of different substrates was also assayed. Our results showed that CH-MTX-Ti substrates not only significantly inhibited the adhesion, proliferation and viability of GCTs, promoted the apoptosis of GCTs, but also prevented the adhesion of the bacteria and the subsequent formation of bacterial biofilms, when compared to other Ti substrates. Thus CH-MTX-Ti substrates are expected to be used as orthopedic prostheses in bone tumor surgery that can inhibit both osteoclastoma formation and bacterial infections.
Introduction
Limb salvage surgery for malignant or aggressive benign bone tumor has been widely accepted 1 and shown to improve the quality of life compared to amputation. Titanium (Ti) and its alloys2–4 are the most commonly used implants in bone tumor surgery as replacements for surgical reconstruction due to their excellent biocompatibility and mechanical properties in recent years5. However, unmodified Ti-based implants are susceptible to bacterial infection and tumor recurrence in orthopedic tumor operation. Such bacterial infection may lead to inflammation, delay wound healing and cause complex complications6. Meanwhile, the tumor recurrence and metastasis in bone are still challenges and pose problems for both tumor treatments and bone repair. Therefore, there is an urgent need in bone implants that have both anti-cancer and anti-bacterial properties. Towards this end, one promising approach is to modify the surface chemistry of the implants such that they inhibit the adhesion of bacterial cells and induce the death of bone cancer cells when cells are in close proximity with the implant surface7.
In recent years, the development of antibacterial Ti substrates has attracted considerable attention. To overcome bacterial infections, previous studies have focused on surface modification, such as coating with silver nanoparticles8, conjugation with antibiotics9, and physical adsorption of cationic antibacterial agents10. Chitosan (poly- β −1, 4-glucosamine, CH) is a natural nontoxic polymer, which possesses a higher antibacterial activity and broader spectrum of activity11, 12. In this work, CH was chosen as an important layer to functionalize Ti substrates for its unique characteristic, such as non-toxicity, biocompatibility, antibacterial activity and highly positively-charged properties. Nevertheless, even though the modification of bone implants by CH may reduce or minimize the risk of bacterial infection, the recurrence of tumor on the implants was still unsolved.
Methotrexate (MTX) is one of the most widely used cancer chemotherapeutics13, 14. It has been reported that MTX can be used as chemotherapeutics in treating bone tumors, especially the giant cell tumor of bone14 and osteosarcoma15. However, there has been no work reported on the immobilization of chemotherapeutics, especially MTX, on the Ti surfaces for the purpose of inhibiting bone tumor formation.
Hence, in this work we propose to introduce both CH and MTX to the surface of Ti implants in order to develop anti-cancer/antibacterial dual functional implants (Figure 1). To achieve this goal, we propose an electrostatic layer-by-layer assembly method to functionalize the Ti substrates (Fig. 1). Firstly dopamine (DA) is deposited onto the Ti substrates to form DA-Ti because DA is known capable of coating Ti substrates 9. In order to deposit the positively charged CH and MTX to DA-Ti, we used an intermediate layer of anionic nanoparticles made of polylysine (PLL) and heparin (Hep), which is sandwiched between the CH/MTX layers and DA-Ti substrates16, 17. It is known that electrostatic interaction between cationic PLL and anionic Hep can drive them to be assembled into nanoparticles (PLL/Hep)18. Thus PLL/Hep nanoparticles, made through the electrostatic assembly of PLL and Hep, were immobilized on the DA-Ti substrates to form PLL/Hep-Ti substrates, through the Schiff base conjugation reaction 19 between PLL on the PLL/Hep nanoparticles and DA on DA-Ti. Subsequently, a cationic layer made of CH and MTX was deposited onto PLL/Hep-Ti to form CH-MTX-Ti substrates through the electrostatic interaction between anionic Hep on PLL/Hep-Ti and cationic CH and MTX. By evaluating the adhesion, proliferation, apoptosis and necrosis of osteoclastoma (Giant cell tumor of bone) cancer cells (GCTs) and adhesion and growth of Staphylococcus aureus (S. aureus) bacteria in the presence of CH-MTX-Ti, we found that CH-MTX-Ti could not only inhibit the adhesion and proliferation of GCTs and promote their apoptosis due to the presence and release of MTX from the substrates, but also prevent the adhesion and growth of bacteria due to the presence of CH on the substrates. Overall, this work suggests that CH-MTX-Ti is a functionalized bone implant that can inhibit both osteoclastoma formation and bacterial infection.
Figure 1. Schematic illustration of the general idea of this study.
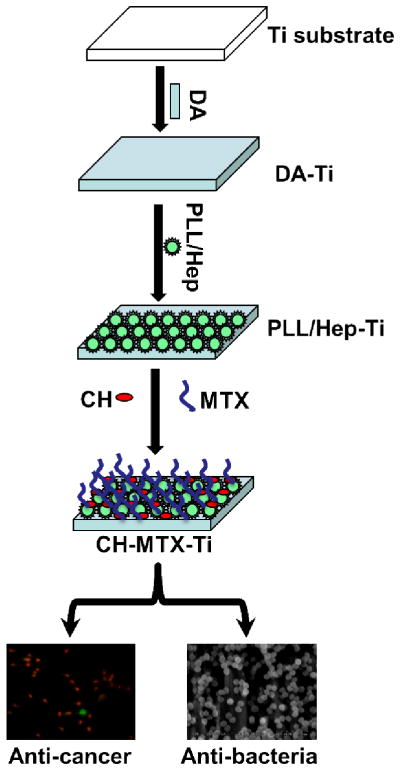
Dopamine (DA) is first deposited onto pristine Ti to form DA-Ti substrate. Then a suspension of anionic nanoparticles, formed through the electrostatic interaction between poly-lysine (PLL) and heparin (Hep), is deposited onto DA-Ti to form PLL/Hep-Ti, through the Schiff base conjugation reaction between PLL and DA as well as the electrostatic interaction between Hep and DA. Finally, cationic chitosan (CH) and methotrexate (MTX) are simultaneously immobilized onto the anionic PLL/Hep-Ti to form CH-MTX-Ti substrates through electrostatic interaction. The CH- MTX-Ti substrates are interacted with GTCs and bacteria to confirm their anti-cancer and anti-bacteria capability, respectively. Abbreviation in the schematic illustration: Ti=Titanium, DA=Dopamine, PLL/Hep=Poly-lysine/Heparin, CH=Chitosan, MTX=Methotrexate
Materials and Methods
Materials and Reagents
Ti foils (purity≥99.6%) of 0.2 mm thickness were purchased from Baoji Qichen New Metal Co., LTD. 3,4-dihydroxyphenylalanine (dopamine, DA), heparin sodium (Hep), poly-D-lysine hydrobromide (PLL, MW: 1000–5000), chitosan (CH, ≥75%, deacetylated, MW:1.6×105) were purchased from Sigma-Aldrich. MTX was purchased from Shanghai Fusheng Industrial Co., LTD. Peptone, yeast extract, agar, and beef extract were purchased from Guangdong Huan Kai Microbial technology Co., LTD. Staphylococcus aureus 25923 bacteria was purchased from ATCC. Ultra-pure water (18.4 MΩ) was generated by Millipore Milli-Q system. Acetic acid (HOAc), tris (hydroxymethyl) aminomethane (Tris), sodium chloride (NaCl), ethanol, and acetone were all AR grade and purchased from Guangzhou chemical reagent Co., LTD.
Preparation of Dopamine-modified Titanium surfaces (DA-Ti)
The Ti foils were cut into 1.0 cm×1.0 cm pieces and polished using #600 grid sandpaper followed by ultrasonication in ultra-pure water for 30 min. The substrates were then washed with acetone, ethanol, and ultra-pure water, respectively, for 30 min in ultrasonic baths to remove residual surface impurities and blown dry with purified nitrogen gas. To produce DA-Ti, the substrates were incubated in a DA solution (2mg/ml DA in pH 5.5 Tirs-HCl) at 20°C for 12 h, then rinsed with distilled water for three times and dried with purified nitrogen gas. This step was repeated for three times.
Preparation of PLL/Hep-Ti, CH-Ti, and CH-MTX-Ti Substrates
25 mg Hep and 5 mg PLL were separately dissolved in a 10 ml phosphate buffered solution (PBS) to form two solutions. To prepare a suspension of PLL/Hep nanoparticles through the electrostatic interaction betweenthe cationic PLLand anionic Hep, the Hep solution was added into the PLL solution dropwise, and then mixed under ultrasonication for 30s. To immobilize PLL/Hep nanoparticles onto the surface of DA-Ti substrates by means of Schiff base reaction, the DA-Ti substrates were immersed into the prepared PLL/Hep nanoparticle suspension solution at 20°C for 12 h with gentle shaking, the substrate was then rinsed with PBS. This process was repeated for three times. The resultant substrates were then dried with nitrogen to get PLL/Hep-Ti. The Schiff base reaction19 between PLL and DA in DA-Ti will enable the conjugation of PLL/Hep nanoparticles onto DA-Ti to form the PLL/Hep-Ti.
To prepare CH and MTX modified Ti substrates, the PLL/Hep-Ti substrates were immersed into a solution made of a mixture of cationic CH (1.5mg/ml) and MTX (200, 300, or 400 μg/ml) in 0.1 M HOAc containing 0.14 M NaCl, pH=3.8) for 12 h at 20 °C. The solution containing the substrates was subjected to gentle shaking overnight to generate CH-MTX-Ti substrates due to the strong electrostatic interaction between the anionic Hep in PLL/Hep nanoparticles and the cationic CH and MTX, the functionalized Ti substrates were then rinsed with ultra-pure water and blown dry with nitrogen gas. The same procedure was carried out without MTX to obtain a control substrate denoted as CH-Ti. The drug density of CH and MTX on the substrates was calculated by the change of concentrations of CH and MTX in CH/MTX solutions.
Surface Characterization
The size of PLL/Hep nanoparticles was determined by dynamic light scattering (DLS). The zeta potential and polydispersity index of the nanoparticles were measured using an Acoustic and Electroacoustic spectrometer (Dispersion Technologies, Inc) equipped with titration unit, pH, conductivity and electroacoustic probes. The PLL/Hep nanoparticles on the Ti surface were characterized using atomic force microscope (AFM, Shimadzu SPM-9600, Japan). The chemical composition of the surfaces was analyzed by X-ray photoelectron spectroscopy (XPS) following a reported procedure 20 on an AXIS His spectrometer (Kratos Axis Ultra DLD, Britain) with an AlKα X-ray source (1486.6 eV photons). The C1s hydrocarbon peak at 284.84 eV was used as the reference for all binding energies. The area of each peak was normalizedto the total peak area of all atomic elements to calculate surface atomic percentages.
In vitro Release of CH and MTX
To evaluate the amount of CH and MTX released from CH-Ti and CH-MTX-Ti, the substrates were immersed in 5 ml of PBS buffer (pH 7.4) and 5 ml of Tris-HCl buffer (pH 5.5) containing 0.01% (w/v) sodium azide and gently shaken at 100 rpm at 37 °C. At predetermined time intervals of 0 h, 8 h, 16 h, 1 d, 2 d, 5 d, 8 d, 10 d, 15 d, 20 d, 25 d, and 30 d, the amount of released MTX was determined by measuring optical density (OD) at 306 nm ultraviolet (UV) -Visible spectrophotometer (UV 1601, Shimadzu, Japan). The amount of released CH was determined by reacting with ninhydrin solution and measuring the OD at 570 nm.
Cell Culture
GCTs (Primary cultures obtained after the ninth passage) were cultured by following a published protocol21. Briefly, they were cultured in Dulbecco’s minimum essential medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum, containing 100 U/ml penicillin and 100 mg/ml streptomycin. The cells were incubated at 37 °C in 5% CO2 and 95% air and passaged every 2 d. Cultured cells were detached by trypsinization (0.25% trypsin-EDTA), suspended in fresh H-DMEM culture media and used for the experiments described below.
Cell Imaging
Cell morphology on differently functionalized substrates was examined by scanning electron microscopy (SEM, ZEISS Ultra 55, Germany). After 24 h of culture, the substrates were taken out of the culture plates and fixed with 3% glutaradehyde for 6 h. After rinsed for three times with PBS for 10 min, each substrate was dehydrated sequentially in a series of ethanol (50%, 70%, 95% and 100%) at each concentration twice for 10 min each time. Then the substrates were allowed to air dry in a fume hood. The adhesion and spreading of the cells on different substrates were observed by SEM.
Cell Proliferation
The proliferation of GCTs on different substrates was characterized by using 3-(4, 5-Dimethylthiazol-2-yl)- diphenyltetrazolium bromide (MTT) assay after the cells were cultured for 1, 3, 5 and 7 days. Cells were seeded onto the substrates at a density of 1×104cells/cm2. For MTT assay, at each of the designated time points (Day 1, 3, 5, and 7), 30 μL of MTT (Sigma, USA) solution (5 mg/ml in PBS) was added to 300 ml of culture medium and incubated for 4 h at 37°C. After the culture medium was removed, the formazan reaction products were dissolved in 300 μL dimethylsulfoxide (DMSO) for 20 min. The optical density of the formazan solution was read on an ELISA plate reader (Thermo, Multiskn Go) at 490 nm.
Cell Viability
For viability staining studies, cells were seeded in 24-well plates at a concentration of 1 × 104 cells/cm2 at 37°C with 5% CO2. The substrates were added in 24-well plates before cells were seeded and incubated for 24 h. At the end of incubation, the media was removed and the adherent cells were subjected to Live/Dead staining following the manufacturer’s protocol (Sigma, USA). Briefly, 1 μM calcien AM and 2 μM ethidium homodimer-1 solutions were prepared in PBS. After the removal of the culture medium, the cells were rinsed once in PBS, followed by addition of 100 μL of 1 μM calcein AM and 2 μM ethidium homodimer-1 solution. Cells were then photographed using a fluorescence microscope.
Immunofluorescence Staining
The GCTs cultured on the functionalized Ti substrates for 24 h were fixed in 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton after washed with PBS twice. Non-specific binding was blocked with 1% BSA in PBS for 30 min. Focal adhesion was imaged using a commercial kit (FAK 100 and AP124F, Millipore) according to the manufacture’s protocol. The samples were then washed, mounted and examined by fluorescent electronic microscope (Olympus, BX50) with a 20× objective.
Cell Apoptosis and Necrosis
Apoptosis is a form of programmed cell death occurring through the activation of a cell-intrinsic suicide machinery22. To analyze changes in nuclear morphology, GCTs were measured using Guava Nexin Reagent (Millipore, USA). Cells on the substrates were washed with PBS and centrifuged at 200g for 5 min. The cell pellets were resuspended in 100 μL DMEM medium supplemented with 1% FBS, then incubated with 100 μL of Annexin V-PE and 7-AAD labeling solution for 20 min at room temperature. Cells were finally analyzed on Guava EasyCyte 5HT flow cytometer (Millipore, USA) by using 488 nm excitation and a 575 nm bandpass filter for Annexin V-PE detection and using 546 nm excitation and a 647 nm filter for 7-AAD detection. The data were analyzed by Guava Nexin Software v2.2.2.
Determination of Antibacterial Efficacy
The antibacterial assays were carried out with S. aureus, the most common microbial pathogen encountered in orthopedic infections. S. aureus were cultivated in yeast-dextrose broth at 37 °C. The bacteria-containing broth was centrifuged at 2700 rpm for 10 min. After the removal of the supernatant, the bacteria were washed twice with PBS and resuspended in PBS at a concentration of 107cfu/ml. All glassware and Ti substrates were sterilized with UV irradiation for 1 h before the experiments. One milliliter of the bacteria suspension was then added to each Ti substrate in a 24-well plate and incubated at 37 °C for 8 h without stirring. After the designated incubation time points, the substrates were washed twice with PBS before fixed with 3% glutaraldehyde for 1 h at room temperature. After stepwise dehydration with serial ethanol for 10 min each and coated with platinum, the surfaces of the substrates were imaged using SEM. Quantification of bacteria adhesion after 24 h of incubation was also carried out using the spread plate method as reported23. All experiments were performed in triplicate with three substrates and the mean values were calculated.
Statistical Analysis
All statistical analysis was performed by analysis of variance (ANOVA) using SPSS 13.0. Results were considered statistically significant if the p-value was <0.05.
Results
Surface Characterization of Functionalized Ti Implants
The zeta potential of PLL/Hep nanoparticles in PBS was measured to be −22.8 mV at pH 7.4, suggesting that the surface of PLL/Hep nanoparticle layer was negativelycharged and thus could be sandwiched betweenthe cationic DA-Ti and CH/MTX layer Fig. 1). The average diameter of PLL/Hep nanoparticles was determined to be 217.2 nm by DLS (Fig. 2). The polydispersity index was found to be 0.053. The results showed that the PLL/Hep nanoparticles were fairly monodisperse. The size of nanoparticles determined from AFM (Fig. 2) was consistent with DLS data.
Figure 2.

Characterization of PLL/Hep nanoparticles by DLS (A) and AFM (B).
XPS is a surface-sensitive analysis technique, which can provide both qualitative and quantitative information about the presence of different elements on the surfaces. We employed XPS to investigate the surface chemistryof different Ti substrates. Table 1 listed the surface compositions of different substrates derived from the XPS spectra (Fig. S1). Successful DA immobilization on the pristine Ti substrates was confirmed by an increase of the N content and C content compared to pristine Ti. The appearance of S element, unique for Hep, in XPS spectrum confirmed the successful immobilization of PLL/Hep nanoparticles on DA-Ti to form PLL/Hep-Ti. The deposition of CH and/or MTX on the surface of PLL/Hep-Ti to form CH-MTX-Ti was further confirmed by a decrease in S compared to PLL/Hep-Ti.
Table 1.
Quantitative surface composition of titanium substrates terminated with different functional molecules determined by XPS spectra.
| Groups | Chemical compositions (at.%)
|
||||
|---|---|---|---|---|---|
| O | Ti | N | C | S | |
| Ti | 41.98 | 13.36 | 4.91 | 39.76 | 0 |
| DA-Ti | 21.09 | 0.29 | 10.82 | 67.8 | 0 |
| PLL/Hep-Ti | 24.61 | 0 | 9.78 | 64.5 | 1.12 |
| CH-Ti | 23.29 | 0 | 8.61 | 68.00 | 0.09 |
| CH-MTX-Ti | 26.38 | 0 | 9.52 | 64.1 | 0 |
In vitro Loading and Release of CH and MTX
We generated CH/MTX solutions with a CH concentration of 1.5 mg/ml and three different MTX concentrations (200, 300 and 400 μg/ml). Then the PLL/Hep-Ti substrates were immersed into these CH/MTX solutions to get CH-MTX-Ti substrates loaded with different amount of MTX. The resultant substrates were termed CH/200MTX-Ti, CH/300MTX-Ti and CH/400MTX-Ti. The surface loading density of both CH and MTX at different solution concentrations was shown in Fig. S2, which showed that immersion of PLL/Hep-Ti substrates in a solution of 300 μg/ml MTX and 1.5 mg/ml CH resulted in a proper drug density on Ti substrates. The release of MTX from three substrates with different drug loading density (CH/200MTX-Ti, CH/300MTX-Ti and CH/400MTX-Ti) into PBS (pH 7.4) (Fig. S3) showed that there was no significant difference in drug release profile between CH/300MTX-Ti and CH/400MTX-Ti. Therefore, we used CH/300MTX-Ti as CH-MTX-Ti for the subsequent studies.
A comparison between CH and MTX release from both CH-Ti and CH-MTX-Ti substrates in PBS (Fig. 3) showed that the release of MTX from the CH-MTX-Ti substrates exhibited a sustained release profile for up to 30 days. For example, 6.06 μg of MTX was released from 1 cm2 of CH-MTX-Ti substrates on Day 1, and the released amount of MTX reached 12.98 μg per cm2 of the substrates in 8 days. On Day30, the released amount reached about 16 μg/cm2. The CH release from both CH-Ti and CH-MTX-Ti showed a similar profile (Fig. 3). Although the CH release was not burst, it was faster than the MTX release from CH-MTX-Ti.
Figure 3. In vitro release of CH from CH-Ti and CH-MTX-Ti substrates (A) and MTX from CH-MTX-Ti substrates (B) at 37 °C.

The data represent means±S.D. (n=3).
In order to understand the effect of pH value on the CH and MTX release, we studied the release from CH-MTX-Ti and CH-Ti in 0.1 M Tris-HCl (pH 5.5) and PBS (pH 7.4). The results (Fig. S4) showed that the pH value indeed influenced the CH release from both CH-MTX-Ti and CH-Ti, probably due to the difference in solubility of CH at different pH values. CH release in PBS (pH7.4) reached equilibrium faster than in 0.1 M Tris-HCl (pH 5.5) solution. However, in both PBS (pH7.4) and 0.1 M Tris-HCl (pH 5.5) solution, MTX release showed a similar sustained release profile.
Cell adhesion
We used SEM to investigate the adhesion of GCTs onto different Ti substrates. It can be seen from Fig. 4, GCTs spread out and exhibited a spindle cell body on pristine Ti substrates as well as those cultured on DA-Ti, PLL/Hep-Ti and CH-Ti substrates. However, the cells cultured on CH-MTX-Ti substrates tended to atrophy rather than spread out, which was totally different from other substrates, because of the presence of MTX on the substrates. The results indicated that cell-surface interactions affected cell morphology and adhesion and CH-MTX-Ti inhibited the adhesion of GCTs onto the substrates.
Figure 4.
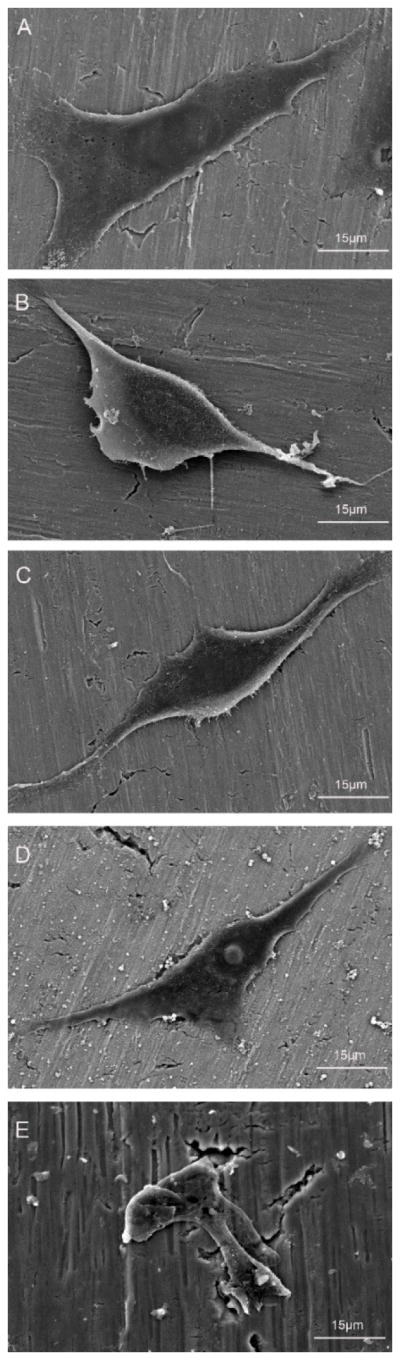
SEM images of different Ti substrates after exposure to GCT cell suspension (104 cells/cm2) for 24 h: (A) pristine Ti; (B) DA-Ti; (C) PLL/Hep-Ti; (D) CH-Ti; (E) CH-MTX-Ti.
Cell Proliferation
The effect of different Ti substrates on the proliferation of GCTs was examined by MTT assay (Figs. S5 and 5). Cell proliferation on CH-MTX-Ti substrates was significantly lower (p<0.01) than the other four substrates at each time point (Fig. 5) and CH-MTX-Ti derived from different concentrations of MTX (300 and 400 μg/ml) did not show significant difference (Fig. S5). The cells on all substrates showed a significant increase (p<0.01) on Day 3 compared to Day 1. The number of cells cultured on Ti, DA-Ti, PLL/Hep-Ti or CH-Ti substrates increased significantly at each incubation interval. However, cells cultured on CH-MTX-Ti substrates showed inconspicuous proliferation from Day 3 to Day 7 when compared to those on Ti, DA-Ti, PLL/Hep-Ti and CH-Ti substrates. These results indicated that the CH-MTX- Ti can inhibit the proliferation of GCT cells through cell-surface interactions.
Figure 5. MTT reduction activity of GCTs cultured on differently functionalized Ti substrates for 1, 3 and 8 days.

Symbol denotes significant differences as determined by one way ANOVA. One symbol, p<0.05; 2 symbols, p<0.01. #, compared to substrates modified with CH-MTX-Ti group on the same day. *, compared to the same substrates on Day 3.
Live/Dead Cell Staining Assay
The effects of different substrates on cell survival were also evaluated using Live/Dead Viability/Cytotoxicity staining (Fig. 6). Most of the GCTs were alive (green) on Ti, DA-Ti, PLL/Hep-Ti, and CH-Ti substrates after cultured for 24 h. However, most of the GCTs cultured on MTX-CH-Ti were vague and dead (red). These results supported the idea that the MTX-CH-Ti can inhibit GCTs growth and kill them.
Figure 6. Fluorescence micrographs of live/dead dye-stained GCTs after cultured on differently functionalized Ti substrates for 24 h.
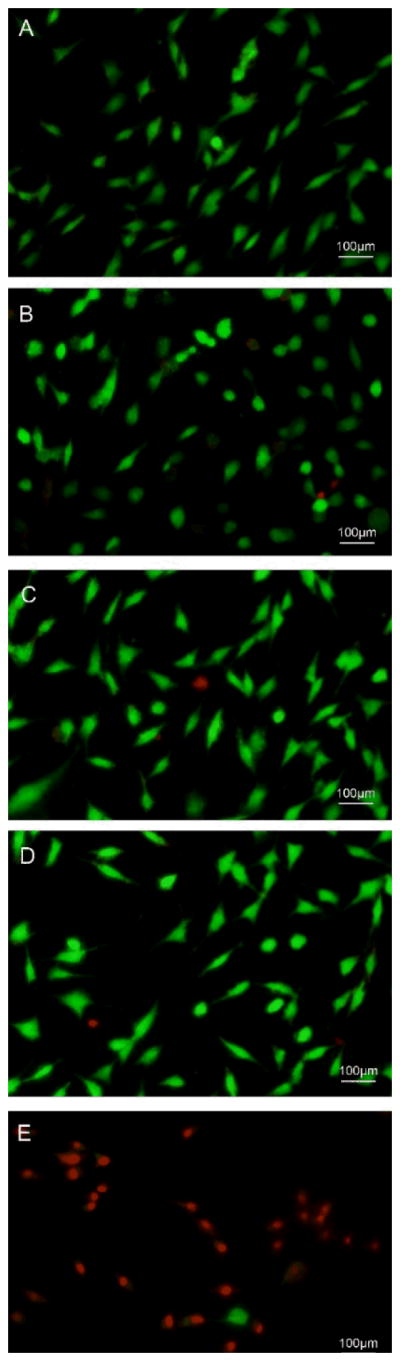
(A) Pristine Ti; (B) DA-Ti; (C) PLL/Hep-Ti; (D) CH-Ti; (E) CH-MTX-Ti. Live cells were dyed by calcein AM and thus are green whereas red cells were dyed by ethidium homodimer and thus are red.
Cell Cytoskeleton Assay
The actin cytoskeleton is a dynamic structure that rapidly changes its shape and organization in response to stimuli and cell cycle progression. The disruption of its normal regulation maylead to cell transformations. Thetransformed cells will haveless F-actin than untransformed cells and exhibit atypical coordination of F-actin levels through the cell cycle. We used focal adhesion analysis to confirm the cell spreading on different substrates (Fig. 7). Actin (stained to be red) was partially colocalized with focal adhesion protein vinculin (stained to be green) in the adhesion plaques. The nuclei showed blue color due to the staining by DAPI. The focal adhesion formation and the cell morphology were totally different for the GCTs cultured on CH-MTX-Ti substrates from other substrates (Fig. 7). Cells cultured on MTX-CH-Ti substrate became less well-defined and smaller (Fig. 7E) whereas those on other Ti substrates were larger and showed a well-defined cytoskeleton (Fig. 7A–D). Furthermore, much less focal adhesion protein vinculin (green) and actin (red) were expressed on MTX-CH-Ti substrates than other Ti substrates. The results suggested that the immobilization of MTX onto Ti substrates would inhibit the adhesion and spreading GCTs.
Figure 7. The focal adhesion of GCTs after cultured on differently functionalized Ti substrates for 24 h.

(A) pristine Ti; (B) DA-Ti; (C) PLL/Hep-Ti; (D) CH-Ti; (E) CH-MTX-Ti. Focal adhesion protein vinculin and actin were stained to be red and green, respectively, showing cytoskeleton of GCTs on different Ti substrates. Cell nuclei were stained by DAPI and thus blue.
Cell Apoptosis and Necrosis
To analyze the effects of functional groups on cell apoptosis and necrosis, GCTs cultured on different Ti substrates were harvested for apoptosis analysis using Annexin V-PE and 7-AAD. As shown in Fig. 8, the CH-MTX-Ti directed about 10% and 4% cells into apoptosis and necrosis, respectively, whereas other substrates did not show statistical difference in terms of the number of cells undergoing apoptosis and necrosis. These results indicated that CH-MTX-Ti promoted the cell apoptosis.
Figure 8. Apoptosis and necrosis of GCT cells after cultured on differently functionalized Ti substrates for 24 h.

(A) Pristine Ti; (B) DA-Ti; (C) PLL/Hep-Ti; (D) CH-Ti; (E) CH-MTX-Ti; (F) The apoptosis and necrosis rate of GCT cells cultured on different Ti substrates. Results are represented by means±SD of 3 independent experiments.
Antibacterial Assay
Figure 9 showed the representative SEM images of different Ti substrates after incubated with S. aureus suspension of 1×107 cfu/ml in PBS for 8 h. Fewer bacteria were found on PLL/Hep-Ti substrates than on the pristine Ti (Fig. 9A) and DA-Ti (Fig. 9B), probably because poly-lysine24, 25 and heparin26, 27 could inhibit the adhesion of bacterial cells. Moreover, the number of the bacteria on CH-Ti and CH-MTX-Ti was dramatically decreased compared to those on pristine Ti, DA-Ti and PLL/Hep-Ti (Fig. 9D and 9E).
Figure 9. SEM images of differently functionalized Ti substrates after exposure to S. aureus suspension (107 cfu/ml) in PBS for 8 h.

(A) Pristine Ti; (B) DA-Ti; (C) PLL/Hep-Ti; (D) CH-Ti; (E) CH-MTX-Ti. (F) The number of S. aureus (cfu) in culture normalized to the area of the substrates after the bacterial cell culture was interacted with various Ti substrates for 24 h. ** denotes significant differences compared to substrates modified with CH-MTX-Ti group. The error bars represent the standard deviations calculated from three independent experiments.
Furthermore, there was no obvious difference between the number of the bacteria on CH-Ti and CH-MTX-Ti. Thus the antibacterial effect of Ti substrates mainly depended on the presence of CH. The antibacterial effect of the Ti substrates was also evaluated using the reported spread plate method23. Briefly, we co-cultured different substrates with S. aureus suspension of 1×106cfu/ml in PBS for 24 h. The suspension was diluted for 100 times and plated onto culture medium to determine the number of bacteria (Fig. 9F). The number of bacteria in the culture after interacted with CH-Ti and CH-MTX-Ti was significantly lower than that with other Ti substrates due to the release of CH only from these two substrates (Fig. 3). This result was in agreement with the findings from SEM imaging (Fig. 9A–E).
Discussion
Orthopedic implants played a very important role in operations for the recovery of patients. However, the failure of orthopedic implants in tumor operation often occurs due to the bacterial infections and tumor recurrence. Thus there is a pressing need in the development of novel and functional orthopedic implants capable of both inhibiting tumor cell growth and minimizing bacteria growth. Here we have showed that the introduction of dual drugs (CH and MTX) to the Ti implants is a practical method towards this goal. In order to securely immobilize CH and MTX onto Ti implants, we first chemically conjugated an anionic PLL/Hep nanoparticle layer to the surface of DA-Ti through Schiff base reaction and electrostatic interaction, and then deposited the cationic CH/MTX onto the nanoparticle layer to form CH-MTX-Ti. Namely, the anionic PLL/Hep nanoparticle layer served as a “linker” to ensure the tight connection between CH/MTX layer and the DA-Ti substrates. DA was as the first layer because it was proved that it could not only well coat the pristine Ti substrates but also electrostatically bind to heparin10, 28, 29. CH was introduced along with MTX to form the last layer on Ti because as a biocompatible polymer rich in amine groups, it can bind to various drugs30, 31 or growth factors32, 33, regulate their release, and also inhibit bacterial adhesion. The immobilization of CH/MTX on PLL/Hep-Ti substrates can be easily achieved via the electrostatic interactions between the amine groups of CH/MTX and the carboxylic/sulfate groups of heparin. The formation of CH-MTX-Ti substrates was successfully demonstrated by XPS (Table 1) and the controlled release of CH and MTX from the substrates (Fig. 3).
From drug delivery perspective, our CH-MTX-Ti is actually a drug carrier that shows a sustained MTX release profile (Fig. 3). In the development of drug delivery systems, sustained (instead of burst) drug release is highly desired and our functionalized Ti substrates satisfied this criterion. Inorganic or organic materials such as chitosan34, colloid gold 35 or polymers36 have been used as drug carriers. However, some of these systems have problems of controlling the drug release rates. In this study, the MTX was released from CH-MTX-Ti over an extended period of time in a sustained manner (Fig. 3). The sustained and extended release pattern of MTX was probably due to the strong electrostatic interactions between MTX and Heparin.
We conducted a series of studies to evaluate the response of GCTs to different Ti substrates. It is well established that cell adhesion and morphology can influence the subsequent activity of cells37. We investigated cell adhesion by SEM (Fig. 4) and cell cytoskeleton assay (Fig. 7). Cells cultured on CH-MTX-Ti substrates were smaller and more vague (Fig. 4) and displayed a significantly lower level of focal adhesion protein (vinculin) expression than those adhered to other Ti substrates (Fig. 7). The results clearly suggested that CH-MTX-Ti inhibited the adhesion of GCTs. Cell adhesion is essentially important to mediate subsequent cellular behaviors, including proliferation, cell cytotoxicity and cell apoptosis38. The proliferation of GCTs cultured on CH-MTX-Ti substrates was significantly lower (p<0.01) than that on the other Ti substrates (Fig. 5) from Day 1 to Day 7, which was consistent with the adhesion studies. The inhibition of cell growth by CH-MTX-Ti was also accompanied by an increase in cell toxicity (Fig. 6). Apoptosis is an important and regulatory pathway of cell growth and proliferation39. Our results indicated that the CH-MTX-Ti substrates encouraged cell apoptosis (Fig. 8). Taken together, our studies showed that CH-MTX-Ti could inhibit the adhesion and proliferation of GCTs and promote their apoptosis, suggesting that CH-MTX-Ti substrates have a great potential in inhibiting tumor formation.
The tumor operation always lasts for a longer period of time than common implant surgery in orthopedics, and thus is more susceptible to bacterial infection. Once bacterial cells adhere to the surface of the implants, bacteria can form a biofilm, which acts as a barrier to host defense mechanisms and resists to antimicrobial agents40, 41. CH and its derivatives have received significant scientific interests in surface modification for antibacterial, medical and pharmaceutical applications30, 42–44. Due to the presence of CH in CH-Ti and CH-MTX-Ti substrates, these substrates show significant resistance to the adhesion of S. aureus (Fig. 9), the bacteria commonly associated with infections of tumor operation, orthopedic implants and wounds. We also found that the CH released from CH-Ti and CH-MTX-Ti substrates (Fig. 3) could inhibit the growth of S. aureus bacteria (Fig. 9). The antibacterial activity of CH-Ti and CH-MTX-Ti substrates probably arose from the capability of CH in altering the bacteria cell membrane’s permeability through the interaction between its positively charged amino groups and the negatively charged cell membranes45.
Collectively, the in vitro antibacterial and anti-cancer activity of all of the functionalized Ti substrates shows that the CH-MTX-Ti substrates can not only promote GCTs apoptosis, inhibit their adhesion and proliferation, but also resist to bacterial adhesion and prevent the bacteria growth. Hence, they can serve as ideal implants to solve the two daunting challenges, tumor recurrence and bacterial infection, in orthopedic tumor surgery.
Conclusion
In summary, CH and MTX were chemically immobilized onto the surfaces of DA-functionalized titanium substrates by an intermediate “linker” layer of PLL/Hep nanoparticles. The resultant CH-MTX-Ti substrates could significantly inhibit the adhesion and proliferation of GCTs and promote their apoptosis due to the presence and sustained release of MTX when compared to other Ti substrates. They also significantly resisted to the adhesion of the bacteria commonly met in bone and tumor surgery, inhibited the growth of bacteria, and thus prevented the formation of bacterial biofilms due to the presence of chitosan. As a result, this study provides an approach to the development of biofunctionalized implants that can be used in bone tumor surgery.
Supplementary Material
Acknowledgments
This work was financed by National Natural Science Foundation of China (Grant No.81271957), National Basic Research Program of China (Grant No.2012CB619106) and Guangdong Key Lab of Orthopedic Technology and Implant Materials Construction Grant (No. 2011233-32). C.B.M. would also like to thank the financial Support from National Science Foundation (DMR-0847758, CMMI-1234957, CBET-0854414), National Institutes of Health (1R21EB015190), Department of Defense Peer Reviewed Medical Research Program (W81XWH-12-1-0384), Oklahoma Center for the Advancement of Science and Technology (HR11-006) and Oklahoma Center for Adult Stem Cell Research (434003).
Contributor Information
Yu Zhang, Email: luck_2001@126.com.
Chuanbin Mao, Email: cbmao@ou.edu.
Notes and references
- 1.Postma A, Kingma A, De Ruiter JH, Schraffordt Koops H, Veth RP, Goeken LN, Kamps WA. Journal of surgical oncology. 1992;51:47–51. doi: 10.1002/jso.2930510113. [DOI] [PubMed] [Google Scholar]
- 2.Mao CB, Li H, Cui F, Feng Q, Ma C. J Crystal Growth. 1999;206:308–321. [Google Scholar]
- 3.Mao CB, Li H, Cui F, FQ, Ma C. J Mater Chem. 1999;9:2573–2582. [Google Scholar]
- 4.Mao CB, Li H, Cui F, Feng Q, Wang H, Ma C. J Mater Chem. 1998;8:2795–2800. [Google Scholar]
- 5.Roberts P, Chan D, Grimer RJ, Sneath RS, Scales JT. The Journal of bone and joint surgery. 1991;73:762–769. doi: 10.1302/0301-620X.73B5.1894662. British volume. [DOI] [PubMed] [Google Scholar]
- 6.Thomas P, Thomas M, Summer B, Dietrich K, Zauzig M, Steinhauser E, Krenn V, Arnholdt H, Flaig MJ. The Journal of bone and joint surgery. 2011;93:e61. doi: 10.2106/JBJS.J.00317. American volume. [DOI] [PubMed] [Google Scholar]
- 7.Hetrick EM, Schoenfisch MH. Chemical Society reviews. 2006;35:780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 8.Zhao L, Wang H, Huo K, Cui L, Zhang W, Ni H, Zhang Y, Wu Z, Chu PK. Biomaterials. 2011;32:5706–5716. doi: 10.1016/j.biomaterials.2011.04.040. [DOI] [PubMed] [Google Scholar]
- 9.Lee DW, Yun YP, Park K, Kim SE. Bone. 2012;50:974–982. doi: 10.1016/j.bone.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Xu D, Yang W, Hu Y, Luo Z, Li J, Hou Y, Liu Y, Cai K. Colloids and surfaces B, Biointerfaces. 2013;110:225–235. doi: 10.1016/j.colsurfb.2013.04.050. [DOI] [PubMed] [Google Scholar]
- 11.Jing YJ, Hao YJ, Qu H, Shan Y, Li DS, Du RQ. Acta biologica Hungarica. 2007;58:75–86. doi: 10.1556/ABiol.57.2007.1.7. [DOI] [PubMed] [Google Scholar]
- 12.Chua PH, Neoh KG, Kang ET, Wang W. Biomaterials. 2008;29:1412–1421. doi: 10.1016/j.biomaterials.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 13.Kusunoki-Nakamoto F, Matsukawa T, Tanaka M, Miyagawa T, Yamamoto T, Shimizu J, Ikemura M, Shibahara J, Tsuji S. Internal medicine. 2013;52:623–628. doi: 10.2169/internalmedicine.52.8785. [DOI] [PubMed] [Google Scholar]
- 14.Kirchen ME, Menendez LR, Lee JH, Marshall GJ. Clinical orthopaedics and related research. 1996:294–303. [PubMed] [Google Scholar]
- 15.Bacci G, Picci P, Ruggieri P, Mercuri M, Avella M, Capanna R, Brach Del Prever A, Mancini A, Gherlinzoni F, Padovani G, et al. Cancer. 1990;65:2539–2553. doi: 10.1002/1097-0142(19900601)65:11<2539::aid-cncr2820651125>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Singh VK, Yang VC. Journal of biomedical materials research. 1998;42:182–187. doi: 10.1002/(sici)1097-4636(199811)42:2<182::aid-jbm3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 17.Liu T, Liu Y, Chen Y, Liu S, Maitz MF, Wang X, Zhang K, Wang J, Wang Y, Chen J, Huang N. Acta biomaterialia. 2013 doi: 10.1016/j.actbio.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Na K, Kim S, Park K, Kim K, Woo DG, Kwon IC, Chung HM, Park KH. Journal of the American Chemical Society. 2007;129:5788–5789. doi: 10.1021/ja067707r. [DOI] [PubMed] [Google Scholar]
- 19.Lee H, Dellatore SM, Miller WM, Messersmith PB. Science. 2007;318:426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai K, Frant M, Bossert J, Hildebrand G, Liefeith K, Jandt KD. Colloids and surfaces B, Biointerfaces. 2006;50:1–8. doi: 10.1016/j.colsurfb.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 21.Cheng YY, Huang L, Lee KM, Xu JK, Zheng MH, Kumta SM. Calcified tissue international. 2004;75:71–77. doi: 10.1007/s00223-004-0120-2. [DOI] [PubMed] [Google Scholar]
- 22.Galan A, Garcia-Bermejo L, Troyano A, Vilaboa NE, Fernandez C, de Blas E, Aller P. European journal of cell biology. 2001;80:312–320. doi: 10.1078/0171-9335-00159. [DOI] [PubMed] [Google Scholar]
- 23.Shi Z, Neoh KG, Kang ET, Wang W. Biomaterials. 2006;27:2440–2449. doi: 10.1016/j.biomaterials.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 24.Shima S, Matsuoka H, Iwamoto T, Sakai H. The Journal of antibiotics. 1984;37:1449–1455. doi: 10.7164/antibiotics.37.1449. [DOI] [PubMed] [Google Scholar]
- 25.Tome JP, Neves MG, Tome AC, Cavaleiro JA, Soncin M, Magaraggia M, Ferro S, Jori G. Journal of medicinal chemistry. 2004;47:6649–6652. doi: 10.1021/jm040802v. [DOI] [PubMed] [Google Scholar]
- 26.Chhatwal GS, Preissner KT, Muller-Berghaus G, Blobel H. Infection and immunity. 1987;55:1878–1883. doi: 10.1128/iai.55.8.1878-1883.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu L, Wang H, Guo YL, Zen K. Biochemical and biophysical research communications. 2008;369:1061–1064. doi: 10.1016/j.bbrc.2008.02.160. [DOI] [PubMed] [Google Scholar]
- 28.Chien CY, Tsai WB. ACS applied materials & interfaces. 2013;5:6975–6983. doi: 10.1021/am401071f. [DOI] [PubMed] [Google Scholar]
- 29.Ball V, Del Frari D, Toniazzo V, Ruch D. Journal of colloid and interface science. 2012;386:366–372. doi: 10.1016/j.jcis.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 30.Mitra S, Gaur U, Ghosh PC, Maitra AN. Journal of controlled release : official journal of the Controlled Release Society. 2001;74:317–323. doi: 10.1016/s0168-3659(01)00342-x. [DOI] [PubMed] [Google Scholar]
- 31.Yang X, Zhang Q, Wang Y, Chen H, Zhang H, Gao F, Liu L. Colloids and surfaces B, Biointerfaces. 2008;61:125–131. doi: 10.1016/j.colsurfb.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 32.Parajo Y, D’Angelo I, Welle A, Garcia-Fuentes M, Alonso MJ. Drug delivery. 2010;17:596–604. doi: 10.3109/10717544.2010.509357. [DOI] [PubMed] [Google Scholar]
- 33.Yilgor P, Tuzlakoglu K, Reis RL, Hasirci N, Hasirci V. Biomaterials. 2009;30:3551–3559. doi: 10.1016/j.biomaterials.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 34.Luo Y, Wang Q. International journal of biological macromolecules. 2014;64:353–367. doi: 10.1016/j.ijbiomac.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 35.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L. Drug delivery. 2004;11:169–183. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 36.Pitt CG, Jeffcoat AR, Zweidinger RA, Schindler A. Journal of biomedical materials research. 1979;13:497–507. doi: 10.1002/jbm.820130313. [DOI] [PubMed] [Google Scholar]
- 37.Ren YJ, Zhang H, Huang H, Wang XM, Zhou ZY, Cui FZ, An YH. Biomaterials. 2009;30:1036–1044. doi: 10.1016/j.biomaterials.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 38.Grigoriou V, Shapiro IM, Cavalcanti-Adam EA, Composto RJ, Ducheyne P, Adams CS. The Journal of biological chemistry. 2005;280:1733–1739. doi: 10.1074/jbc.M402550200. [DOI] [PubMed] [Google Scholar]
- 39.Fadeel B, Orrenius S. Journal of internal medicine. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 40.Costerton JW, Stewart PS, Greenberg EP. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 41.Hall-Stoodley L, Costerton JW, Stoodley P. Nature reviews Microbiology. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 42.Hussein-Al-Ali SH, El Zowalaty ME, Hussein MZ, Ismail M, Webster TJ. International journal of nanomedicine. 2014;9:549–557. doi: 10.2147/IJN.S53079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez A, Arana P, Fernandez A, Olmo R, Teijon C, Blanco MD. Journal of microencapsulation. 2013;30:398–408. doi: 10.3109/02652048.2012.746747. [DOI] [PubMed] [Google Scholar]
- 44.Sanzgiri YD, Blanton CD, Jr, Gallo JM. Pharmaceutical research. 1990;7:418–421. doi: 10.1023/a:1015887910184. [DOI] [PubMed] [Google Scholar]
- 45.Fu J, Ji J, Yuan W, Shen J. Biomaterials. 2005;26:6684–6692. doi: 10.1016/j.biomaterials.2005.04.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.