

Abstract
The androgen receptor–transcriptional intermediary factor 2 (AR-TIF2) positional protein–protein interaction (PPI) biosensor assay described herein combines physiologically relevant cell-based assays with the specificity of binding assays by incorporating structural information of AR and TIF2 functional domains along with intracellular targeting sequences and fluorescent reporters. Expression of the AR-red fluorescent protein (RFP) “prey” and TIF2-green fluorescent protein (GFP) “bait” components of the biosensor was directed by recombinant adenovirus constructs that expressed the ligand binding and activation function 2 surface domains of AR fused to RFP with nuclear localization and nuclear export sequences, and three α-helical LXXLL motifs from TIF2 fused to GFP and an HIV Rev nucleolar targeting sequence. In unstimulated cells, AR-RFP was localized predominantly to the cytoplasm and TIF2-GFP was localized to nucleoli. Dihydrotestosterone (DHT) treatment induced AR-RFP translocation into the nucleus where the PPIs between AR and TIF2 resulted in the colocalization of both biosensors within the nucleolus. We adapted the translocation enhanced image analysis module to quantify the colocalization of the AR-RFP and TIF2-GFP biosensors in images acquired on the ImageXpress platform. DHT induced a concentration-dependent AR-TIF2 colocalization and produced a characteristic condensed punctate AR-RFP PPI nucleolar distribution pattern. The heat-shock protein 90 inhibitor 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) and antiandrogens flutamide and bicalutamide inhibited DHT-induced AR-TIF2 PPI formation with 50% inhibition concentrations (IC50s) of 88.5±12.5 nM, 7.6±2.4 μM, and 1.6±0.4 μM, respectively. Images of the AR-RFP distribution phenotype allowed us to distinguish between 17-AAG and flutamide, which prevented AR translocation, and bicalutamide, which blocked AR-TIF2 PPIs. We screened the Library of Pharmacologically Active Compounds (LOPAC) set for compounds that inhibited AR-TIF2 PPI formation or disrupted preexisting complexes. Eleven modulators of steroid family nuclear receptors (NRs) and 6 non-NR ligands inhibited AR-TIF2 PPI formation, and 10 disrupted preexisting complexes. The hits appear to be either AR antagonists or nonspecific inhibitors of NR activation and trafficking. Given that the LOPAC set represents such a small and restricted biological and chemical diversity, it is anticipated that screening a much larger and more diverse compound library will be required to find AR-TIF2 PPI inhibitors/disruptors. The AR-TIF2 protein–protein interaction biosensor (PPIB) approach offers significant promise for identifying molecules with potential to modulate AR transcriptional activity in a cell-specific manner that is distinct from the existing antiandrogen drugs that target AR binding or production. Small molecules that disrupt AR signaling at the level of AR-TIF2 PPIs may also overcome the development of resistance and progression to castration-resistant prostate cancer.
Introduction
Prostate cancer (CaP) is the most common solid tumor and the second leading cause of cancer death among men in Western countries, with ∼20% of patients developing metastatic castration-resistant prostate cancer (CRPC), resulting in ∼30,000 deaths annually in the United States.1–5 Front-line androgen ablation therapies (AATs) target either the production or the action of androgens that provide the critical growth and survival signals to prostate cells.1–5 Standard AATs include orchiectomy or chronic administration of gonadotrophin-releasing hormone agonists, estrogens, and antiandrogens such as bicalutamide (Casodex™), flutamide (Eulexin™), and nilutamide (Nilandron™).6–8 More recently approved CRPC therapies include Abiraterone™, which is an inhibitor of CYP17 and androgen biosynthesis, and Enzalutamide™, which reduces androgen receptor (AR) nuclear translocation, DNA binding, and coactivator recruitment.9–11 Although initial responses to AAT are usually favorable, the disease transforms and progresses to metastatic CRPC and patients develop resistance to antiandrogen drugs.6–8 Currently, there is no cure for metastatic CRPC. The AR is a member of the nuclear hormone receptor (NR) family of ligand-dependent and DNA sequence-specific transcriptional regulators that are required for the normal growth, terminal differentiation, and function of the prostate gland.1–5,12–14 A number of mechanisms have been proposed for the emergence of resistance in CRPC, including overexpression of the AR and/or its coactivators, shifts in the balance between coactivators and corepressors, AR mutations, expression of constitutively active AR splice variants, intracrine androgen synthesis, alternative methods of AR activation, or alternate pathway signaling that by-passes AR.1–5,15–17 CRPC exhibits an enhanced sensitivity to androgens that is correlated with increased AR expression and/or activity achieved through gene amplification or improved AR stability.3,4 CaP cells are resistant to apoptosis and can be activated by other steroid hormones, antiandrogens, growth factors, or PKA/PKC modulators.1–5 AR mutations may provide a growth advantage that facilitates disease progression by increasing ligand promiscuity or may alter coregulator interactions, thereby enhancing ARs ability to function in low androgen environments.3,4,15–17 However, since only 10% of CRPCs exhibit AR mutations, CaP growth may require abrogation of normal AR function, either directly through changes in AR structure and function or indirectly through changes in AR transcriptional activity (AR-TA) and signaling.1,2,5,8,15,16
Over 300 proteins have been reported to coregulate AR-TA; coactivators enhance agonist-dependent activity and corepressors suppress transcription in the absence of androgens or in the presence of antiandrogens.1,2,18,19 Coactivators amplify AR transcription complex assembly and context-specific gene expression, and coactivator recruitment profiles influence the tissue-specific spatiotemporal gene expression responses to ligands.13,14 Increased coactivator levels produce a more rapid transcriptional response and reduce ligand concentration requirements.14 Elevated expression levels have been observed in relapsed CaP for a number of the AR coregulators, including TIF2, SRC-1, RAC3, p300, CBP, Tip60, MAGE-11, and ARA70.3–5,15,16,18,20–22 Transcriptional intermediary factor 2 (TIF2/SRC-2) is a member of the steroid receptor coactivator SRC/p160 family that may be particularly relevant to CRPC. TIF2 stabilizes AR-ligand binding, enhances receptor stability, facilitates AR N/C interactions, and promotes both the recruitment of chromatin remodeling coactivators and the assembly of the transcriptional machinery on the promoters of AR target genes.1,2,13,14,23 Numerous studies have implicated TIF2 in normal and CaP cell function, and there is a significant correlation between tumor TIF2 expression and CaP aggressiveness.5,20,24,25 TIF2 levels are significantly higher in recurrent CaP after AAT when compared with androgen-dependent CaP or benign prostatic hyperplasia tissue.5 Increased TIF2 mRNA expression is associated with early recurrence, and the highest TIF2 expression is found in patients with relapsed CaP after AAT.20 Transient TIF2 overexpression increased AR responses to adrenal androgens and non-AR ligands, whereas depletion of TIF2 with antisense oligos or siRNAs reduced AR target gene expression and slowed the proliferation of androgen-dependent and androgen-independent CaP cells.5,20 TIF2 expression in CaP cells is associated with increased proliferation, and overexpression of TIF2 can compensate for reduced androgen levels. Interleukin 6 (IL-6) is elevated in the sera of patients with metastatic CaP, and IL-6 levels correlate with tumor burden, serum prostate-specific antigen levels, metastases, and CRPC.24 IL-6-treated LNCaP cells have significantly higher TIF2 levels and develop resistance to bicalutamide.24 Reduction of TIF2 levels by shRNAi restores bicalutamide sensitivity, whereas overexpression of TIF2 increases resistance to bicalutamide.24 In the CWR22R and C4-2 cell line models of CRPC, increased AR-TA is associated with elevated TIF2 recruitment together with decreased corepressor expression and recruitment.25
Ligand-activated AR signaling promotes AR binding to DNA response elements and the subsequent assembly of coactivators, DNA and/or histone modifying enzymes, scaffolding proteins, and the core transcriptional machinery into a multiprotein complex that orchestrates the transcription activity of target genes.1,2,15,16 Prolonged AR localization on the promoters of AR target genes combined with TIF2 recruitment has been proposed as a mechanism for the development of CRPC, and it was suggested that small molecules that block AR interactions with SRC coactivators might have therapeutic value.5,15,16,20–22,26–28 We describe here the development and optimization of an AR-TIF2 positional biosensor high-content screening (HCS) assay that can be used to identify compounds that can induce, inhibit the formation of, or disrupt preexisting protein–protein interactions (PPIs) between the AR and the TIF2 coactivator, a key component of AR signaling implicated in the development and progression of CRPC (Fig. 1). The AR-TIF2 protein–protein interaction biosensor (PPIB) recapitulates the ligand-induced translocation of AR from the cytoplasm into the nucleus, and the recruitment of AR-red fluorescent protein (RFP) into TIF2-green fluorescent protein (GFP)-positive nucleoli reflects the PPIs between AR and TIF2. We anticipate that novel AR-TIF2 PPI inhibitor/disruptor hits will provide chemical probes to investigate the development and progression of CRPC that may have potential for development as novel CaP therapeutics.
Fig. 1.

Androgen receptor–transcriptional intermediary factor 2 (AR-TIF2) protein–protein interaction biosensor (PPIB) components, principle, and characterization. (A) Cartoon schematic representation of the recombinant adenovirus (rAV) PPIB components. An rAV expression construct was created to express residues 662–919 encoding the AR ligand binding domain (AR-LBD) and activation function 2 (AF2) surface of AR as a chimeric fusion protein with red fluorescent protein (RFP) and both a nuclear localization sequence (NLS) and a nuclear export sequence (NES). The AR-RFP prey protein interaction partner shuttles between the cytoplasm and nucleus in a ligand-dependent manner. The nuclear export and import sequences are part of the chimera and not specific to AR. Likewise, an rAV construct was created to express residues 725–840 from the central region of TIF2 as a chimeric fusion protein with green fluorescent protein (GFP). TIF2 residues 725–840 contain the three α-helical LXXLL motifs responsible for interacting with ligand-bound AR. The TIF2-GFP biosensor component is targeted and anchored in the nucleolus by the inclusion of a high-affinity NLS/nucleolar localization sequence (NoLS) derived from Rev. (B) Grayscale and color composite images of U-2 OS cells infected with TIF2-GFP rAV alone. 20×grayscale and color composite images of U-2 OS cells infected with the TIF2-GFP adenovirus alone, cultured overnight, and then treated±20 nM dihydrotestosterone (DHT) for 30 min were sequentially acquired on the ImageXpress Ultra (IXU) platform in three fluorescent channels: Ch1 Hoechst, blue; Ch2 TIF2-GFP, green; and Ch3 AR-RFP, red. (C) Grayscale and color composite images of U-2 OS cells infected with the AR-RFP rAV alone. 20× grayscale and color composite images of U-2 OS cells infected with the AR-RFP adenovirus alone, cultured overnight, and then treated±20 nM DHT for 30 min were sequentially acquired on the IXU platform in three fluorescent channels: Ch1 Hoechst, blue; Ch2 TIF2-GFP, green; and Ch3 AR-RFP, red. (D) Grayscale and color composite images of U-2 OS cells coinfected with both the TIF2-GFP and AR-RFP rAVs. 20× grayscale and color composite images of U-2 OS cells coinfected with both the TIF2-GFP and AR-RFP adenoviruses, cultured overnight, and then treated±20 nM DHT for 30 min were sequentially acquired on the IXU platform in three fluorescent channels: Ch1 Hoechst, blue; Ch2 TIF2-GFP, green; and Ch3 AR-RFP, red. Adenovirus infected cells were seeded at 2,500 cells per well in 384-well Greiner collagen-coated assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity and were then treated for 30 min with 0.5% dimethyl sulfoxide (DMSO) or 20 nM DHT in 0.5% DMSO before formaldehyde fixation and the Hoechst staining as described in the Materials and Methods section. Individual grayscale images of three fluorescent channels (Hoechst Ch1, FITC Ch2, and Texas Red Ch3) of U-2 OS cells individually infected (B, C) or coinfected (D) with the biosensor adenoviruses were sequentially acquired on the IXU automated imaging platform using a 20×0.45 NA objective, the 405 Ch1, 488 Ch2, and 561 Ch3 laser lines, and a Quad emission filter set as described previously. Images from a single representative experiment of numerous experiments are presented. (E) Cartoon schematic representation of the AR-TIF2 PPIB assay formats and predicted phenotypes. In untreated cells, AR-RFP expression is localized predominantly in the cytoplasm and TIF2-GFP is restricted to nucleoli within the nucleus. Upon exposure to an AR agonist such as DHT, the AR-RFP biosensor translocates to the nucleus where it forms bright fluorescent puncta colocalized with the TIF2-GFP partner in the nucleolus. The AR-TIF2 PPIB assay therefore recapitulates the ligand-induced translocation of AR from the cytoplasm to the nucleus, and colocalization of the AR-RFP with the TIF2-GFP in the nucleolus reflects the protein–protein interactions (PPIs) between AR and the TIF2 coactivator. The AR-TIF2 PPIB assay can therefore be screened in three formats: (1) to screen for novel AR agonists, that is, compounds that induce the formation of AR-TIF2 PPIs; (2) to screen for compounds that block DHT-induced formation of AR-TIF2 PPIs; and (3) to identify compounds capable of disrupting preexisting AR-TIF2 complexes. Compounds that behave as AR agonists will induce an AR-TIF2 PPI phenotype similar to DHT. Compounds that antagonize DHT binding to AR, inhibit the heat-shock protein (Hsp) 90/70 chaperone complex, or block AR trafficking to the nucleus will produce a phenotype where the AR-RFP remains localized in the cytoplasm. Compounds that block DHT-induced AR-TIF2 PPI formation or disrupt AR-TIF2 PPIs will produce a phenotype where the AR-RFP is diffusely distributed throughout the nucleus rather than in puncta colocalized with the TIF2-GFP partner in the nucleolus.
Materials and Methods
Reagents
Formaldehyde, dihydrotestosterone (DHT), flutamide, bicalutamide, mifepristone, budesonide, estrone, N-p-tosyl-L-phenylalanine chloromethyl ketone (TPCK), Bay 11-7085, nilutamide, Z-L-phechloromethyl ketone (ZPCK), (Z)-gugglesterone, parthenolide, 17-alpha-hydroxyprogesterone (17-α-H-PG), 2-methoxyestradiol (2-MOED), 4-phenyl-3-furoxancarbonitrile (4-P-3-FOCN), spironolactone, cortexolone, and cyproterone acetate were all purchased from Sigma-Aldrich (St. Louis, MO). Hoechst 33342 was purchased from Invitrogen (Carlsbad, CA). Dimethyl sulfoxide (DMSO) (99.9% high-performance liquid chromatography grade, under argon) was from Alfa Aesar (Ward Hill, MA). The Dulbecco's Mg2+- and Ca2+-free phosphate-buffered saline (PBS) was purchased from Corning (Tewksbury, MA). The rabbit monoclonal anti-human fibrillarin antibody was purchased from Thermo Fisher (Waltham, MA), and the Cy5-conjugated donkey anti-rabbit IgG was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
Cells and Tissue Culture
The U-2 OS osteosarcoma cell line was acquired from American Type Culture Collection and was maintained in the McCoy's 5A medium with 2 mM l-glutamine (Invitrogen) supplemented with 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA) and 100 U/mL penicillin and streptomycin (Invitrogen) in a humidified incubator at 37°C, 5% CO2, and 95% humidity.
Compound Libraries
The 1,280 compound Library of Pharmacologically Active Compounds (LOPAC) was purchased from Sigma-Aldrich and was handled and stored as described previously.29–33 Three hundred eighty-four-well master plates were prepared from four 96-well LOPAC master plates mapped into the quadrants of a single 384-well plate using the Bravo automated liquid handling platform (Agilent Technologies, Santa Clara, CA) outfitted with a 96-well transfer head. Daughter plates containing 2 μL of 10 mM compounds in DMSO were prepared and replicated from the 384-well LOPAC master plates using the Bravo outfitted with a 384-well transfer head. Aluminum adhesive plate seals were applied with an Abgene Seal-IT 100 (Rochester, NY) plate sealer, and the plates were stored at −20°C in a Matrical MatriMinistore™ (Spokane, WA) automated compound storage and retrieval system. For screening, LOPAC daughter plates were withdrawn from −20°C storage, thawed to ambient temperature, centrifuged 1–2 min at 50 g, and the plate seals were removed before the transfer of 98 μL of serum-free tissue culture medium (SFM) into wells using the BioTek Microflo liquid handler (BioTek, Winooski, VT) to generate a 200 μM intermediate stock concentration of library compounds (2.0% DMSO). The diluted compounds were mixed by repeated aspiration and dispensation using a 384-well P30 dispensing head on the Evolution P3 (EP3) liquid handling platform (PerkinElmer, Waltham, MA) and then 5 μL of diluted compounds was transferred to the wells of assay plates. In the LOPAC screen, compounds were individually tested at a final concentration of 20 μM (0.2% DMSO). After the diluted compounds had been transferred from the daughter plates to the primary HCS assay plates, aluminum adhesive plate seals were applied and the diluted daughter plates were stored frozen at −20°C.
To confirm active compounds identified in the primary screen, we withdrew the LOPAC daughter plates containing the 200 μM intermediate concentration of library compounds (2.0% DMSO) from −20°C storage, thawed them to ambient temperature, centrifuged them for 1–2 min at 50 g, and removed the plate seals. A work list was generated to direct the Multiprobe Janus liquid handling platform (PerkinElmer) to cherry pick 75 μL from the wells of the diluted daughter plates that were flagged active in the primary screen and to reformat these into a 384-well plate hit confirmation daughter plate. Five microliters of primary HCS active compounds was then transferred into triplicate wells of assay plates using a 384-well P30 dispensing head on the Evolution P3 (EP3) liquid handling platform (PerkinElmer).
For the determination of the 50% inhibition concentrations (IC50s), 10-point twofold serial dilutions of test compounds in 100% DMSO were performed using a 384-well P30 dispensing head on the EP3 liquid handling platform. Daughter plates containing 2 μL of the serially diluted compounds in DMSO were prepared and replicated from the 384-well serial dilution master plates using the Bravo outfitted with a 384-well transfer head. Aluminum adhesive plate seals were applied and plates were stored at −20°C. For testing in the bioassays, daughter plates were withdrawn from −20°C storage, thawed to ambient temperature, centrifuged for 1–2 min at 50 g, and the plate seals were removed before the transfer of 38 μL of SFM into wells using the BioTek Microflo liquid handler (BioTek) to generate an intermediate stock concentration of library compounds ranging from 1.95 to 500 μM (5.0% DMSO). The diluted compounds were mixed by repeated aspiration and dispensation using a 384-well P30 dispensing head on the EP3 and then 5 μL of diluted compounds was transferred to the wells of assay plates to provide a final concentration response ranging from 0.195 to 50 μM (0.5% DMSO).
Image Acquisition on the ImageXpress Ultra Confocal Automated Imaging Platform
The ImageXpress Ultra (IXU) platform (Molecular Devices LLC, Sunnyvale, CA) is a fully-integrated, point-scanning, confocal automated imaging platform configured with four independent solid-state lasers providing four excitation wavelengths of 405, 488, 561, and 635 nm. The IXU was equipped with a Quad filter cube providing emission ranges of 417–477, 496–580, 553–613, and 645–725 nm and four independent photomultiplier tubes (PMTs) each dedicated to a single detection wavelength.32 The IXU utilizes a dedicated high-speed infra-red laser auto-focus system, has a four-position automated objective changer with air objectives (10×, 20×, 40×, and 60×), and the detection pinhole diameter of the confocal optics was configurable in the software. For the AR-TIF2 HCS assays, the IXU was set up to acquire two images per well using a 20×0.45 NA ELWD objective in each of three fluorescent channels, which were acquired sequentially. In the Hoechst channel (Ch1), the 405 laser was set at 10% power and the PMT gain was 550. In the TIF2-GFP channel (Ch2), the 488 laser was set at 10% power, and the PMT gain was 625. In the AR-RFP channel (Ch3), the 561 laser was set at 10% power, and the PMT gain was 625. On average, the IXU scanned a single 384-well plate, two images per channel, in 90 min using these settings.
The ImageXpress Micro (IXM) is an automated field-based high-content imaging platform integrated with the MetaXpress Imaging and Analysis software. The IXM optical drive includes a 300-W Xenon lamp as broad-spectrum white light source and 2/3″ chip cooled CCD camera and optical train for standard field of view imaging and an IXM transmitted light option with phase contrast. The IXM is equipped with a 4× Plan Apo 0.20 NA objective, a 10× Plan Fluor 0.3 NA objective, a 20× Ph1 Plan Fluor ELWD DM objective, a 20×, S Plan Fluor ELWD 0.45 NA objective, a 40×, S Plan Fluor ELWD 0.60 NA objective, and a single slide holder adaptor. The IXM is equipped with the following ZPS filter sets: DAPI, FITC/ALEXA 488, CY3, CY5, and Texas Red.
Image Analysis Using the Multiwavelength Translocation and Translocation Enhanced Modules
We used the multiwavelength translocation (MWT) image analysis module to quantify the expression of the AR-RFP and TIF2-GFP biosensors in the digital images of infected U-2 OS cells acquired on the IXU as described previously. Hoechst 33342 was used to stain and identify the nucleus of the U-2 OS cells, and this fluorescent signal in Ch1 was used by the MWT module to define a nuclear mask. Objects in Ch1 that exhibited the appropriate fluorescent intensities above background and size (width, length, and area) characteristics were identified and classified by the image segmentation as nuclei and used to create nuclear masks for each cell. The nuclear mask was eroded by 2 μm in from the edge of the detected nucleus to reduce cytoplasmic contamination within the nuclear area, and the reduced inner mask was used to quantify the amount of target channel fluorescence within the nuclear region of Ch2 (TIF2-GFP) and Ch3 (AR-RFP). The outer cytoplasm mask was then established 2 μm out from the edge of the detected nucleus and the width of the outer mask was set at 5 μm to cover as much of the cytoplasm region as possible without going outside the cell boundary. The outer mask was used to quantify the amount of target channel fluorescence within the cytoplasm region of Ch2 (TIF2-GFP) and Ch3 (AR-RFP). The MWT image analysis module outputs quantitative data, including the average and integrated fluorescent intensities of the Hoechst-stained objects (compartments) in Ch1, the number of compartments or total cell count in Ch1, and the integrated and average fluorescent intensities of the Ch2 and Ch3 signals in the nucleus (inner) or cytoplasm (outer) regions of the cell. We used the average fluorescent intensity output of the MWT module for the Ch2 and Ch3 signals in the nucleus (inner) or cytoplasm (outer) regions of the cell to quantify the relative expression levels of the TIF2-GFP and AR-RFP biosensors in U-2 OS cells infected with their respective recombinant adenoviruses (rAVs).
We utilized the translocation enhanced (TE) image analysis module to analyze and quantify the AR-TIF2 PPIs from the digital images acquired on the IXU as described previously. We used the TIF2-GFP biosensor component in Ch2 to create a translocation mask of the nucleoli within the Hoechst-stained nucleus. The TIF2-GFP remains localized to bright fluorescent puncta anchored within the nucleolus of the nucleus, and objects in Ch2 that had TIF2-GFP fluorescent intensities above background with appropriate morphologic characteristics (width, length, and area) were classified by the image segmentation as nucleoli and used to create a TIF2 mask and counted as the number of TIF2-GFP-positive nucleoli. Objects that met these criteria were used to create masks of the nucleoli within the Hoechst-stained nuclei of each cell. AR-RFP images from Ch3 were segmented into an inner nucleolus region with a mask set using the edge of the detected TIF2-GFP-positive nucleoli in Ch2. The TE image analysis module outputs quantitative data, such as the average fluorescent intensities of the TIF2-GFP-stained objects in Ch2, the selected object or nucleoli count in Ch2, and the integrated and average fluorescent intensities of the AR-RFP Ch3 signal in the TIF2-positive nucleolus (inner) region. The mean average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli output of the TE image analysis module was used to quantify AR-TIF2 PPIs.
Automated AR-TIF2 PPIB HCS Assay Protocol
The automated AR-TIF2 PPIB HCS assay protocol is presented in Table 1. rAV expression constructs bearing the individual TIF2-GFP (TagGFP; Evrogen, Inc., Moscow, Russia) and AR-RFP (Tag RFP; Evrogen, Inc.) PPI partners were utilized to infect U-2 OS cells according to the manufacturer's (Cyprotech US, Watertown, MA) instructions. Typically, 1×107 U-2 OS cells were coinfected with the TIF2-GFP and AR-RFP adenovirus expression constructs by incubating cells with the manufacturer's recommended volume of virus (usually 5 μL/106 U-2 OS cells) in 1.0 mL of culture medium for 1 h at 37°C, 5% CO2, and 95% humidity with periodic inversion (every 10 min) to maintain cells in suspension. Coinfected cells were then diluted to 6.25×104 cells/mL in culture media, and 40 μL (2,500 cells) was seeded in each well of a 384-well collagen-coated barcoded microplate (#781956; Greiner Bio-One, Kremsmünster, Austria) using a BioTek Microflo liquid handler (BioTek), and plates were incubated overnight at 37°C, 5% CO2, and 95% humidity. Prediluted compounds or DMSO (5 μL) were added to appropriate wells using the Bravo automated liquid handling platform (Agilent Technologies) or an Evolution P3 (PerkinElmer) for a final screening concentration of 20 μM and 0.2% DMSO. Compound-treated assay plates were incubated at 37°C, 5% CO2, and 95% humidity for either 60 or 180 min. DHT (5 μL, maximum plate controls and compound-treated wells) or media (5 μL, to minimum plate controls) were added to the appropriate wells using the Bravo automated liquid handling platform (Agilent Technologies) or an Evolution P3 (PerkinElmer) for a final DHT concentration of 20 nM. Assay plates were incubated at 37°C, 5% CO2, and 95% humidity for 30 min, and the samples were fixed by the addition of 50 μL of prewarmed (37°C) 7.4% formaldehyde and 2 μg/mL Hoechst 33342 in PBS using a BioTek ELx405 (BioTek) and incubated at room temperature for 30 min, as described previously. Liquid was then aspirated and plates were then washed twice with 85 μL PBS using the BioTek ELx405 (BioTek) plate washer and sealed with adhesive aluminum plate seals (Abgene) with the last 85 μL wash of PBS in place. Fluorescent images were then acquired on an IXU automated HCS platform (Molecular Devices LLC), and images were analyzed with the TE image analysis module of the MetaXpress software (Molecular Devices LLC) as described previously.
Table 1.
AR-TIF2 PPIB HCS Assay Protocol in U-2 OS Cells
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Harvest and centrifuge U-2 OS cells | 5 min, 500 g | Aspirate medium, wash with PBS, trypsinize cells, add the McCoy's 5A medium+10% FBS, and centrifuge |
| 2 | Viable U-2 OS cell count | Viable cell count | Resuspend cells in the culture medium and count the number of trypan blue excluding viable cells in a hemocytometer |
| 3 | Coinfect U-2 OS cell with the TIF2-GFP and AR-RFP adenovirus biosensors | 5 μL rAVs per 106 cells | Incubate rAVs and U-2 OS cells in 1.0 mL culture medium for 1 h at 37°C, 5% CO2, and 95% humidity with periodic inversion (every 10 min) |
| 4 | Adjust U-2 OS cells to the required density and seed into 384-well assay plate | 40 μL of 6.25×104 cells/mL, 2,500 cells per well | Seed assay plates with 2,500 cells per well and incubate overnight at 37°C, 5% CO2, and 95% humidity |
| 5 | Transfer library compounds/DMSO to control wells | 5 μL | 20 μM final concentration in well, 0.2% DMSO |
| 6 | Incubate assay plates | 1 or 3 h | At 37°C, 5% CO2, and 95% humidity |
| 7 | Add DHT to compound treated and Max controls, media to Min controls | 5 μL | 20 nM DHT final in well of compound treated and Max controls, media to Min controls |
| 8 | Incubate assay plates | 30 min | At 37°C, 5% CO2, and 95% humidity |
| 9 | Fix cells | 50 μL | 7.4% formaldehyde containing 2 μg/mL Hoechest 33342 in Ca2+- and Mg2+-free PBS prewarmed to 37°C |
| 10 | Incubate plates | 10–30 min | Ambient temperature |
| 11 | Aspirate fixative and wash 2× with PBS | 85 μL | Aspirate fixative and wash twice with 85 μL Ca2+ and Mg2+ free PBS, 50 μL PBS in well |
| 12 | Seal plates | 1× | Sealed with adhesive aluminum plate seals |
| 13 | Acquire images | 20×, 0.4 NA objective | Images of the Hoechst (Ch1), TIF2-GFP (Ch2), and AR-RFP (Ch3) were sequentially acquired on the ImageXpress Ultra platform using the 405, 488, and 561 nm excitation laser lines, a Quad filter cube set, and individual PMTs for each channel |
| 14 | Image analysis assay readout | Average inner intensity AR-RFP in TIF2-GFP-positive nucleoli | Images were analyzed using the translocation enhanced image analysis module using the average inner intensity parameter to quantify the AR-RFP within TIF2-GFP-positive nucleoli |
Step Notes
1. U-2 OS cells maintained in the McCoy's 5A medium with 2 mM l-glutamine supplemented with 10% FBS and 100 U/mL penicillin and streptomycin in a humidified incubator at 37°C, 5% CO2, and 95% humidity. Cell monolayers (<70% confluent) were washed 1× with PBS and then exposed to trypsin-EDTA until they detach from the surface of the tissue culture flasks. Cells pelleted at 500 g for 5 min in the Sorvall ST 16 Centrifuge with a TX-400 Rotor.
2. Aspirate medium, resuspend pelleted cells in tissue culture medium+FBS, and count the number of trypan blue excluding viable cells in a hemocytometer.
3. U-2 OS cells were coinfected with the TIF2-GFP and AR-RFP adenovirus expression constructs by incubating cells with the manufacturer's recommended volume of virus, typically 5 μL/106 cells, in 1.0 mL culture medium for 1 h at 37°C, 5% CO2, and 95% humidity with periodic inversion (every 10 min) to maintain cells in suspension.
4. U-2 OS cells coinfected with the rAV biosensors were seeded into 384-well black-walled clear-bottom plates, Greiner Bio-One Catalogue No. 781956, BioTek Microflo (BioTek, Winooski, VT), at 2,500 cells per well and incubated for 24 h at 37°C, 5% CO2, and 95% humidity in the McCoy's 5A medium with 2 mM l-glutamine supplemented with 10% FBS and 100 U/mL penicillin and streptomycin.
5. Twenty micromolars of compounds added to wells in columns 3–22 using a Bravo (Agilent Technologies, Inc., Santa Clara, CA) or an Evolution P3 (PerkinElmer, Waltham, MA) automated liquid handler outfitted with a 384-well transfer head.
6. Incubate treated coinfected U-2 OS cells 1–3 h at 37°C, 5% CO2, and 95% humidity.
7. DHT (20 nM final in well) added to Max controls and compound wells, media to Min control wells using a Bravo (Agilent Technologies, Inc.) or an Evolution P3 (PerkinElmer) automated liquid handler outfitted with a 384-well transfer head.
8. Incubate treated coinfected U-2 OS cells±DHT 30 min at 37°C, 5% CO2, and 95% humidity.
9. Aspiration of media and fixative addition automated on BioTek ELx405 (BioTek) plate washer.
10. Ten to thirty minutes incubation at ambient temperature to fix cells and stain nuclei with Hoechst.
11. Aspiration of fixative and PBS wash steps automated on BioTek ELx405 (BioTek) plate washer.
12. Plates sealed with adhesive aluminum plate seals using the Abgene Seal-IT 100 plate sealer (Abgene, Rochester, NY).
13. Plates loaded into the ImageXpress Ultra confocal HCS platform (Molecular Devices LLC, Sunnyvale, CA) for scanning using a Catalyst robotic plate handler (Thermo Fisher Scientific, Waltham, MA).
14. Images analyzed using the translocation enhanced image analysis module of MetaXpress (Molecular Devices LLC).
AR, androgen receptor; DHT, dihydrotestosterone; DMSO, dimethyl sulfoxide; FBS, fetal bovine serum; GFP, green fluorescent protein; HCS, high-content screening; PBS, phosphate-buffered saline; PMT, photomultiplier tube; PPIB, protein–protein interaction biosensor; rAV, recombinant adenovirus; RFP, red fluorescent protein; TIF2, transcriptional intermediary factor 2.
Fibrillarin Immunofluorescence
U-2 OS cells infected with the TIF2-GFP adenovirus were cultured overnight and then fixed in prewarmed (37°C) 3.7% paraformaldehyde containing 2 μg/mL Hoechst 33342. After 30 min at room temperature, fixed cell monolayers were washed 2× with PBS and then the fixed cells were permeabilized by the addition of 95% ice-cold methanol. After 30 min on ice, the 95% methanol was removed and the permeabilized cell monolayers were washed 2× with Tween 20 blocking buffer, and the cells were incubated in Tween 20 blocking buffer at room temperature for 15 min before a 1:100 dilution of rabbit anti-human fibrillarin primary antibody was added to 384-well assay plates and incubated for 1 h in the dark at room temperature. Cell monolayers were then washed 1× with Tween 20 blocking buffer and then 1:500 dilution of donkey anti-rabbit IgG secondary antibody conjugated to Cy5 in Tween 20 blocking buffer was added to 384-well assay plates and incubated for 1 h in the dark at room temperature. Cell monolayers were then washed 2× with PBS and the plates were sealed with aluminum foil seals and imaged with a 40×, S Plan Fluor ELWD 0.60 NA objective, on the IXM automated imaging platform.
Data Processing, Visualization, Statistical Analysis, and Curve Fitting
HCS data processing for the LOPAC screening assays were performed using ActivityBase™ (IDBS, Guildford, United Kingdom) and CytoMiner (UPDDI, Pittsburgh, PA). Processed data and HCS multiparameter features were visualized using Spotfire™ DecisionSite™ (Somerville, MA) software. An ActivityBase primary HTS template was created that automatically calculated percent inhibition together with plate control signal-to-background (S:B) ratios and Z′-factor coefficients. For the LOPAC screen, we utilized the mean average inner intensity values of the AR-RFP biosensor in TIF-GFP-positive nucleoli of the 0.2% DMSO minimum plate control wells (n=32) and the mean average inner intensity values of the 20 nM DHT maximum plate control wells (n=32) to normalize the mean average inner intensity values in the compound-treated wells and to represent 0% and 100% of AR-TIF2 PPIs, respectively.
We also constructed an ActivityBase concentration–response template to calculate percent inhibition together with plate control S:B ratios and Z′-factor coefficients for quality control purposes.33–35 IC50 values were calculated using an XLfit four parameter logistic model, also called the sigmoidal dose–response model: y=(A+((B−A)/(1+((C/x)^D)))), where y was the percent activation and x was the corresponding compound concentration. The fitted C parameter was the IC50 and defined as the concentration giving a response half way between the fitted top (B) and bottom (A) of the curve. For normalized data, the A and B parameters were locked as 0 and 100, respectively. For nonnormalized concentration–response data, we used GraphPad Prism 5 software to plot and fit data to curves using the sigmoidal dose–response variable slope equation,
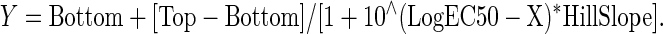 |
Results
AR-TIF2 PPIB Design, Subcellular Distribution Phenotypes, and Potential Assay Formats
The two chimeric AR and TIF2 biosensor components were cloned into rAV expression constructs, a high efficiency coexpression system that we have previously exploited for similar PPIB assays.34,35 The AR component of the biosensor was created to express AR residues 662–919 as a chimeric fusion protein with RFP and includes both a nuclear localization sequence (NLS) and a nuclear export sequence (NES) (Fig. 1A). The TIF2 component of the biosensor was created to express residues 725–840 from the central region of TIF2 as a chimeric fusion protein with GFP and a high affinity NLS/nucleolar localization sequence (NoLS) derived from Rev (Fig. 1A). In Figure 1, 20× grayscale and color composite images of three fluorescent channels (Hoechst-DNA Ch1, TIF2-GFP Ch2, and AR-RFP Ch3) are presented that were sequentially acquired on the IXU automated confocal HCS platform. In U-2 OS cells infected with just the TIF2-GFP rAV alone, TIF2-GFP exhibited a bright punctate expression pattern in Ch2 that was unaltered by exposure to 20 nM DHT (Fig. 1B). The colocalization of the green and blue signals apparent in the color composite images of U-2 OS cells infected with the TIF2-GFP rAV alone indicated that TIF2-GFP expression was localized to nucleoli within the Hoechst-stained nuclei (Fig. 1B). The associated Ch3 images of U-2 OS cells infected with the TIF2-GFP rAV alone indicated that there was no appreciable bleed through of the Hoechst (Ch1) or TIF2-GFP (Ch2) fluorescent signals into Ch3 (Fig. 1B). In U-2 OS cells infected with the AR-RFP rAV alone, the AR-RFP signal was predominantly localized in a diffuse pattern throughout the cytoplasm of cells in Ch3, with little or none apparent in the nuclei (Fig. 1C). In color composite images of U-2 OS cells infected with the AR-RFP rAV alone, the cytoplasm is red and the Hoechst-stained nuclei are blue (Fig. 1C). However, exposure of U-2 OS cells infected with the AR-RFP rAV alone to DHT induced an appreciable accumulation of AR-RFP within the nuclei of these cells apparent in the predominant mauve color of nuclei in composite images (Fig. 1C). The associated Ch2 images of U-2 OS cells infected with the AR-RFP rAV alone indicated that there was no appreciable bleed through of the Hoechst (Ch1) or AR-RFP (Ch3) fluorescent signals into Ch2 (Fig. 1C). In untreated U-2 OS cells coinfected with both rAVs, AR-RFP expression is localized predominantly to the cytoplasm and TIF2-GFP expression is localized only in the nucleolus as indicated by the diffusely red cytoplasm and blue nuclei containing bright green TIF2-GFP puncta of the corresponding composite images (Fig. 1D). Upon exposure of U-2 OS cells coinfected with both rAVs to DHT, the AR-RFP formed bright fluorescent puncta colocalized with the TIF2-GFP partner in the nucleolus indicated by the bright yellow of the AR-TIF2 puncta within the blue-stained nuclei of the corresponding composite images (Fig. 1D). The AR-TIF2 biosensor therefore recapitulates the ligand-induced translocation of AR from the cytoplasm to the nucleus, and the PPIs between AR and TIF2 results in the colocalization of AR-RFP and TIF2-GFP within the nucleolus. Based on the subcellular localizations of the AR-RFP and TIF2-GFP interacting partners and their responses to DHT, the AR-TIF2 PPIB assay could therefore be configured to screen in three distinct formats: (i) to discover novel AR agonists capable of inducing the formation of AR-TIF2 PPIs, (ii) to screen for compounds that block DHT-induced formation of AR-TIF2 PPIs, and (iii) to identify compounds that disrupt preexisting AR-TIF2 complexes (Fig. 1E).
TIF2-GFP Localization to the Nucleolus and AR-TIF2 PPIs
To demonstrate that the TIF2-GFP biosensor was targeted to and expressed in the nucleoli of infected U-2 OS cells, we used specific antibodies to fibrillarin36 to stain the nucleolar compartment by immunofluorescence and acquired 40× images on the IXM to evaluate the colocalization of the TIF2-GFP and fibrillarin (Fig. 2A and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/adt). TIF2-GFP and fibrillarin exhibited bright punctate expression patterns in the grayscale images of Ch2 and Ch3, respectively, which appeared to coincide with the less brightly stained areas of the Hoechst-stained nuclei in Ch1 images (Fig. 2A). The bright yellow/orange puncta of the color composite images indicated that TIF2-GFP and fibrillarin were colocalized within the nucleoli of the Hoechst-stained nuclei (Fig. 2A). Some cells in the field of view presented were not infected by the TIF2 rAV or expressed low levels of TIF-GFP, and the corresponding fibrillarin puncta were represented in mauve/red in color composite images (Fig. 2A). In the absence of the fibrillarin primary antibody, the TIF2-GFP puncta were bright green within the blue Hoechst-stained nuclei of composite images (Supplementary Fig. S1). These data demonstrate that the TIF2-GFP biosensor was expressed in the nucleolar compartment.
Fig. 2.

TIF2-GFP localization in the nucleolus and AR-TIF2 PPIs. (A) Colocalization of TIF2-GFP with fibrillarin in nucleoli. 40×grayscale and color composite images of U-2 OS cells infected with the TIF2-GFP adenovirus alone, which were cultured overnight and then fixed and stained with a specific antifibrillarin antibody, were sequentially acquired on the ImageXpress Micro (IXM) platform in three fluorescent channels: Ch1 Hoechst, blue; Ch2 TIF2-GFP, green; and Ch3 fibrillarin, red. Representative images from one of the two independent experiments are shown. (B) Control grayscale and color composite images of U-2 OS cells coinfected with both the TIF2-GFP and AR-RFP rAVs. 40× grayscale and color composite images of U-2 OS cells coinfected with both the TIF2-GFP and AR-RFP adenoviruses, cultured overnight and then fixed and stained with Hoechst, were sequentially acquired on the IXM platform in three fluorescent channels: Ch1 Hoechst, blue; Ch2 TIF2-GFP, green; and Ch3 AR-RFP, red. Representative images from one of the two independent experiments are shown. (C) Grayscale and color composite images of U-2 OS cells coinfected with both the TIF2-GFP and AR-RFP rAVs and the treated with DHT. 40× grayscale and color composite images of U-2 OS cells coinfected with both the TIF2-GFP and AR-RFP adenoviruses, cultured overnight, treated±20 nM DHT for 30 min and then fixed and stained with Hoechst, were sequentially acquired on the IXM platform in three fluorescent channels: Ch1 Hoechst, blue; Ch2 TIF2-GFP, green; and Ch3 AR-RFP, red. Representative images from one of the two independent experiments are shown. (D) Pairwise coinfection experiments with the AR-RFP, TIF2-GFP, hDM2-RFP, and p53-GFP adenovirus biosensors. 40× color composite images of U-2 OS cells coinfected with pairwise combinations of the AR-RFP, TIF2-GFP, hDM2-RFP, and p53-GFP adenoviruses, cultured overnight, treated±20 nM DHT or 10 μM Nutlin-3 for 30 min and then fixed and stained with Hoechst, were sequentially acquired on the IXM platform in three fluorescent channels: Ch1 Hoechst, blue; Ch2 TIF2-GFP or p53-GFP, green; and Ch3 AR-RFP or hDM2-RFP, red. The following pairwise combination of the biosensor adenoviruses were tested: AR-RFP plus TIF2-GFP, AR-RFP plus p53-GFP, hDM2-RFP plus TIF2-GFP, and hDM2 plus p53-GFP. Representative images from one of the two independent experiments are shown.
To more clearly demonstrate the DHT-induced colocalization of the AR-RFP biosensor within TIF2-GFP expressing nucleoli, we acquired high-resolution 40× images of coinfected U-2 OS cells on the IXM (Fig. 2B, C). As described previously (Fig. 1), in untreated cells, AR-RFP expression was diffusely localized throughout the cytoplasm and TIF2-GFP expression was localized only to puncta within the nucleolus, and in the corresponding color composite of the grayscale images, the cytoplasm was diffusely red and the blue nuclei contained bright green puncta (Fig. 2B). Exposure to DHT induced a redistribution of the AR-RFP biosensor to bright fluorescent puncta colocalized with the TIF2-GFP biosensor in the nucleolus, as indicated by the bright yellow puncta within the blue-stained nuclei of the corresponding color composite of the grayscale images (Fig. 2C).
To demonstrate that the DHT-induced colocalization AR-RFP and TIF2-GFP within nucleoli depended on the PPIs between AR and TIF2, we conducted pairwise coinfection experiments with the hDM2-RFP and p53-GFP rAV biosensors34,35 (Fig. 2D). The p53-hDM2 PPI biosensor is designed such that p53-GFP bait is anchored in the nucleolus, whereas the hDM2-RFP prey can shuttle between the nucleus and cytoplasm.34,35 Coinfection and expression of the p53-GFP and hDM2-RFP biosensors results in their colocalization within the nucleolus, and exposure to the p53-hDM2 PPI disruptor Nutlin-3 induces the redistribution of hDM2-RFP into the cytoplasm.34,35 In cells coinfected with the AR-TIF2 PPI biosensors, exposure to DHT, but not to Nutlin-3, induced the colocalization of AR-RFP within TIF2-GFP-positive nucleoli (Fig. 2D). In cells coinfected with the p53-hDM2 PPI biosensors, exposure to Nutlin-3, but not to DHT, induced the redistribution of hDM2 into the cytoplasm (Fig. 2D). In cells coinfected with the hDM2-RFP and TIF2-GFP biosensors and exposed to DHT or Nutlin-3, hDM2-RFP expression remained diffusely localized throughout the cytoplasm and TIF2-GFP expression remained localized to the nucleolus, indicating that the biosensors failed to form productive PPIs under these conditions (Fig. 2D). In cells coinfected with the AR-RFP and p53-GFP biosensors and exposed to Nutlin-3, AR-RFP expression remained diffusely localized throughout the cytoplasm and p53-GFP expression remained localized to the nucleolus (Fig. 2D). Exposure of cells coexpressing the AR-RFP and TIF2-GFP biosensors to DHT induced an appreciable accumulation of AR-RFP within the nuclei of these cells that was apparent in the predominant mauve color of nuclei in composite images (Fig. 2D). However, there was no evidence of recruitment and colocalization of AR-RFP within p53-GFP-positive nucleoli (Fig. 2D). Although DHT induced the translocation of the AR-RFP biosensor from the cytoplasm into the nucleus, the absence of effective PPIs between AR and p53 failed to result in the accumulation of AR-RFP within p53-GFP-positive nucleoli (Fig. 2D). These data indicate that the DHT-induced colocalization of AR-RFP within TIF2-GFP-positive nucleoli depends on two things: the ligand-induced translocation of AR ligand binding domain (AR-LBD) containing biosensor from the cytoplasm to the nucleus and the PPIs with the TIF2-LXXLL containing biosensor anchored in the nucleolus that drives their colocalization.
Image Analysis Module to Quantify AR-TIF2 PPIs
To analyze and quantify AR-TIF2 PPIs in the confocal digital images acquired on the IXU platform, we utilized the TE image analysis module (Fig. 3). The bright fluorescent TIF2-GFP puncta in Ch2 were used to define a translocation mask of the nucleoli within the nuclei of infected U-2 OS cells (Fig. 3A). Objects in Ch2 that had fluorescent intensities above background with appropriate morphologic characteristics (width, length, and area) were classified by the image segmentation as nucleoli and used to create a TIF2 mask (Fig. 3A, red and green masks). The following settings typically proved effective; objects defined as TIF2-GFP-positive nucleoli exhibited fluorescent intensities >750 gray levels over background, had an approximate width of 4.0 μm with a minimum area of 5.0 μm2, and did not exceed a maximum area of 150 μm2. Objects that met these criteria were used to create TIF2 masks within the nuclear regions of each cell (Fig. 3A). AR-RFP images from Ch3 were then segmented into an inner nucleolus region using masks established from the edge of the detected TIF2-GFP-positive nucleoli in Ch2 (Fig. 3A). The red or green color of the masks indicate whether the correlation coefficient for colocalization of the AR-RFP signal within the established TIF2-GFP nucleoli masks was below (red) or above (green) a preestablished threshold (typically ≥0.25) set in the software (Fig. 3A). The TE image analysis module outputs quantitative data, including the selected compartment or nucleoli count in Ch2 (Fig. 3B), the average fluorescent intensities of the TIF2-GFP-positive compartments in Ch2 (Fig. 3C), and the average (Fig. 3D) and integrated (Fig. 3E) fluorescent intensities of the AR-RFP signals in Ch3 within the TIF2-GFP-defined nucleoli masks. Treatment of U-2 OS cells coinfected with both rAVs±20 nM DHT for 30 min did not significantly affect either the number of TIF2-GFP-positive nucleoli detected (Fig. 3B) or their average fluorescent intensities (Fig. 3C). In marked contrast, however, 30 min exposure to DHT significantly increased the average (Fig. 3D) and integrated (Fig. 3E) fluorescent intensities of AR-RFP within the TIF2-defined masks compared with untreated controls. DHT treatment increased the average fluorescent intensity of AR-RFP within the TIF2-defined masks by 7.5-fold over untreated cells and produced a Z′-factor coefficient of 0.7 (Fig. 3E). Similarly, DHT treatment increased the integrated fluorescent intensity of AR-RFP within the TIF2-defined masks by 6.4-fold over untreated cells and exhibited a Z′-factor coefficient of 0.4 (Fig. 3E). Based on the slightly larger dynamic range and better Z′-factor coefficients, we selected the mean average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli output of the TE image analysis module to quantify DHT-induced AR-TIF2 PPIs.
Fig. 3.

Translocation enhanced (TE) image analysis module. (A) Image segmentation derived TIF2-GFP-positive nucleoli masks in the FITC and Texas Red channels. Enlarged and cropped grayscale images of TIF2-GFP (Ch2) and AR-RFP (Ch3) from U-2 OS cells coinfected with both the TIF2-GFP and AR-RFP adenoviruses, cultured overnight and then treated±20 nM DHT for 30 min. The TE image analysis module utilized the TIF2-GFP biosensor component in Ch2 to create a mask of the nucleoli. The TIF2-GFP was localized to bright fluorescent puncta within the Hoechst-stained nuclei of U-2 OS cells and objects in Ch2 with TIF2-GFP fluorescent intensities >750 gray levels over background, with an approximate width of 4.0 μm, a minimum area of 5.0 μm2, and that did not exceed a maximum area of 150 μm2 were classified by the image segmentation as TIF2-GFP-positive nucleoli and used to create translocation masks within the nucleus of cells. AR-RFP images from Ch3 were segmented into a nucleolus region using the mask derived from the detected TIF2-GFP-positive nucleoli in Ch2. The red or green color of the nucleolus masks indicate where the correlation coefficient between the TIF2-GFP and AR-RFP signals within the nucleoli were below (red) or above (green) a preset threshold (typically≥0.25). Quantitative data extracted by the TE image analysis module: (B) Number of the TIF2-GFP-positive compartments (selected objects) or nucleoli count in Ch2; (C) average fluorescent intensity of the TIF2-GFP-stained nucleoli in Ch2; (D) average fluorescent intensity of the AR-RFP signal in Ch3 within the TIF2-GFP-positive defined nucleolus region; and (E) integrated fluorescent intensity of the AR-RFP signal in Ch3 within the TIF2-GFP-positive defined nucleolus region. The enlarged grayscale images and the derived masks of nucleoli are representative of those obtained in numerous independent experiments. The quantitative data represent the mean±standard deviation (SD) (n=32) of 32 DMSO-treated (0.2%) and DHT-treated (20 nM in 0.2% DMSO) wells from one of numerous independent experiments. The mean average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli mask of Ch3 (D) was selected as the primary output of the TE image analysis module to quantify DHT-induced AR-TIF2 PPIs.
Development and Optimization of the AR-TIF2 PPIB HCS Assay
Since the AR-TIF2 biosensor components are encoded on separate rAVs and effective PPIs require both interaction partners, we needed to optimize the coinfection and expression of the AR-RFP and TIF2-GFP constructs (Fig. 4). We performed a series of individual and coinfection rAV titration experiments in which we increased the volume of adenovirus used to infect 1×106 U-2 OS cells, acquired 10× images 24 h postinfection (Fig. 4A, B), and utilized the MWT image analysis module to quantify the average fluorescent intensities of the AR-RFP and TIF-GFP biosensors in the nuclei and cytoplasm of infected cells (Fig. 4C, D). The MWT image analysis module uses the Hoechst-stained U-2 OS nuclei captured in Ch1 to define a nuclear mask. The Hoechst-stained objects in Ch1 that exhibited the appropriate fluorescent intensities above background and size (width, length, and area) characteristics were identified and classified by the image segmentation as nuclei. For the Hoechst-stained U-2 OS cells, the following settings typically proved effective: objects defined as nuclei exhibited fluorescent intensities >2,500 gray levels over background and they had an approximate minimum width of 8 μm with an approximate maximum width of 35 μm. Objects that met these criteria were used to create nuclear masks for each cell. An outer cytoplasm mask was then established 2 μm out from the edge of the detected nucleus and the width of the outer mask was set at 5 μm to cover as much of the cytoplasm region as possible without going outside the cell boundary. The outer mask was used to quantify the average AR-RFP fluorescence intensity within the cytoplasm region in Ch3 (Fig. 4C). An inner nuclear mask was eroded by 2 μm in from the edge of the detected nucleus to reduce cytoplasmic contamination within the nuclear area, and this inner mask was used to quantify the average TIF2-GFP fluorescence intensity within the nuclear region in Ch2 (Fig. 4D). Increasing the volume of adenovirus used to infect or coinfect 1×106 U-2 OS cells produced higher expression levels apparent in the color composite images of the AR-RFP and TIF2-GFP biosensors (Fig. 4A, B) and in the corresponding quantitative data extracted by the MWT image analysis module (Fig. 4C, D). The average fluorescent intensities of both biosensors increased in a roughly linear manner between 1.25 and 5 μL of rAV per 106 U-2 OS cells, and although infection with 10 and 20 μL of rAV per 106 U-2 OS cells also produced higher signals, the magnitude of the increase in signals was not proportional to the larger amount of virus (Fig. 4C, D). Although similar trends were observed in cells individually infected or coinfected with increasing amounts of the rAVs, the average fluorescent intensities of AR-RFP and TIF2-GFP were on average 13% and 19.3% lower, respectively, in cells coinfected with both rAVs when compared with the same volume of the individual viruses (Fig. 4C, D). Based on the results of several rAV titration experiments (Fig. 4) and the manufacturer's recommendations, we selected coinfection conditions of 5 μL each of the AR-RFP and TIF2-GFP rAVs per 106 U-2 OS cells for all further assay development experiments. Greater than 90% of U-2 OS cells coinfected under these conditions coexpressed both biosensors (Fig. 1D).
Fig. 4.

Titration of the AR-RFP and TIF2-GFP rAV biosensors in U-2 OS cells. (A) Color composite images of U-2 OS cells infected with increasing amounts of the AR-RFP biosensor alone. Increasing volumes (μL) of the AR-RFP rAV biosensor were incubated with 1× 106 U-2 OS cells and 2,500 infected cells were seeded into the wells of 384-well assay plates and cultured overnight at 37°C, 5% CO2, and 95% humidity. Cells were then fixed and stained with Hoechst 33342 and 10× images of three fluorescent channels (Ch1, blue; Ch2, green; and Ch3, red) were acquired on the IXU automated imaging platform as described previously. Representative color composite images of U-2 OS cells infected with the indicated volumes of AR-RFP virus are presented. (B) Color composite images of U-2 OS cells infected with increasing amounts of the TIF2-GFP biosensor alone. Increasing volumes (μL) of the TIF2-GFP rAV biosensor were incubated with 1×106 U-2 OS cells and 2,500 infected cells were seeded in the wells of 384-well assay plates and cultured overnight at 37°C, 5% CO2, and 95% humidity. Cells were then fixed and stained with Hoechst 33342 and 10× images of three fluorescent channels (Ch1, blue; Ch2, green; and Ch3, red) were acquired on the IXU automated imaging platform as described previously. Representative color composite images of U-2 OS cells infected with the indicated volumes of TIF2-GFP virus are presented. (C) Average intensity of AR-RFP in the cytoplasm of U-2 OS cells infected with increasing amounts of the AR-RFP biosensor, alone or in combination with the TIF2-GFP biosensor. The multiwavelength translocation image analysis module derived mean±SD (n=8) average cytoplasm (outer) intensity of AR-RFP in U-2 OS cells infected with the indicated volumes of AR-RFP rAVs, either alone (●) or in combination (○) with TIF2-GFP, are presented. Representative experimental data from one of several independent experiments are shown. (D) Average intensity of TIF2-GFP in the nucleoli of U-2 OS cells infected with increasing amounts of the TIF2-GFP biosensor, alone or in combination with the AR-RFP biosensor. The multiwavelength translocation image analysis module derived mean±SD (n=8) average nuclear (inner) intensity of TIF2-GFP in U-2 OS cells infected with the indicated volumes of TIF2-GFP rAVs, either alone (●) or in combination (○) with AR-RFP, are presented. Representative experimental data from one of several independent experiments are shown.
To optimize the assay signal window and reproducibility of the AR-TIF2 PPIB assay, we conducted a series assay development experiments (Fig. 5). In time course experiments, exposure to 20 nM DHT induced a rapid and linear increase in the average fluorescent intensity of AR-RFP colocalized with TIF2-GFP-positive nucleoli that approached a maximum plateau after 30 min and was maintained through 120 min of continuous DHT exposure (Fig. 5A). In four independent DHT concentration–response experiments, DHT exhibited an EC50 of 5.33±1.0 nM for the induction of AR-TIF2 PPIs (Fig. 5B). Although the maximum average fluorescent intensity of AR-RFP within the TIF2-GFP-positive nucleoli and the dynamic range of the third assay were ∼33% smaller than in the other experiments (Fig. 5B), the 4 nM EC50 for DHT was consistent with the EC50 values from the other experiments. Based on these observations (Fig. 5A, B), we selected 20 nM DHT and 30 min exposure as the conditions for the maximum induction of AR-TIF2 PPIs. Since most compound libraries are dissolved in DMSO, we needed to determine the DMSO tolerance of the AR-TIF2 PPIB assay (Fig. 5C). The AR-TIF2 PPI responses in unstimulated and DHT-treated cells were unaltered at DMSO concentrations of ≤0.625% (Fig. 5C). However, at DMSO concentrations >1.25%, the AR-TIF2 PPI responses of unstimulated and DHT-treated cells increased in a DMSO-dependent manner. At 10% DMSO, however, the DHT-induced AR-TIF2 PPI response decreased dramatically, whereas the unstimulated AR-TIF2 PPI response was marginally higher than at 5% DMSO (Fig. 5C). Based on these data, we selected ≤0.5% as the maximum DMSO concentration to be utilized in the AR-TIF2 PPIB assay. At seeding densities between 625 and 5,000 cells per well of the 384-well plate, the DHT-induced AR-TIF2 PPI S:B ratios were all ≥4.2-fold, but at 10,000 and 20,000 cells per well, the assay signal window collapsed to <4-fold (Fig. 5D). To reduce the cell culture burden and maintain a robust and reproducible assay signal window, we selected a 384-well plate seeding density of 2,500 cells per well for the AR-TIF2 PPIB HCS assay.
Fig. 5.

AR-TIF2 PPIB assay development. (A) DHT activation time course. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then treated with 20 nM DHT for the indicated time periods. Cells were then fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the DHT-induced AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=16) (●) average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli at time 0 and various time points ranging from 10 to 120 min are presented. Representative experimental data from one of the three independent experiments are shown. (B) DHT concentration responses. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then treated with the indicated concentrations of DHT for 30 min. Cells were then fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the DHT-induced AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=3) average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli at concentrations ranging between 0.001 and 100 nM DHT are presented. Representative experimental data from four independent experiments are shown: experiment 1 (●), experiment 2 (○), experiment 3 (■), and experiment 4 (□). (C) DMSO tolerance. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, cells were exposed to the indicated DMSO concentrations for 1 h, and then treated±20 nM DHT for 30 min. Cells were then fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the DHT-induced AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=4) average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli in control (○) and 20 nM DHT-treated (●) cells at DMSO concentrations ranging between 0.001% and 10% DMSO are presented. Representative experimental data from one of the three independent experiments are shown. (D) U-2 OS cell seeding density. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors and then seeded into the wells of 384-well assay plates at the indicated cell densities, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then treated±20 nM DHT for 30 min. Cells were then fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the DHT-induced AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=6) average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli in DMSO control (○) and 20 nM DHT-treated (●) cells at seeding densities ranging between 625 and 20,000 cells per well are presented. Representative experimental data from one of the three independent experiments are shown.
AR-TIF2 PPIB HCS Assay Validation
To validate that the AR-TIF2 PPIB HCS assay could identify known modulators of AR signaling, we conducted concentration–response studies with the heat-shock protein (Hsp) 90 inhibitor 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) and two antiandrogens, flutamide and bicalutamide, that are approved for the treatment of CaP (Fig. 6).6,37 Exposure to DHT and biclautamide, but not to flutamide or 17-AAG, induced a concentration-dependent increase in the mean fluorescent intensity of AR-RFP in TIF2-GFP-positive nucleoli (Fig. 6A). Consistent with the data presented in Figure 5B, DHT exhibited an EC50 of 5.9±0.5 nM for the induction of AR-PPIs. Although bicalutamide induced a concentration-dependent increase in AR-TIF2 colocalization with an EC50 of 7.8±3.6 μM, it only achieved ∼30% the efficacy of a maximal DHT-induced response (Fig. 6A) and it exhibited a diffuse distribution throughout the nucleus rather than the condensed punctate DHT-induced AR-RFP PPI pattern in nucleoli (Fig. 6C). Both 17-AAG and flutamide produced a cytoplasmic AR-RFP distribution phenotype similar to that of media controls, indicating that the AR-RFP biosensor was retained predominantly in the cytoplasm (Fig. 6C). Exposure of cells to the indicated concentrations of these compounds for 1 h before treatment with 20 nM DHT inhibited AR-RFP translocation into TIF2-GFP-positive nucleoli in a concentration-dependent manner (Fig. 6B). 17-AAG exhibited an IC50 of 88.5±12.5 nM and flutamide exhibited an IC50 of 7.6±2.4 μM for DHT-induced AR-TIF2 PPIs. Both 17-AAG and flutamide produced a cytoplasmic AR-RFP distribution phenotype similar to DMSO controls (Fig. 6D). Bicalutamide exhibited an IC50 of 1.6±0.4 μM for DHT-induced AR-TIF2 PPIs (Fig. 6B) and produced a diffuse AR-RFP nuclear distribution phenotype (Fig. 6D). 17-AAG completely inhibited the DHT-induced formation of AR-TIF2 PPIs, whereas flutamide and bicalutamide were only partial inhibitors (Fig. 6B). To more clearly demonstrate the compound-induced AR-TIF2 biosensor phenotypes, we acquired high-resolution 40× images on the IXM of coinfected U-2 OS cells exposed to compounds with and without DHT treatment (Fig. 6E). 17-AAG and flutamide did not affect the distribution of the biosensors by themselves, but they both blocked DHT-induced AR-RFP nuclear translocation (Fig. 6C–E). In contrast, bicalutamide induced AR-RFP translocation into the nuclear compartment but inhibited the PPIs between AR and TIF2 and prevented AR-RFP recruitment into TIF2-GFP-positive nucleoli (Fig. 6C–E).
Fig. 6.

Antiandrogen and Hsp 90 inhibitor concentration responses in the AR-TIF2 PPIB assay. (A) DHT and inhibitor induced AR-TIF2 PPI responses. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to compounds at the indicated concentrations for 1 h. Cells were then fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=3) average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli in cells exposed to the indicated concentrations of DHT (●), bicalutamide (■), flutamide (Δ), or 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) (◊) are presented. Representative experimental data from one of the five independent experiments are shown. (B) Inhibition of DHT induced AR-TIF2 PPIs. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to compounds at the indicated concentrations for 1 h. Cells were then treated with 20 nM DHT for 30 min, fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=3) average inner intensity of AR-RFP within the TIF2-GFP-positive nucleoli in cells exposed to the indicated concentrations of bicalutamide (■), flutamide (Δ), or 17-AAG (◊) for 1 h and then treated with 20 nM DHT are presented. Representative experimental data from one of the five independent experiments are shown. (C) Images of the AR-RFP phenotype in cells preexposed to compounds for 1 h. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to compounds at a variety of concentrations for 1 h. Cells were then fixed and stained with Hoechst, and 20× images in three fluorescent channels were acquired on the IXU automated imaging platform. Grayscale images of the AR-RFP distribution phenotype of coinfected U-2 OS cells exposed to 0.5% DMSO, 50 μM bicalutamide in 0.5% DMSO, 50 μM flutamide in 0.5% DMSO, or 5 μM 17-AAG in 0.5% DMSO for 1 h. Representative images from one of the five independent experiments are shown. (D) Images of AR-RFP phenotype in cells preexposed to compounds for 1 h and then treated with 20 nM DHT for 30 min. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to compounds at a variety of concentrations for 1 h. Cells were then treated with 20 nM DHT for 30 min, fixed and stained with Hoechst, and 20× images in three fluorescent channels were acquired on the IXU automated imaging platform. Grayscale images of the AR-RFP distribution phenotype of coinfected U-2 OS cells preexposed to 0.5% DMSO, 50 μM bicalutamide in 0.5% DMSO, 50 μM flutamide in 0.5% DMSO, or 5 μM 17-AAG in 0.5% DMSO for 1 h before treatment with 20 nM DHT. Representative images from one of the five independent experiments are shown. (E) 40× color composite images of compound induced AR-TIF2 biosensor phenotypes±20 nM DHT. 40× color composite images of the AR-TIF2 biosensor phenotypes of coinfected U-2 OS cells preexposed to 0.5% DMSO, 50 μM bicalutamide in 0.5% DMSO, 50 μM flutamide in 0.5% DMSO, or 5 μM 17-AAG in 0.5% DMSO for 1 h before 30 treatment±20 nM DHT. Representative images from one of the two independent experiments are shown. Ch1 Hoechst, blue; Ch2 TIF2-GFP, green; Ch3 AR-RFP, red; AR-TIF2 PPIs, yellow.
To validate the compatibility of the AR-TIF2 PPIB HCS assay with screening, we ran our 3-day assay signal window and DMSO validation tests.38 The 3-day assay signal window and Z-factor coefficient determination consisted of three independent experiments of two full 384-well plates each of the minimum (Min, 0.2% DMSO) and maximum (Max, 20 nM DHT in 0.2% DMSO) plate controls conducted on 3 separate days (Supplementary Table S1). The Max and Min plate controls performed very reproducibly and their respective average AR-RFP inner intensity population responses were well separated from each other with S:B ratios of 7.6-, 7.0-, and 6.8-fold on days 1, 2, and 3, respectively (Supplementary Table S1). Overall, the DHT-induced AR-TIF2 PPI assay exhibited intraplate Z-factor coefficients on all 3 days ranging between 0.67 and 0.75, with interplate Z-factors of 0.74, 0.71 and 0.70 on days 1, 2 and 3 respectively (Supplementary Table S1). The day 1 to 2 Z-factor coefficient was 0.65, whereas the day 2 to 3 Z-factor coefficient was negative because the response on the two maximum plates from day 3 were appreciably lower than on day 2, such that the corresponding standard deviations and coefficient of variations for the day-to-day comparison exceeded acceptable limits. Based on the statistical indices of the AR-TIF2 PPIB HCS assay (Supplementary Table S1), we selected a plate controls-based percent inhibition data processing method for the DMSO validation test and set a preliminary active criterion of ≥50% inhibition of DHT-induced AR-TIF2 PPIs. The 3-day 5-plate DMSO validation test mimics 3 days of automated screening operations, and all fifteen 384-well plates exhibited S:B ratios of ≥9-fold and Z′-factor coefficients of ≥0.59 (Supplementary Table S2). All assay plates passed our quality control criteria, and the percent inhibition data from the 4,800 wells of the 15×384-well DMSO plates closely approximated a normal distribution (data not shown) with none of the wells exhibiting ≥50% inhibition of DHT-induced AR-TIF2 PPIs producing an estimated false-positive rate of 0%. An analysis of variance in the DMSO validation data revealed no significant row/column effects or other positional biases (data not shown). Based on the statistical indices of the AR-TIF2 PPIB HCS assay (Supplementary Tables S1 and S2), we elected to continue with a plate controls-based percent inhibition data processing method and set a preliminary active criterion of ≥50% inhibition of DHT induced AR-TIF2 PPIs.
Screening the LOPAC Set in the AR-TIF2 PPIB HCS Assay
To confirm that the optimized AR-TIF2 PPIB assay would perform well in the presence of compounds, we screened the 1,280 member LOPAC at 20 μM to identify compounds that could either inhibit the DHT-induced formation of AR-TIF2 PPIs (Fig. 7A) or disrupt preexisting AR-TIF2 complexes (Fig. 7B). Figure 7A and B shows 4×384-well plate overlay scatter plots of the percent inhibition and percent disruption, respectively, produced by the LOPAC set screened in these two distinct formats of the AR-TIF2 PPIB assay. Each 384-well assay plate had 32× DHT-induced Max plate control wells and 32×0.2% DMSO min plate control wells. We screened the LOPAC set in both formats in two independent experiments, and the average plate Z′-factor coefficients and S:B ratios for the 16 LOPAC screening plates were 0.68±0.06- and 6.65±0.53-fold, respectively, indicating that the Max and Min plate controls provided a robust assay signal window and the automated AR-TIF2 PPIB HCS assay was stable at an HTS scale (Fig. 7A, B). Although the majority of compounds exhibited AR-TIF2 PPI responses that coincided with those of the Max controls, indicating that they were inactive, 24 compounds achieved ≥50% inhibition of DHT-induced AR-TIF2 PPI formation (Fig. 7A) compared with only three compounds that disrupted preexisting AR-TIF2 complexes by ≥50% (Fig. 7B). An additional 13 compounds disrupted preformed AR-TIF2 complexes by ≥30%. To qualify compounds flagged as actives in either format, we used parameters output by the image analysis algorithm to identify and eliminate compounds that were acutely cytotoxic (z-score cell counts <−4) or that were average of integrated fluorescent intensity outliers (z-scores <−4 or >4) in the Hoechst (Ch1), GFP (Ch2), and RFP (Ch3) channels. To confirm the remaining qualified actives, we cherry picked the compounds flagged as inhibitors of AR-TIF2 formation (>50%) or disruptors of preexisting AR-TIF2 complexes (>30%) into a 384-well hit confirmation daughter plate and rescreened them at 20 μM in triplicate wells in both assay formats. A total of 17 (1.3%) compounds were confirmed to reproducibly inhibit the DHT-induced formation of AR-TIF2 PPIs by ≥50% at 20 μM. Eleven of the inhibitors are known modulators of steroid family nuclear hormone receptors (NRs): nilutamide and cyproterone acetate are antiandrogens; mifepristone, 17alpha-hydroxy-4-pregnene-3,20-dione, and allopregnan-3-alpha-ol-20-one are progesterone receptor (PR) modulators; spironolactone and cortexolone are mineralocorticoid receptor (MR) modulators; 2-hydroxyestradiol 2-methyl ether and estrone are estrogen receptor (ER) modulators; guggulsterone is a farnesoid X receptor (FXR) antagonist; and budesonide is a glucocorticoid receptor (GR) modulator (Supplementary Fig. S2A). The remaining six inhibitors are not NR ligands (Supplementary Fig. S2B): 1-chloro-3-tosylamido-4-phenyl-2-butanone (TPCK), Bay 11-7085, N-Carbobenzyloxy-L-phenylalanyl chloromethyl ketone (ZPCK), parthenolide, 4-phenyl-3-fluroxane carbo-nitrile, and (±) Bay K 8644. Although none of the qualified actives achieved ≥50% disruption of preexisting AR-TIF2 complexes, nine compounds were confirmed to reproducibly disrupt preformed AR-TIF2 PPIs by between 32% and 42% at 20 μM. Five compounds inhibited DHT-induced formation of AR-TIF2 PPIs by ≥50% and also disrupted preformed AR-TIF2 PPIs by ≥34% at 20 μM.
Fig. 7.

Library of Pharmacologically Active Compounds (LOPAC) screen to identify compounds that block DHT-induced AR-TIF2 PPI formation or that disrupt preexisting AR-TIF2 complexes. (A) Overlay scatter plot of the percent inhibition of DHT-induced AR-TIF2 PPI formation from the 4×384-well assay plates of the 1,280 compound LOPAC set. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 4×384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to the LOPAC compounds at 20 μM (0.2% DMSO) for 1 h. Cells were then treated with 20 nM DHT for 30 min, fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean average inner intensity values of AR-RFP within the TIF2-GFP-positive nucleoli of the 0.2% DMSO minimum (Min) plate control wells (n=32) and the 20 nM DHT maximum (Max) plate control wells (n=32) were used to normalize the mean average inner intensity values of AR-RFP within the TIF2-GFP-positive nucleoli of compound-treated wells and to represent 100% and 0% inhibition of DHT-induced AR-TIF2 PPI formation, respectively. Max (green) controls, Min (red) controls, inactive (<50% inhibition) compound-treated wells (dark blue), and active (≥50% inhibition) compound-treated wells (light blue). Representative experimental data from one of the two independent LOPAC screens are presented. (B) Overlay scatter plot of the percent disruption of preexisting AR-TIF2 PPI complexes from the 4×384-well assay plates of the 1,280 compound LOPAC set. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 4×384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then treated with 20 nM DHT for 30 min to induce AR-TIF2 PPI formation. Cells were then exposed to the LOPAC compounds at 20 μM (0.2% DMSO) for 1 h, fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean average inner intensity values of AR-RFP within the TIF2-GFP-positive nucleoli of the 0.2% DMSO Min plate control wells (n=32) and the 20 nM DHT Max plate control wells (n=32) were used to normalize the mean average inner intensity values of AR-RFP within the TIF2-GFP-positive nucleoli of compound-treated wells and to represent 100% and 0% disruption of preexisting AR-TIF2 PPI complexes, respectively. Max (green) controls, Min (red) controls, inactive (<30% inhibition) compound-treated wells (dark blue), and active (≥30% inhibition) compound-treated wells (light blue). Representative experimental data from one of the two independent LOPAC screens are presented.
To determine the potencies of confirmed actives in the two AR-TIF2 PPIB assay formats, we purchased dry powder samples for 15 compounds and conducted three independent 10-point concentration–response experiments starting at a maximum of 50 μM (Fig. 8 and Table 2). We increased the compound exposure period from 1 to 3 h for these experiments because we had observed that the disruption of preexisting AR-TIF2 complexes was more pronounced after 3 h, whereas compounds that inhibited DHT-induced AR-TIF2 formation exhibited similar IC50s after 1 and 3 h exposures (data not shown). Exposure of U-2 OS cells to the indicated concentrations of the 15 compounds for 3 h before treatment with 20 nM DHT inhibited AR-RFP translocation into TIF2-GFP-positive nucleoli in a concentration-dependent manner with IC50s ranging from 12 nM to 3 μM (Fig. 8 and Table 2, and Supplementary Fig. S3). The five non-NR ligand AR-TIF2 PPI inhibitors exhibited high-quality curve fits (Fig. 8B) with IC50s in the 0.5–1.0 μM range (Table 2), and a predominant cytoplasm AR-RFP distribution phenotype (Supplementary Fig. S3B). Five of the NR ligand AR-TIF2 PPI inhibitors also produced high-quality curve fits (Fig. 8A) with IC50s in the 0.7–3.0 μM range (Table 2): 17alpha-hydroxy-4-pregnene-3,20-dione, nilutamide, cortexolone, 2-hydroxyestradiol 2-methyl ether, and estrone. However, the activity of the other five NR ligand AR-TIF2 PPI inhibitors was not fully reduced in the concentration range tested, and their corresponding IC50s were extrapolated from incomplete curve fits (Supplementary Fig. S3A and Table 2): mifepristone, guggulsterone, spironolactone, budesonide, and cyproterone. The corresponding AR-RFP distribution phenotypes for some of these NR ligand AR-TIF2 PPI inhibitors were also less definitive (Supplementary Fig. S3C). Guggulsterone displayed a predominant cytoplasm AR-RFP distribution phenotype similar to flutamide and 17-AAG, whereas both spironolactone and budesonide produced a diffuse AR-RFP nuclear distribution phenotype similar to bicalutamide (Supplementary Fig. S3C). However, the other NR ligand AR-TIF2 PPI inhibitors exhibited a mixed and diffuse AR-RFP distribution phenotype in both the cytoplasm and nuclear compartments (Supplementary Fig. S3C and Table 2). Exposure of U-2 OS cells to the indicated concentrations of compounds for 3 h after a 30 min pretreatment with 20 nM DHT disrupted the preexisting AR-TIF2 complexes in a concentration-dependent manner (Fig. 8C and Supplementary Fig. S4A, and Table 2). Although none of the compounds achieved >90% disruption of the preformed AR-TIF2 complexes and their corresponding IC50s were typically ≥10-fold less potent than for the inhibition of DHT-induced AR-TIF2 PPIs (Table 2), these data indicate that the AR-TIF2 biosensor is reversible. All 10 disruptors of preexisting AR-TIF2 complexes produced a mixed and diffuse AR-RFP distribution phenotype in both the cytoplasm and nuclear compartments (Supplementary Fig. S4B).
Fig. 8.

Concentration-dependent confirmation of LOPAC actives that inhibit the DHT-induced formation of AR-TIF2 PPIs or disrupt preexisting AR-TIF2 PPI complexes. (A) Concentration-dependent inhibition of DHT-induced AR-TIF2 PPI formation by NR ligands. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to compounds at the indicated concentrations for 3 h. Cells were then treated with 20 nM DHT for 30 min, fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=3) percent inhibition of DHT-induced AR-TIF2 PPIs in cells exposed to the indicated concentrations 17-alpha-hydroxyprogesterone (17-α-H-PG) (○), nilutamide (■), 2-methoxyestradiol (2-MOED) (▼), or estrone (▽) are presented. Representative experimental data from one of the three independent experiments are shown. (B) Concentration-dependent inhibition of DHT-induced AR-TIF2 PPI formation by non-NR ligands. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to compounds at the indicated concentrations for 3 h. Cells were then treated with 20 nM DHT for 30 min, fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=3) percent inhibition of DHT-induced AR-TIF2 PPIs in cells exposed to 4-phenyl-3-furoxancarbonitrile (4-P-3-FOCN) (●), Bay 11-7085 (■), parthenolide (Δ), 1-chloro-3-tosylamino-4-phenyl-2-butanone (TPCK) (▼), or N-Carbobenzyloxy-L-phenylalanyl chloromethyl ketone (ZPCK) (○) are presented. Representative experimental data from one of the three independent experiments are shown. (C) Concentration-dependent confirmation of disruptor actives identified in the AR-TIF2 PPIB LOPAC screens. U-2 OS cells were coinfected with the AR-RFP and TIF2-GFP rAV biosensors, 2,500 cells were seeded into the wells of 384-well assay plates, cultured overnight at 37°C, 5% CO2, and 95% humidity, and then exposed to 20 nM DHT for 30 min. Cells were then treated with compounds at the indicated concentrations for 3 h fixed and stained with Hoechst, 20× images in three fluorescent channels were acquired on the IXU automated imaging platform, and the AR-TIF2 PPIs were quantified using the TE image analysis module as described previously. The mean±SD (n=3) percent disruption of preformed AR-TIF2 PPI complexes in cells exposed to the indicated concentrations of mifepristone (●), 4-phenyl-3-fluroxane carbo-nitrile (4-P-3-FOCN) (□), and gugglesterone (■) are presented. Representative experimental data from one of the three independent experiments are shown.
Table 2.
IC50 Summary of AR-TIF2 PPIB LOPAC Hits
| Inhibition of DHT-induced AR-TIF2 PPI formation | Disruption of Preexisting AR-TIF2 PPIs | |||||
|---|---|---|---|---|---|---|
| Compounds | Mean IC50 μM | SD | AR-RFP phenotypeb | Mean IC50 μM | SD | AR-RFP phenotypeb |
| NR-ligands (structure No.a) | ||||||
| Mifepristone (1) | 0.012 | 0.015 | Nuc and Cyt | 0.955 | 0.427 | Nuc and Cyt |
| 17-Alpha-hyroxy-pregnene-3,20-dione (4) | 1.280 | 1.474 | Nuc and Cyt | 11.761 | 5.303 | Nuc |
| Nilutamide (2) | 0.715 | 0.919 | Nuc and Cyt | 4.771 | 2.423 | Nuc and Cyt |
| Spironolactone (7) | 0.497 | 0.629 | Nuc | 6.004 | 0.460 | Nuc |
| Guggulsterone (3) | 0.202 | 0.267 | Cyt | 3.550 | 2.066 | Nuc and Cyt |
| Cyproterone (11) | NC | Nuc | NT | Nuc | ||
| 2-Methoxy-estradiol (6) | 2.484 | 2.383 | Nuc and Cyt | 24.170 | 13.481 | Nuc |
| Budesonide (8) | 0.311 | 0.077 | Nuc | 16.913 | 10.080 | Nuc |
| Cortexolone (10) | 2.963 | 2.564 | Nuc | 33.357 | 17.873 | Nuc |
| Estrone (9) | 1.718 | 2.072 | Nuc and Cyt | 19.810 | n=1 | Nuc |
| Non-NR ligands (structure No.) | ||||||
| 4-Phenyl-3-furoxancarbonitrile (16) | 0.598 | 0.157 | Cyt | 12.093 | 4.174 | Nuc |
| Bay 11-7085 (13) | 0.922 | 0.043 | Cyt | NT | ||
| Parthenolide (15) | 1.170 | 0.340 | Cyt | NT | ||
| TPCK (12) | 0.764 | 0.178 | Cyt | NT | ||
| ZPCK (14) | 0.559 | 0.145 | Cyt | NT | ||
To determine the IC50s for compounds that inhibit the formation of DHT-induced AR-TIF2 PPIs, U-2 OS cells were preexposed to compounds for 3 h before treatment with 20 nM DHT for 30 min. To determine the IC50s for compounds that disrupt preexisting AR-TIF2 PPIs, U-2 OS cells were treated with 20 nM DHT for 30 min and then exposed to compounds for an additional 3 h. The IC50 values represent the mean and SD (n=3) of three independent concentration–response assays each conducted in a 10-point dilution series with triplicate wells per concentration. Representative curve fits used to generate the IC50 values from one of the three independent experiments presented in Table 2 are shown in Figure 8 and Supplementary Figures 2A and 3A.
The numbers in parentheses refer to the compound structures presented in Supplementary Figure S1A and B.
The AR-RFP biosensor distribution phenotype was designated as predominantly nuclear (Nuc), predominantly in the cytoplasm (Cyt), or in both compartments (Nuc and Cyt) based on representative images consistent with those presented in Supplementary Figures S2B, C and S3B.
IC50, 50% inhibition concentration; LOPAC, Library of Pharmacologically Active Compounds; NC, not calculable; NR, nuclear hormone receptor; NT, not tested; PPI, protein–protein interaction; SD, standard deviation; TPCK, 1-chloro-3-tosylamino-4-phenyl-2-butanone; ZPCK, N-Carbobenzyloxy-L-phenylalanyl chloromethyl ketone.
Discussion
PPIs are obligatory for all cellular functions and represent potential therapeutic targets to be exploited for drug discovery.39–43 The enormous potential of PPI inhibitors/disruptors as therapeutics has prompted the development and implementation of many biochemical and cell-based assay formats compatible with HTS and/or HCS.34,35,39–41,44 Nevertheless, the relative paucity of bona fide PPI inhibitor/disruptor drugs indicates that the discovery of such molecules remains challenging. Indeed, the prevailing perception is that PPI targets, and perhaps most especially transcriptional regulators, are essentially undruggable.27 However, the structural elucidation of several protein–protein complexes has revealed that protein-binding interfaces contain discrete hot spots that may preferentially facilitate binding interactions.39,40,42,43 It has been proposed that a relatively small number of amino acids at the PPI interface contribute to the majority of the binding energy and that the contact surfaces exhibit some degree of flexibility with cavities, pockets, and grooves available for small-molecule binding.39,40,42,43 Small-molecule PPI inhibitors, which bind with drug-like potencies to hotspots, appear to bind with much higher efficiencies and deeper within the target protein than do the contact atoms of the native protein partner.39,40,42,43 This article describes the development and validation of a high-content positional biosensor assay that can be configured to screen for compounds that can induce the establishment of, block the formation of, or disrupt preexisting AR-TIF2 PPIs (Fig. 1E). TIF2 is an AR coactivator that is a key component of the AR transcriptional complex, which has been implicated in the development and progression of CRPC.5,20,24,25 The positional biosensor approach described herein combines physiologically relevant cell-based assays with the specificity of binding assays by incorporating structural information about AR and TIF2 functional domains along with intracellular targeting sequences and fluorescent reporters and uses high-content imaging and analysis to quantify AR-TIF2 PPIs (Figs. 1–3).
In vitro biochemical PPI assay formats use small interacting peptides, protein subdomains, or full-length proteins purified from Escherichia coli, mammalian cell lines, or insect cells engineered to express them or from cells that express the proteins endogenously.39,40 Although biochemical assays are typically easier to develop and implement in HTS, relatively large quantities of the PPI partners may be required for these assays, and the expression and purification of sufficient material to screen a large compound library can sometimes be prohibitive.39,40 In addition, some PPI formats, such as ELISAs and cell surface binding assays, require multiple transfer and wash steps that can be difficult to automate. There are also concerns about the biological relevance of biochemical PPI assays conducted under nonphysiological conditions where competing interaction partners and/or contributing proteins are absent. In cell-based formats, the interacting partners are generated in situ and PPIs occur within the cellular milieu where cofactors or post-translational modifications are available.34,35,41,45–51 However, cell permeability and/or cytotoxicity can limit the chemotypes identified in cell-based assays, and cell wall permeability is a major concern for yeast genetic and two-hybrid PPI screens.39,40 Imaging-based formats provide valuable information on the subcellular localization of PPI partners, but it should be shown that the fluorescent fusion protein component of the chimeras does not alter the behavior of the protein of interest.34,35,45–47,51 Similarly, nonphysiological PPIs could arise from either the overexpression of interacting partners or the coexpression of proteins that normally reside in different cellular compartments.34,35,45–47,51 Whenever possible, fusion protein expression should be titrated to the levels of their endogenous counterparts and PPI assays should be validated with specific small-molecule disruptors or by mutations that eliminate the PPIs.34,45–47,51 Fluorescence resonance energy transfer PPI assays measure the separation between and orientation of donor and acceptor molecules that have been inserted into appropriate site(s) of the interacting polypeptides, and the design of such biosensors can be difficult to predict and/or implement.52 The association of fluorescent protein fragments in protein complementation assays (PCA) is irreversible, and this limits their use to the discovery of compounds that block the formation of PPIs.48,50,51 The lack of reversibility means that PCA formats cannot be used to screen for molecules that disrupt preexisting PPI complexes. There are also concerns that the irreversible association of the fluorescent protein fragments may lead to the trapping of transient interactions in the absence of specific interactions between the attached fusion partners or that complementation occurs independent of fusion partner PPIs.48,50,51 PCA PPI assays therefore need to be rigorously validated by mutations that eliminate the PPIs and/or with specific small-molecule PPI inhibitors that prevent the development of complementation.48,50,51 The localization of macromolecules to specific subcellular compartments is a tightly regulated process.53–55 Although small molecules passively diffuse through nuclear pore complexes (NPCs) in the nuclear envelope separating the nucleus and cytoplasm, cargos larger than ∼40 kDa require an active import and export process facilitated by specific receptor proteins.53–55 Nuclear entry of proteins ≥40 kDa requires assembly of a trimeric complex with an importin-α adaptor receptor that recognizes the NLS of the cargo protein and an importin-β transport receptor that facilitates docking interactions with the nucleoporins lining the NPC.53–55 Similarly, protein export from the nucleus is facilitated by the assembly of a complex between exportin-1 (CRM-1), Ran-GTP, and protein cargos bearing a leucine-rich NES.53–55 The steady-state localization of a nucleocytoplasmic shuttling protein will therefore be a function of the balance between the operational strengths and/or accessibility of the NLS and NES sequences of that protein.53–55 Since the nucleus and cytoplasm regions of cells can be readily identified, separated, and quantified by image analysis methods, investigators often exploit the regulated nucleocytoplasmic transport of proteins to develop subcellular protein distribution HCS assays as readouts for signal transduction pathways.32,33,56–59 Furthermore, the specific intracellular targeting sequences that direct proteins to specific subcellular sites or compartments have also been exploited to design positional biosensors to measure PPIs.34,35,41,54,55 Positional PPI biosensors employ two components: a “bait” biosensor that is targeted and anchored to a specific location within cells and a “prey” biosensor designed to shuttle between distinct cellular compartments.34,35,41,54,55 Colocalization of both biosensor components to a specific cellular site denotes the formation of productive PPI complexes. Stauber et al. pioneered the design of positional PPI biosensors that target bait PPI partners to the nucleolus using expression constructs that make target protein chimeras with a NES-deficient HIV-1 Rev sequence and a fluorescent reporter protein.54,55,60,61 The corresponding prey PPI biosensors are designed to be capable of shuttling between the cytoplasm and nucleus by incorporating an SV40 NLS sequence, an HIV-1 Rev NES sequence, and a fluorescent reporter protein in the chimera for the other protein interacting partners.54,55,60,61 Utilizing this design strategy, Stauber et al. have developed positional biosensor assays for a number of PPI pairs, including p53-mdm2, Myc-Max, and Jun-Fos.54,55,60,61 We employed a similar design strategy for a p53-hDM2 biosensor that used rAV constructs to coinfect cells and produce homogeneous expression of the two protein interacting components.34,35 Since that assay was successfully implemented in high throughput,34,35 we were confident of extending the approach to a bait:prey pair in which the prey molecule, AR, is normally induced to enter the nucleus after exposure to ligand.
In the nonligand-bound state, AR resides in the cytoplasm in a complex with Hsp 90 and 70 that maintain the NR in a stable partially unfolded state primed for high-affinity interactions with androgenic ligands.1–4 AR ligand binding induces AR homodimerization, trafficking to the nucleus, binding to specific DNA response element sequences in the promoter/enhancer regions of AR target genes resulting in the recruitment of coactivators, assembly of the core transcriptional machinery, and activation of transcription.1–4 Agonist binding induces a conformational change in the AR-LBD to form the activation function 2 (AF2) surface that binds with the LXXLL motifs of SRC/p160 coactivators.1,2,13,14,23 Expression of the AR-RFP prey interaction partner was directed by an rAV comprising AR residues 662–919 encompassing the LBD and AF2 domains of AR fused to RFP and incorporating NLS and NES sequences (Fig. 1A). The NLS and NES sequences are part of the chimera and not specific to the AR-LBD. In cells not expressing the bait, the AR-RFP prey was predominantly localized in the cytoplasm and exposure to DHT induced its translocation into the nucleus (Figs. 1C, D and 2B, C). SRC/p160 family member coactivators share LXXLL motifs that mediate binding to the LBD and AF2 surfaces of ligand activated steroid family NRs.13,14,23 Expression of the TIF2-GFP bait interaction partner was directed by an rAV with an insert composed of TIF2 residues 725–840 containing three α-helical LXXLL motifs, GFP, and a targeting sequence from HIV Rev that directs and anchors the expression of the TIF2-GFP chimera to the nucleolus of infected cells (Figs. 1 and 2A). TIF2-GFP expression produced discrete and regularly shaped high-intensity fluorescent puncta localized within the nucleus that could be readily imaged on the IXU platform and efficiently segmented by the image analysis module (Figs. 1–3). Using specific antibodies to fibrillarin36 to stain nucleoli by immunofluorescence, we demonstrated that the TIF2-GFP biosensor colocalized with fibrillarin (Fig. 2A and Supplementary Fig. S1). In pairwise coinfection experiments conducted with the hDM2-RFP and p53-GFP rAV biosensors,34,35 we demonstrated that the DHT-induced colocalization of AR-RFP within TIF2-GFP-positive nucleoli depends on two things; the ligand induced translocation of the AR-LBD biosensor from the cytoplasm to the nucleus and the PPIs with the TIF2-LXXLL biosensor, which draw the AR-RFP into the nucleolus (Fig. 2D). Consistent with its role as the bait component of the AR-TIF2 PPI biosensor, the localization of TIF2-GFP in the nucleolus was unaffected by either coexpression of the AR-RFP prey interaction partner or by exposure to AR ligands and library compounds. Anchoring the TIF2-GFP bait biosensor in the nucleolus facilitated both the image acquisition process and the subsequent analysis of the ligand-induced colocalization of the AR-RFP prey biosensor in the nucleolus. The adenovirus expression vectors allowed us to develop a relatively simple coinfection protocol for the AR-RFP and TIF2-GFP components of the biosensor that was scalable for HCS purposes (Figs. 4, 5, and 7 and Table 1, and Supplementary Tables S1 and S2).
The AR-TIF2 PPIB assay was able to identify and quantify the concentration-dependent inhibitory effects of antiandrogens (Fig. 6). The FDA approved anti-androgen flutamide is an AR antagonist.62 Bicalutamide has been reported to induce AR nuclear translocation, DNA binding, and to inhibit AR transcription by recruiting NCoR and SMRT corepressors.63,64 It has also been reported that bicalutamide-bound-AR fails to recruit coactivators.65 The AR-TIF2 assay was also inhibited by 17-AAG, an inhibitor of Hsp 90, which prevents AR from assuming a folded state primed for high-affinity interactions with androgenic ligands.37 The images of the AR-RFP phenotype allowed us to distinguish between compounds that block AR translocation and those that block AR-TIF2 PPIs, that is, 17-AAG and flutamide inhibited DHT-induced AR-RFP nuclear translocation, whereas bicalutamide blocked the PPIs between AR and TIF2 and prevented recruitment into the nucleolus (Fig. 6C–E). The EC50 values for DHT and the IC50 values for 17-AAG, flutamide, and bicalutamide in the AR-TIF2 PPIB assay correlate closely with published values from other assay formats,62,66,67 indicating that the rAV expression system does not significantly alter the concentration responses of known AR modulators and that the AR and TIF2 subdomains of the biosensors faithfully recapitulate the responses of the full-length proteins.
The combined statistical indices from the 3-day assay signal window and five-plate DMSO validation tests (Supplementary Tables S1 and S2) together with the performance of the automated AR-TIF2 PPIB assay in the LOPAC screens (Fig. 7) indicated that the HCS assay was compatible with HTS.38 Hits from the LOPAC set were identified in both screening formats and 15 compounds that were purchased for confirmation studies blocked DHT-induced AR-TIF2 PPI formation in a concentration-dependent manner with IC50s ranging from 12 nM to 3 μM (Fig. 8 and Table 2). None of these LOPAC hits were active in a p53-hDM2 PPIB HCS assay, so the hits are therefore unlikely to be nonspecific PPI inhibitors or compounds that interfere with the PPIB assay format. Eleven of the AR-TIF2 PPIB LOPAC hits are known modulators of steroid family NRs (Supplementary Fig. S2A), and the six non-NR ligand inhibitors have a variety of known pharmacological actions, including chymotrypsin inhibition; NFκB inhibition; serotonin antagonism, Ca2+ channel agonist; and nitric oxide donors (Supplementary Fig. S2B). Guggulsterone and the five non-NR hits all exhibited predominant cytoplasm AR-RFP distribution phenotypes, whereas the remaining inhibitors produced a mixed AR-RFP distribution phenotype diffusely spread throughout both the nuclear and the cytoplasm compartments (Supplementary Fig. S3B, C). The 11 steroid hits and 4-P-3-FOCN exhibited AR antagonist activity in a commercially available AR ligand binding assay (data not shown, manuscript in preparation), and this is likely to be the basis for their activity in the AR-TIF2 PPIB assay. Although TPCK, ZPCK, Bay 11-7085, and parthenolide were inactive in the AR ligand binding assay (data not shown), they are active in a dexamethasone-induced GR nuclear translocation HCS assay.33,68 Based on their cytoplasm AR-RFP distribution phenotypes (Supplementary Fig. S3B) and their ability to block Dex-induced GR-GFP nuclear translocation,33,68 it is possible that these nonselective and non-AR antagonist hits interfere with some common component of NR activation and trafficking. For example, Hsp 90 and 70 interact with both AR and GR to maintain the NRs primed for high-affinity interactions with their respective ligands, activated AR and GR are both cargos for dynein-mediated retrograde trafficking through the cytoplasm to the nucleus along microtubules, and passage through the NPC for both AR and GR is mediated by the importin-α/β adaptor system.1–4,33,37,68
In the screen to identify compounds capable of disrupting preexisting AR-TIF2 complexes, none of the LOPAC compounds disrupted AR-TIF2 colocalization by ≥50% after 1 h of exposure; however, with a longer compound exposure of 3 h, 10 of the compounds disrupted preexisting AR-TIF2 complexes in a concentration-dependent manner demonstrating that the AR-TIF2 PPIB assay was reversible and could be used to screen for AR-TIF2 PPI disruptors (Fig. 8C and Supplementary Fig. S3A, and Table 2). All the disruptors hits produced a mixed AR-RFP distribution phenotype diffusely spread throughout the cytoplasm and nuclear compartments (Supplementary Fig. S3B) and exhibited AR antagonist activity in a commercially available AR ligand binding assay (data not shown, manuscript in preparation).
The mapping of NR functional domains for ligand binding, DNA binding, and transactivation has mainly been delineated through the use of reporter assays conducted in cells cotransfected with two DNA expression vectors.12–14 The transvector produces high NR expression levels (full length or subdomains) in cells that do not normally express it, and the cis-vector contains a reporter gene coupled to a NR-responsive promoter with suitable hormone response elements (HREs).12–14,28 Agonist-induced NR activation stimulates the reporter activity in proportion to the proficiency of ligand-bound NR gene transactivation.12–14,28 The transfection-based HRE-reporter assay became the standard for the study of NR molecular function, and several variations of the assay have been developed.12–14,28 Yeast 2 hybrid assays have been used extensively to study the PPIs between NRs and the retinoid receptors (RARs and RXRs) because yeast lack endogenous RXRs and avoid the background issues apparent in mammalian cells due to the ubiquitous expression of RXRs.28,69 A popular mammalian 2 hybrid format utilizes a yeast Gal4-DBD fused to a NR-LBD to generate a chimeric receptor that is cotransfected with a Gal4-responsive reporter.28,70,71 A major benefit of the yeast Gal4 system is that it typically provides a lower reporter background in mammalian cells.28,70,71 The existence of NR coregulators was first indicated by transcriptional interference experiments in yeast and mammalian cotransfection experiments where different ligand-bound NRs squelched or inhibited the transactivation of each other.13,14,72 Coregulators were characterized by their propensity to amplify (coactivators) or silence (corepressors) transcription when transiently transfected and overexpressed with NRs in reporter assays.12–14 However, there are some significant limitations to the use of reporter assays for measuring PPIs. Many of these observations were made in transfected cells containing supraphysiological levels of coregulators, receptors, and promoters.13,14 A comparative quantitative analysis of these reporter assay studies is further complicated because they vary with respect to cell type, DNA amounts, expression plasmid and reporter constructs, and promoter activity readouts.13,14 Reporter assays produce positive outputs and are not reversible, thereby limiting their utility to finding molecules that block PPI formation. Reporter assays measure the cumulative output of multiple cellular processes, including signaling pathway activation, transcription factor trafficking into the nucleus, transcription factor binding to specific DNA response element sequences in the promoter/enhancer regions of target genes, recruitment of coregulators, assembly of the core transcriptional machinery, mRNA transcription, and finally protein translation, maturation, and stability. Reporter assays are therefore subjected to inhibition of any one of these cellular processes, and suitable counter screens and secondary assays need to be incorporated into the hit testing paradigm. Most HTS assays are subject to compound interference, and reporter assays are certainly no exception.73,74 In vitro biochemical assay formats developed to study the ligand-induced recruitment and binding of coregulator peptide sequences by NRs include time-resolved fluorescence intensity, AlphaScreen, and Luminex multiplexed bead-based capture assays.28,70,75,76 These assays provide information on the binding interactions between NRs and specific peptide sequences that may not totally translate to full-length PPIs within the cell. Chromatin immunoprecipitation and fluorescence photobleaching techniques in live cells have been used to investigate the recruitment of coregulators by NRs, but these methods are not compatible with HTS.13,14,77
The AR-TIF2 PPIB assay recapitulates the ligand-induced translocation of AR from the cytoplasm to the nucleus, and the subsequent recruitment of the AR-RFP biosensor into TIF2-GFP nucleoli reflects the PPIs between AR and TIF2. The AR-TIF2 PPIB HCS assay can identify compounds that bind either AR or TIF2, are not limited to a specific method of AR activation, and because the assay is reversible it can be used to screen compound libraries for compounds that block the formation of AR-TIF2 PPIs or that disrupt preexisting AR-TIF2 PPIs. The use of the AR-LBD/AF2 and TIF2-LXXLL subdomains for the biosensors was directed by structural studies that have mapped the AR-TIF2 protein–protein interfaces and identified the major domains of AR and TIF2 that contribute to binding. The biosensors behave like the full-length proteins and the EC50 and IC50 data for known AR modulators are consistent with the published values from other assays using full-length proteins. By targeting the reciprocal binding interaction domains of AR and TIF2, we hope to improve the probability of success in finding small molecules that can inhibit or disrupt these interactions. We have successfully optimized the AR-TIF2 PPIB assay in the PC-3 CaP cell line, and all 15 of the LOPAC hits described above inhibited DHT-induced AR-TIF2 PPI formation in a concentration-dependent manner (manuscript in preparation). However, PC-3 cells required 10-fold higher rAV amounts to achieve only 50% coinfection/expression of the biosensors, and while the S:B ratios were consistently >6-fold, the Z′-factor coefficients were typically >0.4 but <0.5. In addition, PC-3 cells required a three-fold higher cell seeding density. We would propose running the AR-TIF2 PPIB primary HCS in U-2 OS cells because of the better assay performance and the lower requirements for both adenoviruses and cells but confirming the activities in both U-2 OS and PC-3 cells. The LOPAC pilot screens indicate that the assay is capable of hit identification, although none of the preliminary hits survived the triage procedure. The LOPAC set is a small collection of 1,280 compounds assembled over 10 years ago and includes compounds that were selected for pharmacological activity against specific cell signaling and neuroscience targets. Given the restricted biological and chemical diversity inherent in the LOPAC collection and that none of the compounds are annotated as PPI inhibitors/disruptors, it is perhaps not surprising that the AR-TIF2 PPIB assay failed to identify PPI hits in this small set. It is our expectation that screening a much larger and more diverse compound library in the AR-TIF2 PPIB assay will lead to the discovery of AR-TIF2 PPI inhibitor/disruptor hits. The AR-TIF2 PPIB approach offers significant promise for identifying molecules with modalities distal to AR binding and with the potential to modulate AR TA in a cell-specific manner distinct from the traditional antiandrogen drugs that are available for CaP therapy. Small molecules that disrupt AR signaling at the level of AR-TIF2 PPIs may also overcome the development of resistance to AR antagonism. We anticipate that novel AR-TIF2 PPI inhibitor/disruptor hits will provide chemical probes to investigate the development and progression of CRPC that may have potential for development as novel CaP therapeutics.
Supplementary Material
Abbreviations Used
- 17-AAG
17-N-allylamino-17-demethoxygeldanamycin
- 17-α-H-PG
17-alpha-hydroxyprogesterone
- 2-MOED
2-methoxyestradiol
- 4-P-3-FOCN
4-phenyl-3-furoxancarbonitrile
- AAT
androgen ablation therapies
- AF2
activation function 2
- AR
androgen receptor
- CaP
prostate cancer
- Ch1
fluorescent channel 1
- Ch2
fluorescent channel 2
- Ch3
fluorescent channel 3
- CRPC
castration-resistant prostate cancer
- DBD
DNA binding domain
- DHT
dihydrotestosterone
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- GR
glucocorticoid receptor
- HCS
high-content screening
- HRE
hormone response element
- Hsp
heat-shock protein
- LBD
ligand binding domain
- IC50
50% inhibition concentration
- IL-6
interleukin 6
- IXM
ImageXpress Micro
- IXU
ImageXpress Ultra
- LOPAC
Library of Pharmacologically Active Compounds
- MWT
multiwavelength translocation
- NES
nuclear export sequence
- NLS
nuclear localization sequence
- NoLS
nucleolar localization sequence
- NPC
nuclear pore complex
- NR
nuclear hormone receptor
- PBS
phosphate-buffered saline
- PCA
protein complementation assays
- PL-PL
plate to plate comparison
- PMT
photomultiplier tube
- PPI
protein–protein interaction
- PPIB
protein–protein interaction biosensor
- rAV
recombinant adenovirus
- RFP
red fluorescent protein
- S:B
signal-to-background
- SD
standard deviation
- SFM
serum-free tissue culture medium
- TA
transcriptional activity
- TE
translocation enhanced
- TIF2
transcriptional intermediary factor 2
- TPCK
1-chloro-3-tosylamido-4-phenyl-2-butanone
- ZPCK
N-Carbobenzyloxy-L-phenylalanyl chloromethyl ketone
Acknowledgments
The studies reported herein were funded by grant support from the National Institutes of Health (NIH): R21NS073889 Johnston (PI) from the NINDS and R01CA160423 Johnston (PI) from the NCI.
Disclosure Statement
No competing financial interests exist.
References
- 1.Burd C, Morey LM, Knudsen KE: Androgen receptor corepressors and prostate cancer. Endocr Relat Cancer 2006;13:979–994 [DOI] [PubMed] [Google Scholar]
- 2.Chmelar R, Buchanan G, Need EM, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer 2006;120:719–733 [DOI] [PubMed] [Google Scholar]
- 3.Culig Z, Hobisch A, Bartsch G, Klocker H: Androgen receptor—an update of mechanisms of action in prostate cancer. Urol Res 2000;28:211–219 [DOI] [PubMed] [Google Scholar]
- 4.Culig Z, Klocker H, Bartsch G, Hobisch A: Androgen receptors in prostate cancer. Endocr Relat Cancer 2002;9:155–170 [DOI] [PubMed] [Google Scholar]
- 5.Gregory C, Johnson RT, Jr., Mohler JL, French FS, Wilson EM: Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res 2001;61:2892–2898 [PubMed] [Google Scholar]
- 6.Gao W, Dalton JT: Expanding the therapeutic use of androgens via selective androgen receptor modulators. Drug Discov Today 2007;12:241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stavridi F, Karapanagiotou EM, Syrigos KN: Targeted therapeutic approaches for hormone-refractory prostate cancer. Cancer Treat Rev 2010;36:122–130 [DOI] [PubMed] [Google Scholar]
- 8.Yu C, Sawyers CL, Scher HI: Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 2008;8:440–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larsson R, Mongan NP, Johansson M, Shcherbina L, Abrahamsson PA, Gudas LJ, Sterner O, Persson JL: Clinical trial update and novel therapeutic approaches for metastatic prostate cancer. Curr Med Chem 2011;18:4440–4453 [DOI] [PubMed] [Google Scholar]
- 10.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL: Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yap T, Zivi A, Omlin A, de Bono JS: The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol 2011;8:597–610 [DOI] [PubMed] [Google Scholar]
- 12.Evans RM: The steroid and thyroid hormone receptor family. Science 1988;240:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna N, O'Malley BW: Minireview: nuclear receptor coactivators—an update. Endocrinology 2002;143:2461–2465 [DOI] [PubMed] [Google Scholar]
- 14.McKenna N, O'Malley BW: Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002;108:465–474 [DOI] [PubMed] [Google Scholar]
- 15.Culig Z, Santer FR: Androgen receptor co-activators in the regulation of cellular events in prostate cancer. World J Urol 2012;30:297–302 [DOI] [PubMed] [Google Scholar]
- 16.Culig Z, Santer FR: Molecular aspects of androgenic signaling and possible targets for therapeutic intervention in prostate cancer. Steroids 2013;78:851–859 [DOI] [PubMed] [Google Scholar]
- 17.Peacock S, Fahrenholtz CD, Burnstein KL: Vav3 enhances androgen receptor splice variant activity and is critical for castration-resistant prostate cancer growth and survival. Mol Endocrinol 2012;26:1967–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linja M, Porkka KP, Kang Z, Savinainen KJ, Jänne OA, Tammela TL, Vessella RL, Palvimo JJ, Visakorpi T: Expression of androgen receptor coregulators in prostate cancer. Clin Cancer Res 2004;10:1032–1040 [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Hsu CL, Chang C: Androgen receptor corepressors: an overview. Prostate 2005;63:117–130 [DOI] [PubMed] [Google Scholar]
- 20.Agoulnik I, Vaid A, Nakka M, Alvarado M, Bingman WE, 3rd, Erdem H, Frolov A, Smith CL, Ayala GE, Ittmann MM, Weigel NL: Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res 2006;66:10594–10602 [DOI] [PubMed] [Google Scholar]
- 21.Fujimoto N, Miyamoto H, Mizokami A, Harada S, Nomura M, Ueta Y, Sasaguri T, Matsumoto T: Prostate cancer cells increase androgen sensitivity by increase in nuclear androgen receptor and androgen receptor coactivators; a possible mechanism of hormone-resistance of prostate cancer cells. Cancer Invest 2007;25:32–37 [DOI] [PubMed] [Google Scholar]
- 22.Shi X, Xue L, Zou JX, Gandour-Edwards R, Chen H, deVere White RW: Prolonged androgen receptor loading onto chromatin and the efficient recruitment of p160 coactivators contribute to androgen-independent growth of prostate cancer cells. Prostate 2008;68:1816–1826 [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Li Q: Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol 2003;17:1681–1692 [DOI] [PubMed] [Google Scholar]
- 24.Feng S, Tang Q, Sun M, Chun JY, Evans CP, Gao AC: Interleukin-6 increases prostate cancer cells resistance to bicalutamide via TIF2. Mol Cancer Ther 2009;8:665–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakka M, Agoulnik IU, Weigel NL: Targeted disruption of the p160 coactivator interface of androgen receptor (AR) selectively inhibits AR activity in both androgen-dependent and castration-resistant AR-expressing prostate cancer cells. Int J Biochem Cell Biol 2013;45:763–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godoy A, Sotomayor PC, Villagran M, Yacoub R, Montecinos VP, McNerney EM, Moser M, Foster BA, Onate SA: Altered corepressor SMRT expression and recruitment to target genes as a mechanism that change the response to androgens in prostate cancer progression. Biochem Biophys Res Commun 2012;423:564–570 [DOI] [PubMed] [Google Scholar]
- 27.Koehler A: A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol 2010;14:331–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulman I, Heyman RA: The flip side: identifying small molecule regulators of nuclear receptors. Chem Biol 2004;11:639–646 [DOI] [PubMed] [Google Scholar]
- 29.Johnston P, Foster CA, Shun TY, Skoko JJ, Shinde S, Wipf P, Lazo JS: Development and implementation of a 384-well homogeneous fluorescence intensity high-throughput screening assay to identify mitogen-activated protein kinase phosphatase-1 dual-specificity protein phosphatase inhibitors. Assay Drug Dev Technol 2007;5:319–332 [DOI] [PubMed] [Google Scholar]
- 30.Johnston P, Soares KM, Shinde SN, Foster CA, Shun TY, Takyi HK, Wipf P, Lazo JS: Development of a 384-well colorimetric assay to quantify hydrogen peroxide generated by the redox cycling of compounds in the presence of reducing agents. Assay Drug Dev Technol 2008;6:505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston P, Foster CA, Tierno MB, Shun TY, Brummond KM, Wipf P, Lazo JS: Characterization of the Cdc25B dual specificity phosphatase inhibitor hits identified in a high throughput screen of the NIH Compound Library. Assays Drug Dev Technol 2009;7:250–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnston P, Sen M, Hua Y, Camarco D, Shun TY, Lazo JS, Grandis JR: High-content pSTAT3/1 imaging assays to screen for selective inhibitors of STAT3 pathway activation in head and neck cancer cell lines. Assay Drug Dev Technol 2014;12:55–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnston PA, Shinde SN, Hua Y, Shun TY, Lazo JS, Day BW: Development and validation of a high-content screening assay to identify inhibitors of cytoplasmic Dynein-mediated transport of glucocorticoid receptor to the nucleus. Assay Drug Dev Technol 2012;10:432–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudgeon D, Shinde SN, Shun TY, Lazo JS, Strock CJ, Giuliano KA, Taylor DL, Johnston PA, Johnston PA: Characterization and optimization of a novel protein-protein interaction biosensor HCS assay to identify disruptors of the interactions between p53 and hDM2. Assay Drug Dev Technol 2010;8:437–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudgeon D, Shinde SN, Hua Y, Shun TY, Lazo JS, Strock CJ, Giuliano KA, Taylor DL, Johnston PA, Johnston PA: Implementation of a 220,000 compound HCS campaign to identify disruptors of the interaction between p53 and hDM2, and characterization of the confirmed hits. J Biomol Screen 2010;15:152–174 [DOI] [PubMed] [Google Scholar]
- 36.Bártová E, Horáková AH, Uhlírová R, Raska I, Galiová G, Orlova D, Kozubek S: Structure and epigenetics of nucleoli in comparison with non-nucleolar compartments. J Histochem Cytochem 2010;58:391–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saporita A, Ai J, Wang Z: The Hsp90 inhibitor, 17-AAG, prevents the ligand-independent nuclear localization of androgen receptor in refractory prostate cancer cells. Prostate 2007;67:509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shun T, Lazo JS, Sharlow ER, Johnston PA: Identifying actives from HTS data sets: practical approaches for the selection of an appropriate HTS data-processing method and quality control review. J Biomol Screen 2011;16:1–14 [DOI] [PubMed] [Google Scholar]
- 39.Colas P: High-throughput screening assays to discover small-molecule inhibitors of protein interactions. Curr Drug Discov Technol 2008;5:190–199 [DOI] [PubMed] [Google Scholar]
- 40.Lalonde S, Ehrhardt DW, Loqué D, Chen J, Rhee SY, Frommer WB: Molecular and cellular approaches for the detection of protein-protein interactions: latest techniques and current limitations. Plant J 2008;53:610–635 [DOI] [PubMed] [Google Scholar]
- 41.Pagliaro L, Felding J, Audouze K, Nielsen SJ, Terry RB, Krog-Jensen C, Butcher S: Emerging classes of protein-protein interaction inhibitors and new tools for their development. Curr Opin Chem Biol 2004;8:442–449 [DOI] [PubMed] [Google Scholar]
- 42.Reilly M, Cunningham KA, Natarajan A: Protein-protein interactions as therapeutic targets in neuropsychopharmacology. Neuropsychopharmacology 2009;34:247–248 [DOI] [PubMed] [Google Scholar]
- 43.Wells J, McClendon CL: Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007;450:1001–1009 [DOI] [PubMed] [Google Scholar]
- 44.Cummings M, Farnum MA, Nelen MI: Universal screening methods and applications of ThermoFluor. J Biomol Screen 2006;11:854–863 [DOI] [PubMed] [Google Scholar]
- 45.Giuliano K, Taylor DL: Fluorescent-protein biosensors: new tools for drug discovery. Trends Biotechnol 1998;16:135–140 [DOI] [PubMed] [Google Scholar]
- 46.Giuliano K, Chen Y, Haskins JR: Fluorescent protein biosensors. Mod Drug Discov 2003;8:33–37 [Google Scholar]
- 47.Giuliano K, Premkumar D, Taylor DL: Optimal characteristics of protein-protein interaction biosensors for cellular systems biology profiling. In: High Content Screening; Science, Techniques and Applications. Haney S. (ed.), pp. 371–387 John Wiley & Sons, Inc., Hoboken, 2007 [Google Scholar]
- 48.Kerpoola T: Visualization of molecular interactions by fluorescence complementation. Nat Rev Mol Cell Biol 2006;7:449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lundholt B, Heydorn A, Bjorn SP, Praestegaard M: A simple cell-based HTS assay system to screen for inhibitors of p53-Hdm2 protein-protein interactions. Assay Drug Dev Technol 2006;4:679–688 [DOI] [PubMed] [Google Scholar]
- 50.MacDonald M, Lamerdin J, Owens S, Keon BH, Bilter GK, Shang Z, Huang Z, Yu H, Dias J, Minami T, Michnick SW, Westwick JK: Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat Chem Biol 2006;2:329–337 [DOI] [PubMed] [Google Scholar]
- 51.Michnick S, Ear PH, Manderson EN, Remy I, Stefan E: Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov 2007;6:569–582 [DOI] [PubMed] [Google Scholar]
- 52.Li I, Pham E, Truong K: Protein biosensors based on the principle of fluorescence resonance energy transfer for monitoring cellular dynamics. Biotechnol Lett 2006;28:1971–1982 [DOI] [PubMed] [Google Scholar]
- 53.Kumar S, Saradhi M, Chaturvedi NK, Tyagi RK: Intracellular localization and nucleocytoplasmic trafficking of steroid receptors: an overview. Mol Cell Endocrinol 2006;246:147–156 [DOI] [PubMed] [Google Scholar]
- 54.Stauber R, Afonina E, Gulnik S, Erickson J, Pavlakis GN: Analysis of intracellular trafficking and interactions of cytoplasmic HIV-1 Rev mutants in living cells. Virology 1998;251:38–48 [DOI] [PubMed] [Google Scholar]
- 55.Stauber R: Analysis of nucleocytoplasmic transport using green fluorescent protein. Methods Mol Biol 2002;183:181–198 [DOI] [PubMed] [Google Scholar]
- 56.Nickischer D, Laethem C, Trask OJ, Williams RG, Kandasamy R, Johnston PA, Johnston PA: Development and implementation of three mitogen-activated protein kinase (MAPK) signaling pathway imaging assays to provide MAPK module selectivity profiling for kinase inhibitors: MK2-EGFP translocation, c-Jun, and ERK activation. Methods Enzymol 2006;414:389–418 [DOI] [PubMed] [Google Scholar]
- 57.Trask O, Baker A, Williams RG, Nickischer D, Kandasamy R, Laethem C, Johnston PA, Johnston PA: Assay development and case history of a 32K-biased library high-content MK2-EGFP translocation screen to identify p38 mitogen-activated protein kinase inhibitors on the ArrayScan 3.1 imaging platform. Methods Enzymol 2006;414:419–439 [DOI] [PubMed] [Google Scholar]
- 58.Trask O, Nickischer D, Burton A, Williams RG, Kandasamy RA, Johnston PA, Johnston PA: High-throughput automated confocal microscopy imaging screen of a kinase-focused library to identify p38 mitogen-activated protein kinase inhibitors using the GE InCell 3000 analyzer. Methods Mol Biol 2009;565:159–186 [DOI] [PubMed] [Google Scholar]
- 59.Williams R, Kandasamy R, Nickischer D, Trask OJ, Laethem C, Johnston PA, Johnston PA: Generation and characterization of a stable MK2-EGFP cell line and subsequent development of a high-content imaging assay on the Cellomics ArrayScan platform to screen for p38 mitogen-activated protein kinase inhibitors. Methods Enzymol 2006;414:364–389 [DOI] [PubMed] [Google Scholar]
- 60.Knauer S, Moodt S, Berg T, Liebel U, Pepperkok R, Stauber RH: Translocation biosensors to study signal-specific nucleo-cytoplasmic transport, protease activity and protein-protein interactions. Traffic 2005;6:594–606 [DOI] [PubMed] [Google Scholar]
- 61.Knauer S, Stauber RH: Development of an autofluorescent translocation biosensor system to investigate protein-protein interactions in living cells. Anal Chem 2005;77:4815–4820 [DOI] [PubMed] [Google Scholar]
- 62.Luo S, Martel C, LeBlanc G, Candas B, Singh SM, Labrie C, Simard J, Belanger A, Labrie F: Relative potencies of flutamide and casodex: preclinical studies. Endocr Relat Cancer 1996;3:229–241 [Google Scholar]
- 63.Hodgson M, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, Hollenberg AN: The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J Biol Chem 2005;280:6511–6519 [DOI] [PubMed] [Google Scholar]
- 64.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP: Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem 2002;277:26321–26326 [DOI] [PubMed] [Google Scholar]
- 65.Hodgson M, Astapova I, Hollenberg AN, Balk SP: Activity of androgen receptor antagonist bicalutamide in prostate cancer cells is independent of NCoR and SMRT corepressors. Cancer Res 2007;67:8388–8395 [DOI] [PubMed] [Google Scholar]
- 66.Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B: Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci 2005;83:136–148 [DOI] [PubMed] [Google Scholar]
- 67.Festuccia C, Gravina GL, Angelucci A, Millimaggi D, Muzi P, Vicentini C, Bologna M: Additive antitumor effects of the epidermal growth factor receptor tyrosine kinase inhibitor, gefitinib (Iressa), and the nonsteroidal antiandrogen, bicalutamide (Casodex), in prostate cancer cells in vitro. Int J Cancer 2005;115:630–640 [DOI] [PubMed] [Google Scholar]
- 68.Daghestani H, Zhu G, Johnston PA, Shinde SN, Brodsky JL, Day BW: Characterization of inhibitors of glucocorticoid receptor nuclear translocation: a model of cytoplasmic dynein-mediated cargo transport. Assay Drug Dev Technol 2012;10:46–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin C, Pike JW: Human vitamin D receptor-dependent transactivation in Saccharomyces cerevisiae requires retinoid X receptor. Mol Endocrinol 1996;10:196–205 [DOI] [PubMed] [Google Scholar]
- 70.Houck K, Borchert KM, Hepler CD, Thomas JS, Bramlett KS, Michael LF, Burris TP: T0901317 is a dual LXR/FXR agonist. Mol Genet Metab 2004;83:184–187 [DOI] [PubMed] [Google Scholar]
- 71.Tagami T, Lutz WH, Kumar R, Jameson JL: The interaction of the vitamin D receptor with nuclear receptor corepressors and coactivators. Biochem Biophys Res Commun 1998;253:358–363 [DOI] [PubMed] [Google Scholar]
- 72.Meyer M, Gronemeyer H, Turcotte B, Bocquel MT, Tasset D, Chambon P: Steroid hormone receptors compete for factors that mediate their enhancer function. Cell 1989;57:433–442 [DOI] [PubMed] [Google Scholar]
- 73.Johnston P: Redox cycling compounds generate H2O2 in HTS buffers containing strong reducing reagents—real hits or promiscuous artifacts? Curr Opin Chem Biol 2011;15:174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thorne N, Auld DS, Inglese J: Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr Opin Chem Biol 2010;14:315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bramlett K, Burris TP: Effects of selective estrogen receptor modulators (SERMs) on coactivator nuclear receptor (NR) box binding to estrogen receptors. Mol Genet Metab 2002;76:225–233 [DOI] [PubMed] [Google Scholar]
- 76.Iannone M, Consler TG, Pearce KH, Stimmel JB, Parks DJ, Gray JG: Multiplexed molecular interactions of nuclear receptors using fluorescent microspheres. Cytometry 2001;44:326–337 [DOI] [PubMed] [Google Scholar]
- 77.McNally J, Müller WG, Walker D, Wolford R, Hager GL: The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 2000;287:1262–1265 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.