

Abstract
Members of the AFF (AF4/FMR2) family of putative transcription factors are involved in infant acute leukaemia and intellectual disability (ID), although very little is known about their transcriptional targets. For example, deletion of human lymphoid nuclear protein related to AF4/AFF member 3 (LAF4/AFF3) is known to cause severe neurodevelopmental defects, and silencing of the gene is also associated with ID at the folate-sensitive fragile site (FSFS) FRA2A; yet the normal function of this gene in the nervous system is unclear. The aim of this study was to further investigate the function of Laf4 in the brain by focusing on its role in the cortex. By manipulating expression levels in organotypic slices, we demonstrate here that Laf4 is required for normal cellular migration in the developing cortex and have subsequently identified Mdga2, an important structural protein in neurodevelopment, as a target of Laf4 transcriptional activity. Furthermore, we show that the migration deficit caused by loss of Laf4 can be partially rescued by Mdga2 over-expression, revealing an important functional relationship between these genes. Our study demonstrates the key transcriptional role of Laf4 during early brain development and reveals a novel function for the gene in the process of cortical cell migration relevant to the haploinsufficiency and silencing observed in human neurodevelopmental disorders.
Introduction
Mixed lineage leukaemia (MLL) gene rearrangements occur in approximately 70 percent of infant acute lymphoblastic leukaemia (ALL) patients [1]. Lymphoid nuclear protein related to AF4 (LAF4, also known as AFF3) is one of an estimated 40 genes that can form such MLL fusions [2], although LAF4 is one of the few genes that is aberrantly translocated in both B- and T-cell derived leukemia [3]. Moreover, LAF4 has been found to be abnormally expressed in approximately 20 percent of breast cancers, suggesting that it may act as a proto-oncogene [4]. A human microdeletion of 500 kb on chromosome 2q11.1 encompassing only the LAF4 gene has been detected by array comparative genomic hybridization on peripheral lymphocytes [5]. The patient presented with developmental delay, seizures, urogenital and limb defects and died at four months of age after numerous repeated apnoeic episodes [5]. The patient was also noted to be of low birth weight (10th centile), and magnetic resonance imaging (MRI) revealed a dilated ventricular system with cortical and subcortical brain atrophy. In addition, a recent study identified a CGG repeat expansion in the promoter of LAF4 at an autosomal folate-sensitive fragile site (FSFS) named FRA2A that is associated with ID [6]. It was shown that this polymorphic repeat is hypermethylated in FRA2A leading to silencing of LAF4 in the nervous system [6]. However, the functional consequences of such a reduction in LAF4 expression are unknown.
LAF4 is one of four members of the AFF (AF4/FMR2) protein family in higher mammals, consisting of ALL-1 fused gene from chromosome 4 (AF4 or AFF1), Fragile X mental retardation 2 (FMR2 or AFF2) and ALL-fused gene from 5q31 (AF5Q31 or AFF4). Like LAF4, AF4 and AF5Q31 are known to form fusion proteins with MLL [7], [8], whereas FMR2 is silenced in FRAXE (mental retardation, X-linked, associated with fragile site) intellectual disability and is not implicated in ALL [9], [10]. While a mouse Fmr2 null mutant does show some subtle behavioral and electrophysiological deficits related to synaptic plasticity [11], gene knockouts of Af4 and Af5q31 have not revealed a great deal regarding the normal molecular function of AFF proteins [12], [13] and no Laf4 mutants have been reported to date. Interestingly, a dominant point mutation in Af4 in the robotic ataxic mutant mouse reduces the turn-over of the protein by seven in absentia homolog (SIAH) proteins, a family of E3 ubiquitin-protein ligases; these data have revealed the importance of Af4 in the survival of Purkinje neurons in the cerebellum [14], [15].
AFF proteins were originally described as putative transcription factors based on the presence of a conserved transactivation domain [16]. Importantly, subsequent biochemical studies demonstrated the key role that AFF proteins play in mediating transcriptional activity, revealing an association with the positive transcription elongation factor b (P-TEFb) [17]. P-TEFb, AF4 and ENL/AF9 form a large complex capable of interacting with RNA polymerase II (Pol II) and this complex can also interact with disruptor of telomeric silencing (DOT1) to enhance methylation at histone 3 lysine residue 79 (H3K79) [17]. This complex has since become widely studied as a source of aberrant transcriptional activity related to oncogenesis in MLL/AFF protein fusion events [18], [19] and more recently in the direct transcriptional control of integrated HIV genomes [20]. Further in vitro data have shown that all members of the AFF family form nuclear foci, also termed nuclear speckles, which may be important in regulating splicing events through interactions with pre-mRNA factors [15], [21].
Despite these studies, very little is known about the normal transcriptional targets of AFF proteins, as only very few have been confirmed [22], [23]. Furthermore, in view of the relevance of FMR2 and LAF4 to neurodevelopmental disease and cerebellar neurodegeneration in the Af4 mutant mouse robotic, the role of AFF proteins in the CNS warrants further investigation. For example, specific temporal patterns of Fmr2 and Laf4 expression have been described in the developing mouse embryo; whereas Fmr2 expression peaks at late embryonic stages [11], Laf4 was shown to be expressed in the developing cortex as early as embryonic day (E)13.5 [24]. At E13.5, Laf4 is also detected in cartilage tissue in different regions of the embryo as well as in the lung, kidney tubules and bladder [24]. A similar pattern has been confirmed in humans, with very high levels of LAF4 seen in the fetal brain, which then diminished to much lower levels in adults [25].
In light of the reported human LAF4 deletion, association with ID and known expression patterns, the aim of this study was to further investigate the function of Laf4 in the brain, focusing on the role of the protein in the developing cortex. By manipulating expression levels of Laf4 by whole embryo electroporation followed by organotypic culture experiments, we discovered that Laf4 is required for cortical cell migration. We then went on to examine the potential targets of Laf4 transcriptional control and found that Laf4 regulates the expression of Mdga2, an important structural protein in the developing CNS. These data represent the first detailed functional studies of Laf4 and highlight the importance of the gene in the developing nervous system with relevance to human neurodevelopmental disorders.
Materials and Methods
Cloning
A full-length cDNA representing mouse Laf4 (corresponding to NCBI accession number NM_010678.2) with an additional HA-tag at the C-terminus was cloned into the IRES-GFP vector (a gift from Dr Edward Turner [26]) to generate Laf4-HA-IRES-GFP. For Laf4 knockdown, complementary shRNA oligonucleotides were cloned into the LentiLox 3.7 (pLL.3) vector (Science Gateway) using the following primers: 5′-TGGACCTTGCTTGGAGATG GTGGTTCAAGAGAACACCATCTCCAAGCAAGGTCCTTTTTC-3′, 5′-TCGAGAAAAAGGACCTTGCTTGGAGATGGTGTTCTCTTGAACCACC ATCTCCAAGCAAGGTCCA-3′. Site directed mutagenesis was used to introduce three silent single nucleotide mutations into Laf4-HA-IRES-GFP at the shRNA-Laf4 target sequence to generate Laf4-HA-rescue-IRES-GFP (for shRNA primer sequences see Table S1). The scramble construct was designed to match the G:C/A:T ratio of the shRNA-Laf4 knockdown construct and to avoid predicted sequence alignment with the mouse genome. A full-length cDNA representing mouse Mdga2 (NM_001193266) was cloned into the same vector to generate Mdga2-IRES-GFP. The additional shRNAs tested were obtained from Sigma (Mission shRNAs to XM_283603, TRCN0000086608-12).
In situ hybridization
A region of the Laf4 coding sequence corresponding to a ∼300 bp sequence at the 3′ end of the mRNA transcript was amplified by RT-PCR using primers 5′-GATGAACTCTCCAACCGGATC and 5′-AGCCCATGGCACCTCTCTTG. The PCR product was cloned into pCR4-TOPO (Invitrogen) for DIG-labelled riboprobe synthesis from linearised plasmid DNA. Probes were hybridized to 14 µM frozen sections as previously described [14].
Animals
All animal experiments have been approved by the UK Home Office and the University of Oxford Ethical Review Panel.
Cortical cell culture and gene electroporation
E14.5 embryos or P1 neonates were dissected on ice and cortical cells were dissociated and electroporated as per the manufacturer’s protocol. Briefly, after trypsin digestion, trituration and washes, cells (2.7×105/cm2) were mixed with Nucleofector solution (Amaxa), plasmid DNA and electroporated as recommended by the manufacturer. Cells were re-suspended in culture medium (Neurobasal, 5% horse serum, 2% B27 supplement and 2 mM Glutamax (Gibco, UK)) and plated on wells pre-coated with poly-L-lysine. Cells were then cultured for 96 hours or 60 hours, with half the medium being changed every two days.
Western blotting
Total protein extracts from cells were prepared and incubated on ice for 45 minutes in lysis buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.0, 1% Triton X-100, 0.1% SDS) containing protease inhibitors (Sigma, UK). Following centrifugation to clarify lysates, protein levels were then quantified using the BCA assay kit (Pierce, UK). Proteins were run on a 10% SDS-PAGE gel and transferred onto a PVDF membrane (Amersham Biosciences, UK). Membranes were blocked in 5% nonfat-dried milk (Sigma) PBS-Triton and probed with anti-HA (Sigma, 1∶1000) or anti-GAPDH (Covance, 1∶1000) antibodies and secondary HRP-conjugated antibody (1∶5000, Invitrogen). Proteins were visualized using ECL reagent according to the manufacturer’s instructions (Amersham Biosciences) using an ImageQuant LAS4000 imaging system and ImageQuant software (GE Healthcare, UK).
Immunocytochemistry
Cells cultured on coverslips were washed, fixed in 4% PFA for 10 minutes, and permeabilised in 0.4% Triton in PBS for 10 minutes. Coverslips were then blocked in 5% milk-PBS for 30 minutes and anti-HA (Sigma, 1∶1000) or NF-200 (Sigma, 1∶200) was incubated overnight at 4°C. Cells were then washed and incubated in secondary fluorescent-conjugated antibody (1∶1000) (Molecular probes, Invitrogen) for 2 hours and mounted in DAPI-containing Vectorshield mounting media (Vectors lab). Axons, visualized by NF-200 staining, were measured using ImageJ software (NIH). A minimum of 15 cortical neurons was quantified. TUNEL staining for apoptotic cells was carried out as per the manufacturer’s protocol (Roche, UK). Confocal LSM510 (Zeiss) or fluorescence (Leica) microscopes were used to acquire images using Axiovision software (Zeiss).
Organotypic Slice Culture
Organotypic slice cultures were performed from electroporated E14 whole embryos as previously described [27], [28]. Briefly, GFP containing vectors (1 µg/µl) with 0.5% Fast Green (Sigma) were injected using a microinjector (Picospritzer) into the lateral ventricle of the whole embryos. Success of DNA injection was determined by the visualization of Fast Green in the ventricular system. Electroporation was then performed on the whole head (skin and skull intact) with gold-coated electrodes (3×5mm GenePaddles, Havard Apparatus) using an electroporator setting (4×100 ms pulses separated by 100 ms long intervals at 55V) (ECM 830 Square Wave Electroporation system, Harvard Apparatus).
Brains were subsequently dissected from the embryos and placed in a dish of ice cold complete HBSS (HBSS 1x, 2.5 mM Hepes, 0.3 mM D-Glucose, 1 mM CaCl2, 1 mM MgSo4, 4 mM NaHCO3 (Sigma/Gibco)), embedded in low melting point agar (Sigma) and sectioned at 250 µm and transferred to Poly-L-Lysine and laminin (Sigma)-pre-coated membrane inserts (BD Biosciences). Brain slices were cultured in slice culture medium (Basal Medium Eagle 0.7x, Complete HBSS 0.25x, 2.7 mM D-glucose, 0.1 mM L-glutamine and pen/strep (Gibco)) for 6 days. The cultured 250 µm slices were fixed in 4% PFA for 12–24 hours and subsequently embedded in low melt agarose gel (Sigma), re-sectioned at 30 µm thickness using a Vibratome, and slide mounted and imaged using a fluorescent microscope (Leica). Images were captured using Axiovision software (Zeiss).
FACS sorting
Cortical cells electroporated on the day of dissociation and cultured for 4 days were washed with PBS and trypsinised (Gibco), scraped off plates and spun at 400×g for 5 minutes and washed twice in 1% BSA (Sigma) in PBS. They were resuspended in 0.5 ml of 1% BSA/PBS solution and GFP positive (GFP+) cells were immediately sorted for size and GFP signal using FACS on a Beckman Coulter MoFlo XPF machine and were subsequently used for RNA purification, microarray analyses and chromatin immunoprecipitation (ChIP) (see below).
Transcriptional profiling
RNA was extracted from FACS sorted electroporated cortical cells using the RNeasy kit (Qiagen). The quality of the extracted RNA was assessed on a 2100 BioAnalyser using the RNA 6000 Pico Assay (Agilent Technologies). The RNA Integrity numbers were >7 for all samples. Fifteen nanograms of RNA from each of the 4 replicates were reverse transcribed, and the cDNA was amplified using the Ovation Pico system (NuGEN) according to manufacturer’s instruction. Two ‘no template’ controls were processed together with the samples, and no significant amplification due to contamination was found. Amplified double-stranded cDNA was transformed into single-stranded sense cDNA using the Ovation Exon Module (NuGEN). Sense cDNA was fragmented and labeled with biotin using the FL-Ovation cDNA Biotin Module V2 (NuGEN). Successful fragmentation was confirmed for all samples on the BioAnalyzer showing a peak sequence length of around 200 nt. Fragmented and labeled single-stranded sense cDNA was hybridized to Affymetrix Mouse Gene 1.0 ST arrays at 45°C overnight. Arrays were washed and then stained using the GeneChip Hybridization, Wash, and Stain Kit (Affymetrix) according to manufacturer’s instruction. The microarrays were scanned on an Affymetric GeneChip Scanner 3000. Data were normalized in GeneSpring (Agilent Technologies) using robust multi-array average (RMA) and PLIER analysis and differentially expressed genes were identified using the Welch t-test with a p-value cutoff of ≤0.05 and fold-change cut off of 1.5.
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA was purified from primary cortical cells using the RNeasy mini kit (Qiagen) and reversed transcribed using Sensiscript reverse transcriptase (Qiagen). Total cDNA was subject to real-time PCR on an ABI PRISM 7000 sequence detection system using SYBR green PCR master mix (Applied Biosystems) and primers are shown in Table S1. The housekeeping gene Gapdh was used as the internal normalising control. Gapdh has been used as a normalising control because its levels were not changed across all conditions tested as seen on our microarray data.
Chromatin immunoprecipitation
The ChIP-IT enzymatic kit (Active Motif) was used per the manufacturer’s instructions. Briefly, cortical cells electroporated with either HA-IRES-GFP or Laf4-HA-IRES-GFP and cultured for 4 days were fixed with 1% formaldehyde for 20 minutes to cross-link DNA-protein interactions. After cross-linking was stopped with glycine, ∼4×106 cells were washed with PBS and scraped in FACS buffer and cells were FACS sorted based on size and GFP signal as described above. Cross-linked chromatin from ∼1–2×105 GFP+-cells was then enzymatically digested to yield 150–200 bp fragments and incubated overnight with magnetic beads plus 3 µg HA antibody (Sigma) or water at 4°C. This was followed by cross-linking reversal, proteinase K digestion, DNA extraction (PCR purification kit; Qiagen), and quantitative PCR (qPCR) with the primer sets listed in Table S1. The specific binding of HA-tagged Laf4 to promoters of Mdga1 and Mdga2 was calculated by subtracting the value of the ‘no antibody’ control and then normalized to the diluted input (1∶10). The percentage input was then calculated.
Statistical Analysis
Prism GraphPad software was used for 1-way and 2-way ANOVA, with Bonferroni’s multiple comparison test when appropriate. Data are presented as mean ± SEM (*p<0.05; **p<0.01; ***p<0.001).
Results
Laf4 expression during embryonic cortical development in the mouse
Previous studies in the mouse have demonstrated that Laf4 expression is developmentally regulated [24]. However, to obtain a more comprehensive picture of the temporal pattern of Laf4 in the embryonic cerebral cortex, a time course of expression analysis was carried out by in situ hybridization using a riboprobe designed to span all major Laf4 transcripts. These data show that Laf4 is detectable in the developing brain and somites at E11.5 as previously described [6] ( Figure 1A ). By E13.5, Laf4 is highly expressed in the developing brain, and as embryogenesis proceeds, expression becomes localized to the subventricular zone (SVZ) and the dorsal cortex by E15.5 ( Figures 1A and B ); importantly, this corresponds to a developmental window during which intermediate progenitor cells in the SVZ proliferate and then subsequently migrate to form the upper layers of the cortex [29]. These data suggest that Laf4 may be important for the maintenance and development of the embryonic cortex, in particular at key stages of neuronal migration and proliferation that are required to generate the correct patterns of cortical layering.
Figure 1. Laf4 is expressed in the developing mouse cortex.
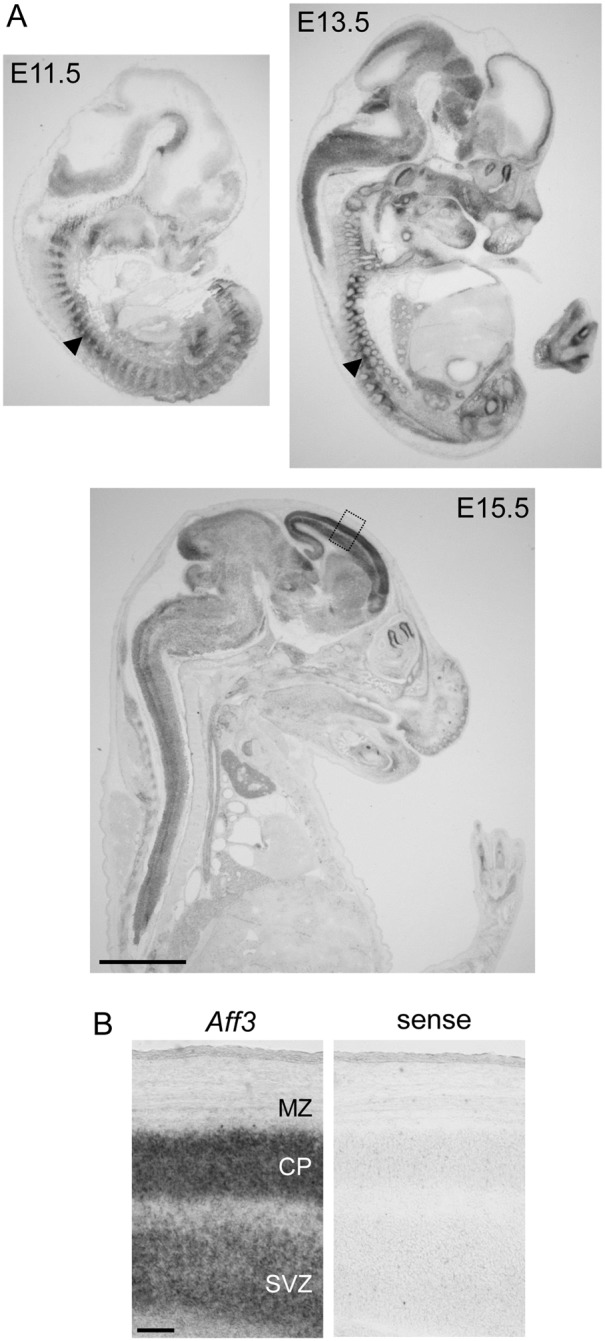
In situ hybridisation of Laf4 in the mouse embryo at E11.5, E13.5 and E15.5 (A). Expression is observed in the brain and somites (arrowheads) at E11.5 and 13.5 (A). By E15.5 expression is observed in the developing cortex below the medial zone (MZ) in the cortical plate (CP) and subventricular zone (SVZ) (B). The boxed region in (A) is shown in (B) with a control sense riboprobe from an adjacent section. Scale bars = 2 mm in (A), 100 µM in (B).
Manipulation of Laf4 expression during cortical development
In order to investigate the potential role of Laf4 in cortical migration, Laf4 expression was manipulated in mouse embryonic brains. Prior to these studies, the selected shRNA construct (shRNA-Laf4) was tested in both N2a and primary cortical cells resulting in a ∼95% or ∼80% knockdown of endogenous Laf4 levels respectively, as quantified by qRT-PCR ( Figures 2B and D ). Importantly, a scramble control shRNA (shRNA-scramble) had no effect on endogenous Laf4 levels ( Figures 2B and D ). The knockdown efficiency of the same shRNA-Laf4 construct was further assessed, demonstrating that even high levels of exogenous Laf4 protein expression (by co-transfection of a Laf4-HA-IRES-GFP construct) could be significantly ablated at the protein level ( Figure 2E ). The specificity of knockdown was also confirmed by co-transfection with a construct that contained a mutated version of Laf4 that would not be recognized by the shRNA (Laf4-rescue-HA-IRES-GFP; Figures 2A–E ).
Figure 2. Knockdown of Laf4 by shRNA.
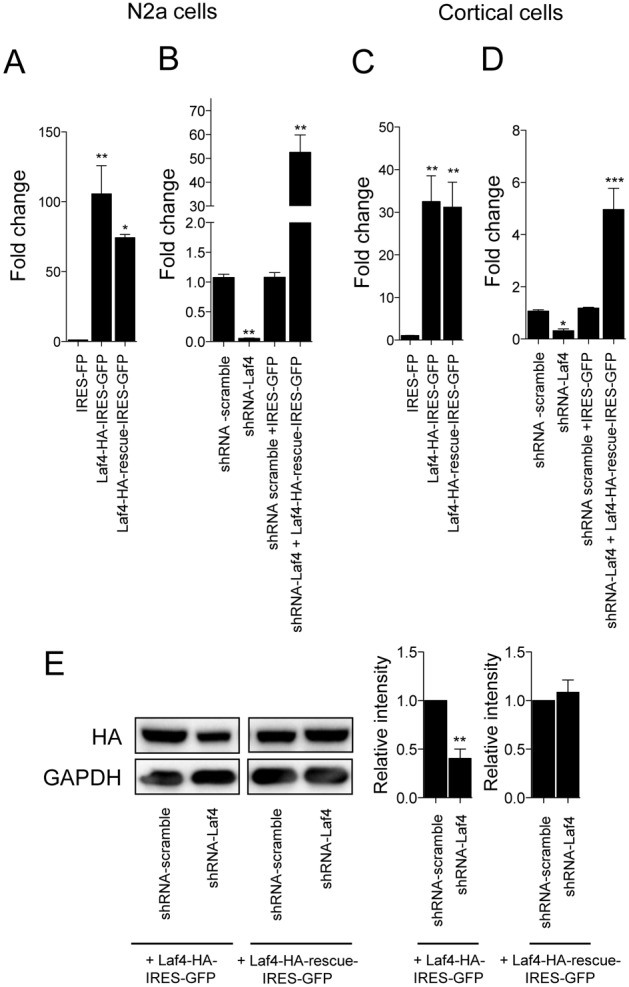
Quantitative RT-PCR of Laf4 in N2a (A and B) and primary cortical cells (C and D). Comparative over-expression of HA-tagged Laf4 (Laf4-HA-IRES-GFP) and the shRNA control (Laf4-HA-rescue-IRES-GFP) occurs at similar levels in both cell types; data are shown versus endogenous Laf4 (negative control HA-IRES-GFP vector) (A and C). Knockdown of endogenous Laf4 in both cell types after transfection (B) or electroporation (D) with shRNA-Laf4 shows the recovery of Laf4 expression using Laf4-HA-rescue-IRES-GFP. (E) Protein levels from over-expression of Laf4 (Laf4-HA-IRES-GFP), but not the control (Laf4-HA-rescue-IRES-GFP) construct is significantly knocked-down by the same shRNA-Laf4 in HeLa cells. The negative control shRNA (shRNA-scramble) has no effect on the levels of either endogenous Laf4 (B and D) or exogenous over-expressed Laf4 (E). In all panels, quantification of three independent repeats in shown (*p<0.05, **p<0.01, ***p<0.001, 1-way ANOVA Bonferroni’s multiple comparison test).
To then study cortical migration, shRNA and control (IRES-GFP) plasmids were injected ex utero into the lateral ventricle of E14.5 whole mouse embryos followed by electroporation and organotypic slice culture from dissected brains. After six days the cortical slices were re-sectioned and the number of GFP+ neurons was counted; the image of each section was divided equally into five bins spanning the cortex using the ventricular and pial borders as anatomical landmarks as previously described ( Figure 3A ) [30]. Embryos electroporated with a negative control scramble shRNA showed a robust migration of GFP+ cells to the cortical pial border as expected ( Figure 3A ). However, a reproducible reduction in migration was observed when the knockdown shRNA-Laf4 construct was expressed ( Figure 3A ); 80% of the cells from knockdown cultures were located in bins 1 and 2 beneath the developing cortical plate, as opposed to a relatively even distribution amongst the layers when the scramble shRNA was electroporated ( Figure 3B ). Importantly, cortical slice cultures co-electroporated with shRNA-Laf4 and Laf4-rescue-HA-IRES-GFP did not show any migration phenotype ( Figures 3A and B ), suggesting that a specific cortical migration deficit is the result of reducing Laf4 expression during early development.
Figure 3. Knockdown of Laf4 causes a defect in the migration of cortical cells.

E14 mouse brains were electroporated with shRNA-Laf4 or shRNA-scramble vectors with a control (IRES-GFP) or the Laf4-rescue-IRES-GFP vector and cultured for six days before re-sectioning for image analysis (A). The average percentage of GFP+ cells in 5 zones (example zones Z1–Z5 shown) spanning from the ventricle (V) to the pial border was quantified (B). Brains electroporated with shRNA-Laf4 construct show a significant reduction in GFP+ cells corresponding to migrating cells to the superficial layers of the cortex; this phenotype is almost completely reversed by co-electroporation with a Laf4-rescue-IRES-GFP construct. (n = 6 brains quantified for each vector combination; *p<0.05, **p<0.01 (2-way ANOVA Bonferroni’s multiple comparison test). Scale bar = 50 µM.
To assess the significance of increasing Laf4 expression at the same point in development, slice cultures were analysed as above from embryos electroporated with the Laf4-HA-IRES-GFP or IRES-GFP constructs ( Figure 4A ). Quantification showed that there was no significant difference in cortical migration ( Figure 4B ), suggesting that the presence of exogenously-expressed Laf4 is not detrimental to cortical development. To further assess the consequences of manipulating Laf4 levels, transfection of over-expression, knockdown and control constructs was carried out in primary cortical culture, showing no alteration in cell survival or axon growth between conditions (Figure S1).
Figure 4. Overexpression of Laf4 does not affect significantly migration of cortical cells.
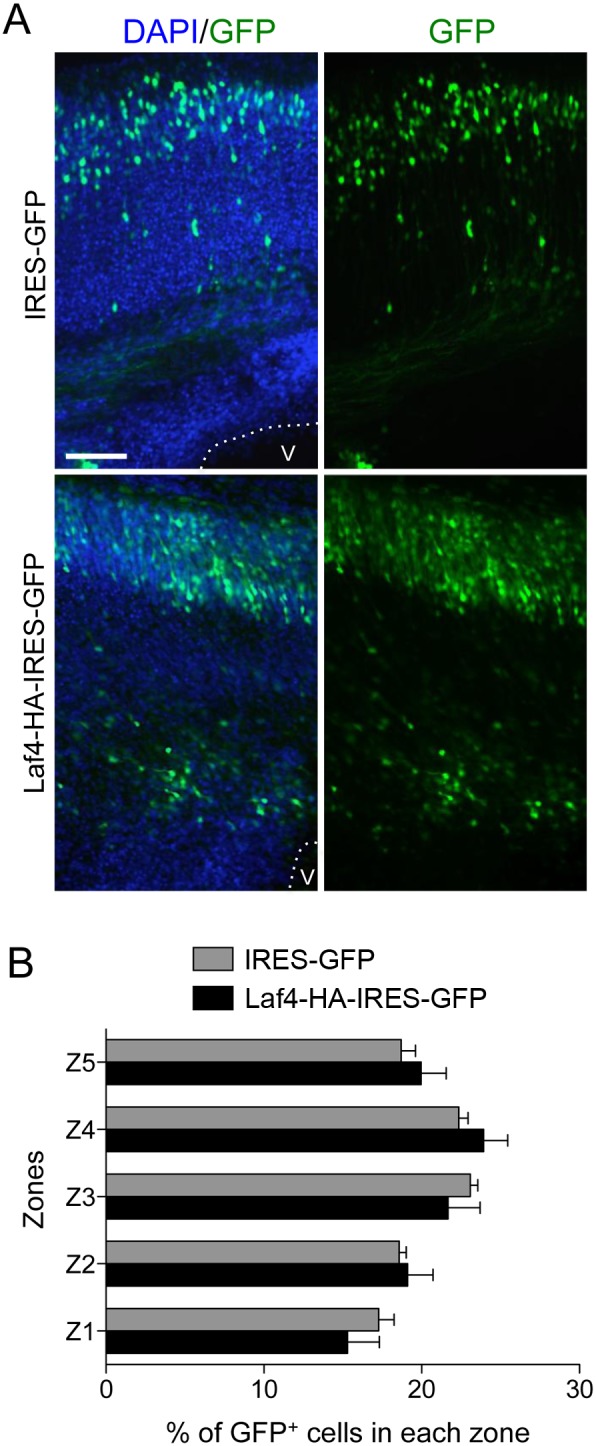
E14 mouse brains were electroporated with Laf4-HA-IRES-GFP or control (HA-IRES-GFP) vectors and cultured for six days before re-sectioning for image analysis (A). The average percentage of GFP+ cells in 5 zones spanning from the ventricle (V) to the pial border was quantified (B) and no significant difference (p = 1.00, 0.27, 0.71, 0.80, 0.63 for Z1 to Z5 respectively) was observed between Laf4-HA-IRES-GFP or the control vector (n = 6 brains quantified for each vector, 2-way ANOVA Bonferroni’s multiple comparison test). Scale bar = 50 µM.
Transcriptional analysis of Laf4 over-expression
Since members of the AFF family are known to act as transcriptional modulators and Laf4 appeared to be influencing cortical development, primary cortical neurons were screened for potential Laf4 target genes related to the observed migration phenotype by expression profiling. It has been shown previously that over-expression of AFF proteins can be used to identify both downstream and direct targets of the AFF transcriptional complex [22]. Therefore, over-expression of Laf4 using the Laf4-HA-IRES-GFP construct was carried out in primary cortical cells and compared with cells expressing the control IRES-GFP vector. To minimize potential non-specific effects of non-transfected cells, FACS sorting was used to select transfected cells on both the basis of size and GFP expression. Transcriptional profiling using Affymetrix microarrays identified a number of genes that were differentially expressed between replicates of Laf4 over-expressing and control cell populations ( Table 1 ). Interestingly, of the transcripts deregulated by more than 1.5-fold, over three-quarters were up-regulated; the most highly over-expressed of these was Aff3/Laf4.
Table 1. Differentially expressed transcripts from Laf4 over-expression in primary cortical cells.
| Gene name | Fold change | P-value | Direction |
| Aff3 | 2.5 | 1.00E-04 | up |
| Gatad1 | 2.0 | 0.00876146 | up |
| Ndufs5 | 2.0 | 0.01630139 | up |
| Snord35a | 1.8 | 0.02089867 | up |
| Zfp780b | 1.8 | 0.03654183 | up |
| Txlng | 1.8 | 0.00685382 | up |
| Txlng | 1.8 | 0.01637254 | up |
| Scn3a | 1.7 | 0.01588485 | up |
| Lemd3 | 1.7 | 0.03688544 | up |
| Rrm2b | 1.7 | 0.01885046 | up |
| Lphn3 | 1.7 | 0.03866969 | up |
| Nop56 | 1.7 | 0.01139775 | up |
| Snord33 | 1.7 | 0.0306814 | up |
| Ptdss1 | 1.6 | 0.00358072 | up |
| Rfesd | 1.6 | 0.02712189 | up |
| Hist1h2bc | 1.6 | 0.04939594 | up |
| Nop16 | 1.6 | 0.02797953 | up |
| Trpm7 | 1.6 | 0.01894537 | up |
| A230046K03Rik | 1.6 | 0.02141088 | up |
| Braf | 1.6 | 0.01453028 | up |
| Samm50 | 1.6 | 0.02693954 | up |
| 1110012L19Rik | 1.6 | 0.0234425 | up |
| Kdm5b | 1.6 | 0.01495683 | up |
| Ercc3 | 1.6 | 0.00651398 | up |
| Mga | 1.6 | 0.00228502 | up |
| Mdga2 | 1.5 | 0.03499631 | up |
| Dcun1d4 | 1.5 | 0.04789757 | up |
| Btaf1 | 1.5 | 0.00433972 | up |
| Gpr137b-ps | 1.5 | 0.01491051 | up |
| Mastl | 1.5 | 0.00317947 | up |
| 4932438A13Rik | 1.5 | 0.01460414 | down |
| Olfr1174-ps | 1.5 | 0.02718545 | up |
| Vps72 | 1.5 | 0.02154237 | up |
| Gm9958 | 1.5 | 0.01058675 | down |
| Gm20091 | 1.5 | 0.00420859 | up |
| Rbbp8 | 1.5 | 0.01891017 | up |
| Gbe1 | 1.5 | 0.03423288 | up |
| Trps1 | 1.5 | 0.01591994 | up |
| 2210408I21Rik | 1.5 | 0.02667212 | up |
| Jhdm1d | 1.5 | 0.02668253 | up |
| Dpy19l4 | 1.5 | 0.01150993 | up |
In order to validate the microarray results, a number of genes were selected for qRT-PCR analysis. Genes were prioritized based on known expression in the brain and in particular overlapping spatial-temporal expression with Laf4. Four genes were selected for further validation: Gatad1, Ndufs5, Mdga2 and Scn3a; independent samples of FACS sorted primary cortical cells were used for these experiments. In agreement with the microarray profiling, all four targets showed up-regulation when Laf4 was over-expressed compared to cells transfected with the control vector ( Figure 5A ).
Figure 5. Identification of Laf4 as a potential transcriptional regulator of Mdga2.
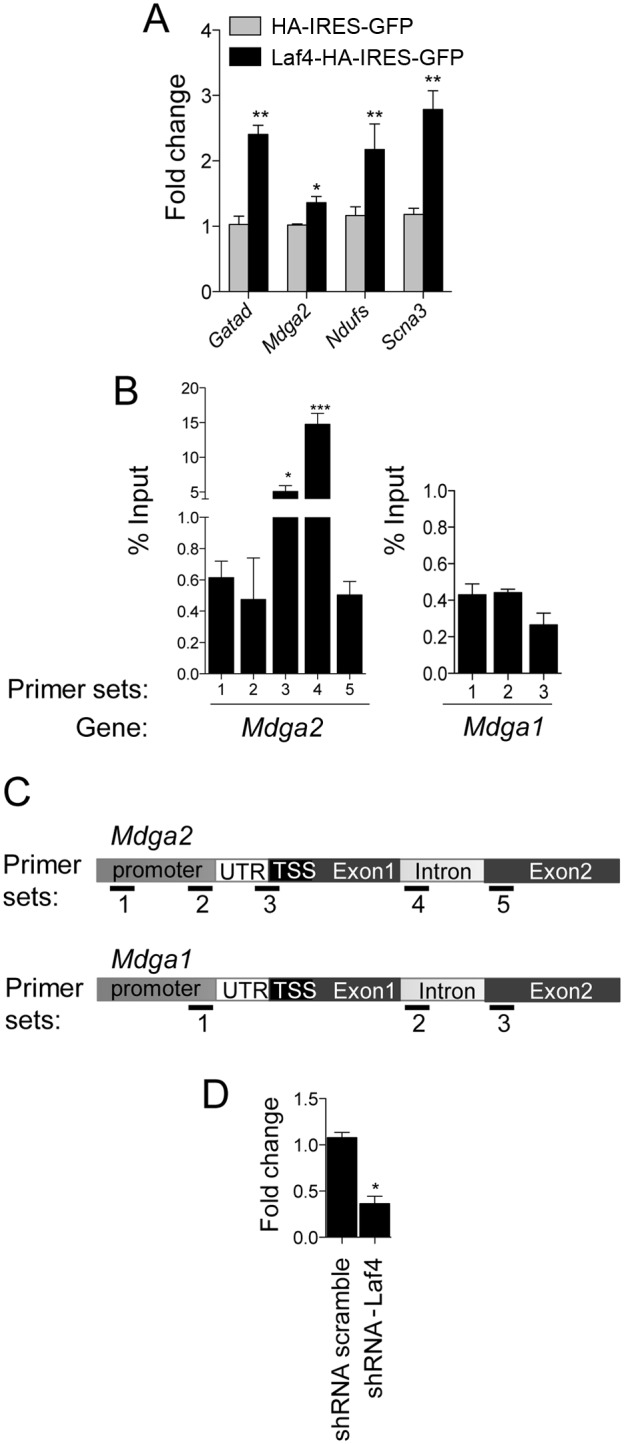
(A) qRT-PCR analysis of four genes originally shown to be up-regulated from microarray transcriptome profiling of FACS-sorted primary cortical cells over-expressing Laf4 (Laf4-HA-IRES-GFP) compared to an empty control (HA-IRES-GFP) vector. Significant up-regulation of Gatad1, Mdga2, Ndufs and Scna3 was confirmed in Laf4 over-expressing cells (n = 3 biological replicates *p<0.05, **p<0.01, 2-way ANOVA Bonferroni’s multiple comparison test). (B) ChIP from cortical cells transfected and FACS-sorted as in (A) was performed using an anti-HA antibody to HA-tagged Laf4-IRES-GFP or empty vector (HA-IRES-GFP). qPCR showed specific binding of Laf4 to Mdga2 at the beginning of the coding region (primer pair 3 and 4 shown in (C)) and at the end of the first exon/beginning of the first intron (primer pair 4). Laf4 did not appear to bind to Mdga1 for the primers tested. (n = 3 biological replicates; *p<0.05, ***p<0.001). 1-way ANOVA Bonferroni’s multiple comparison test used to compare each primer sets to primer 5 (Mdga2) or 3 (Mdga1) used as control primer set. (D) qRT-PCR analysis of Mdga2 levels in cortical cells transfected with shRNA-Laf4 vector (*p<0.01, 1-way ANOVA Bonferroni’s multiple comparison test).
Mdga2 is a potential direct target of Laf4
Of the confirmed microarray targets, Mdga2 (MAM domain containing glycosylphosphatidylinositol anchor 2) is a cell adhesion molecule from an immunoglobulin superfamily that is particularly relevant to cortical development. Mdga2 is highly expressed in the developing rodent brain and has been highlighted as an important factor for normal outgrowth of commissural interneurons in the chick [31]. To determine whether Laf4 or a Laf4-containing complex is able to bind in or around the Mdga2 locus as a potential direct transcriptional regulator [22], ChIP combined with qPCR analysis was performed in primary cortical cells electroporated with either HA-IRES-GFP or Laf4-HA-IRES-GFP. The results indicate that Laf4 binds to the Mdga2 locus, particularly towards the transcription-start site (TSS) and the end of exon 1 ( Figures 5B and C ); a significant increase in Laf4 occupancy was observed from primary cortical cells electroporated with Laf4-HA-IRES-GFP versus a control HA-IRES-GFP construct. We also tested the binding of Laf4 on the Mdga1 promoter, a gene closely related to Mdga2. However, for the primers tested, no binding of Laf4 was detected suggesting that Laf4 might bind specifically to the Mdga2 promoter ( Figure 5B–C ). These data suggest that a Laf4-containing transcriptional complex is able to specifically bind to the Mdga2 locus to regulate its expression. To further analyse the transcriptional relationship between Laf4 and Mdga2, we carried out a reciprocal experiment whereby five independent shRNAs specific to either Laf4 3′-UTR (shRNA Laf4 #08) or various exons (shRNA Laf4 #09 to #12) were tested in N2a cells. In line with the results above, a 50–70% knockdown of endogenous Laf4 resulted in a corresponding 50–60% reduction in Mdga2 expression (Figure S2). In addition, using the shRNA used for the migration assays (shRNA-Laf4) in N2a cells, a 74.5% reduction in Mdga2 expression results from 95.2% knockdown of Laf4 (Figure S2). These data suggest that even the predicted 50% loss of LAF4 expression in humans, either from heterozygous deletion or CCG repeat silencing, is sufficient to influence the transcription of Laf4 target genes.
Over-expression of Mdga2 rescues the Laf4 knockdown-related migration phenotype
As Mdga2 is known to play an important role in neuronal outgrowth and may be regulated by Laf4, it was investigated whether increasing the levels of Mdga2 might influence the migration phenotype observed using Laf4 knockdown. Using the same method as above ( Figure 3 ), cortical slice cultures were co-electroporated with shRNA-Laf4 and Mdga2-IRES-GFP ( Figure 6A–B ). Importantly, these cultures displayed a phenotype almost identical to normal control slices, whereas those electroporated with shRNA-Laf4 alone still showed the stalled migration pattern as observed previously ( Figures 6A and 3A ). This suggests that over-expression of Mdga2 can alleviate the neuronal migration deficit caused by Laf4 knockdown. Therefore not only do these data support a role for Mdga2 in migration, but also provide evidence that regulation of Mdga2 by Laf4 may be important in this process.
Figure 6. Over-expression of Mdga2 rescues the deficit of cortical cells migration generated by Laf4 knockdown.
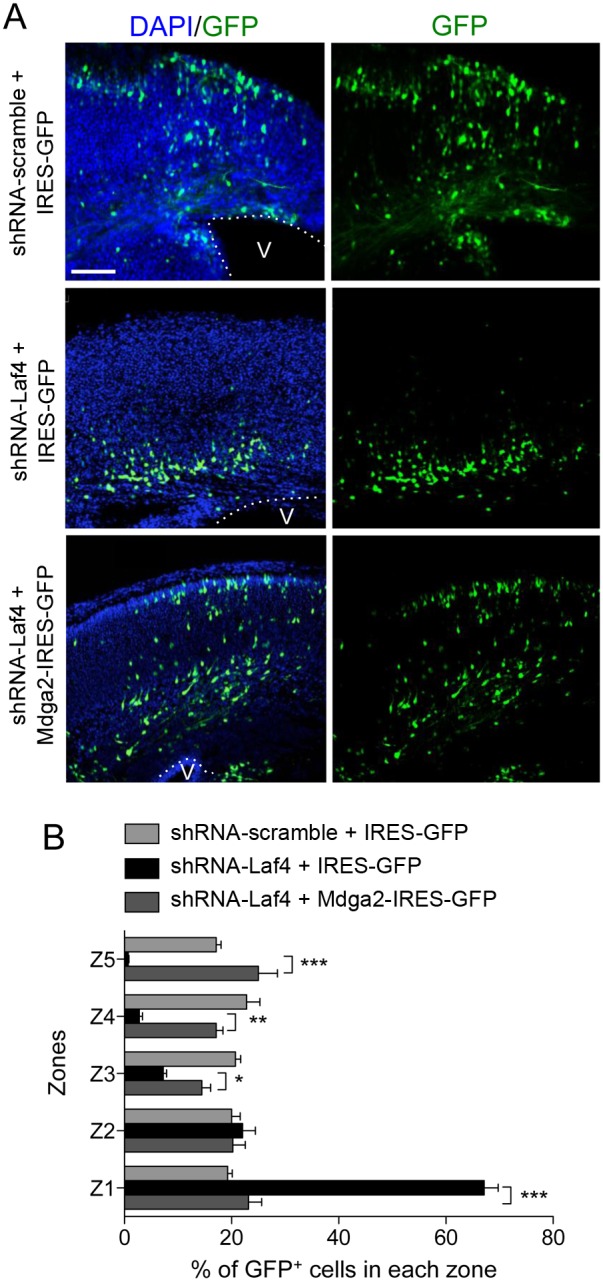
(A) E14 mouse brains were electroporated with shRNA-Laf4 or shRNA-scramble vectors with the control IRES-GFP or an Mdga2-IRES-GFP vector and cultured for six days before re-sectioning for image analysis. The average percentage of GFP+ cells in 5 zones spanning from the ventricle (V) to the pial border was quantified (B). Co-electroporation of shRNA-Laf4 and Mdga2-IRES-GFP constructs results in rescue of the migration defect observed using shRNA-Laf4 alone. (n = 4 brains quantified for each vector combination; *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA Bonferroni’s multiple comparison test).
Discussion
Laf4 is the first AFF member identified as a regulator of cerebral cortex development
Although AF4, AF5Q31 and LAF4 are highly expressed in human embryonic brains [8], [25], a functional role for these genes during neurodevelopment has not been described. We showed here that Laf4 is expressed as early as E13.5 in the mouse neocortex and demonstrated a direct function for this gene in migration of cortical neurons. It is noteworthy that the AFF genes Fmr2 and Af4 are also expressed in the mouse cortical plate, but later in development than Laf4 [32]–[34], suggesting that Laf4 may have a specific and unique role at the earlier embryonic time-points studied here. Given the degree of sequence conservation within the ALF family [35], our present study potentially has implications for AFF-associated disorders of the CNS such as FRAXE mental retardation associated with FMR2 [36]–[38]. Interestingly, Fmr2 knockout mice show a very mild phenotype, displaying memory impairment and abnormalities in nociception [11]. While no cortical migration deficits have been noted in these mice, no detailed anatomical analysis was described [11]. However, more recently Fmr2 has been shown to regulate the transcription of Jun, a gene previously implicated in neuronal migration [23], [39]. Therefore in light of these studies and our data, in addition to LAF4 mutations associated with cortical atrophy and ID, there is increasing evidence that AFF proteins play important roles in neurodevelopment.
AFF proteins were originally described as putative transcription factors based on the presence of a conserved transactivation domain [16]; however, only very few transcriptional targets have been confirmed to date [22]. Our study suggests that Mdga2 gene is under the transcriptional control of Laf4. Interestingly, of the genes deregulated in primary cortical cells by more than 1.5-fold from Laf4 over-expression, 84% were up-regulated. This suggests that Laf4 has a stimulatory role on gene transcription, consistent with data obtained from the Af4-containing complex [17], [40]. This is also in accordance with a previous study showing that LAF4, like AF4 and AF5Q31, interacts with AF9/ENL protein, positive transcription elongation factor b (P-TEFb) and histone-H3 methyltransferase DOT1L [41]. In further support of Laf4 function as a transcription regulator, overexpression of Laf4 in cortical cells led to an increase in BTAF1 RNA polymerase II (Btaf1), part of the pre-initiation transcription factor II D complex [42], and Max gene associated (Mga) which binds to the transcription factor Max, a facultative component of the MLL1 complex [43]. This suggests that Laf4 likely acts in collaboration with other transcription factors or chromatin remodellers to control gene transcription.
Mdga2, a direct transcriptional target for Laf4, is important for cortical development
Here, we demonstrate for the first time that Mdga2 is involved in cell migration during the process of cortical layering; indeed Mdga2 over-expression was able to rescue the migration deficit resulting from Laf4 knockdown. It is noteworthy that while Mdga2 over-expression largely rescued the deficit, there was not a complete rescue. Interestingly, Mdga1, Mdga2 closest homologue, is required for neuronal migration [44], [45]. Thus, while Mdga2 plays a major role in Laf4-regulated migration, it may not be the only effector gene.
Recent studies have shown that reduced levels of Mdga2 in open-book preparations from chicken embryos affect rostral growth of commissural axons [31]. Moreover, Mdga2, like Mdga1, binds to neuroligin-2 [46], [47], and binding of Mdga1 to neuroligin-2 inhibits neuroligin-2 synapse-promoting activity leading to reduction of synapse development and synaptic transmission in culture [46], [47]; importantly, neuroligin-2 is expressed in the mouse brain from E18 to P25 [47]. Together, these studies suggest that, in addition to its role in cellular migration, Mdga2 may also affect axon growth and synapse formation in the developing cortex.
Importance of cell migration deregulation in disease
Our current study has shed some important new light on the potential mechanisms underlying the developmental delay and cortical atrophy observed from a heterozygous LAF4 deletion in humans and repeat expansion silencing of the gene associated with ID [5], [6]. The cell migration data presented here may represent a greater degree of LAF4 knockdown than observed in these human conditions; but importantly, testing of additional shRNAs demonstrated that knockdown of Laf4 to levels that might be expected by haploinsufficiency results in alterations in expression of a transcriptional target of Laf4. Consequently, our results suggest that loss of LAF4 expression may have resulted in defective cell migration during early cortical development. Mutations in genes involved in neuronal cell migration have previously been associated with developmental delay and ID in humans [6], [48], [49]. For example, doublecortin is associated with band heterotopia, a condition with severe intellectual disability and epilepsy, and which has been shown to be a consequence of radial migration deficits during neocortex development [50]. Doublecortin is also associated with lissencephaly, a disease presenting severe ID, seizures and brain with reduced gyration [48]. Similarly, mutations in LIS1, a gene involved in radial cortical neuronal migration, have also been associated with lissencephaly [49], [51], [52]. Moreover, in accordance with a role of Mdga2 in cellular migration during brain development, previous large-scale human genetic analyses have associated variations in MDGA genes with schizophrenia, bipolar disorder and autism spectrum disorder [53]–[56]. Taken together, our study demonstrates the key transcriptional role of Laf4 during cortical cell migration that is relevant to the haploinsufficiency and silencing associated with human neurodevelopmental disorders.
Supporting Information
Modulation of Laf4 levels in cortical cells does not affect survival or axon growth. (A–C) Primary cortical cells were electroporated with the constructs indicated and cultured for 60 hours. (A) Cell death as assessed by TUNEL staining showed no significant difference in the number of apoptotic cells detected across all conditions. (B–C) Axons were visualised by NF-200 immunostaining (examples shown in (B)) and quantified, showing that axon length is not affected by levels of Laf4 or Mdga2. Scale bar: 50 µm.
(TIF)
Levels of Mdga2 are correlated with levels of Laf4. (A–B) N2a cells were transfected for 48 hours with one of five additional shRNA constructs against Laf4 (#8–#12) and levels of Laf4 (A) and Mdga2 (B) were quantified by qRT-PCR. Three out of the five (#11 and #12) additional shRNA constructs led to significant decrease of Laf4 endogenous levels (A) and this was correlated with a significant decrease in Mdga2 level, as also observed using the shRNA construct used for the slice culture experiments (shRNA-Laf4) (B). *p<0.05, 2-way ANOVA Bonferroni’s multiple comparison test.
(TIF)
Primers used in this study.
(DOC)
Acknowledgments
We wish to thank Nigel Rust for assistance with FACs analysis.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All data are included within the manuscript.
Funding Statement
This work is supported by the UK Medical Research Council. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, et al. (2009) New insights to the MLL recombinome of acute leukemias. Leukemia 23: 1490–1499. [DOI] [PubMed] [Google Scholar]
- 2. Von Bergh ARM, Beverloo HB, Rombout P, van Wering ER, van Weel MH, et al. (2002) LAF4, an AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia. Genes, Chromosomes and Cancer 35: 92–96. [DOI] [PubMed] [Google Scholar]
- 3. Chinen Y, Taki T, Nishida K, Shimizu D, Okuda T, et al. (2007) Identification of the novel AML1 fusion partner gene, LAF4, a fusion partner of MLL, in childhood T-cell acute lymphoblastic leukemia with t(2;21) (q11;q22) by bubble PCR method for cDNA. Oncogene 27: 2249–2256. [DOI] [PubMed] [Google Scholar]
- 4. To MD, Faseruk SA, Gokgoz N, Pinnaduwage D, Done SJ, et al. (2005) LAF-4 is aberrantly expressed in human breast cancer. International Journal of Cancer 115: 568–574. [DOI] [PubMed] [Google Scholar]
- 5. Steichen-Gersdorf E, Gassner I, Superti-Furga A, Ullmann R, Stricker S, et al. (2008) Triangular tibia with fibular aplasia associated with a microdeletion on 2q11.2 encompassing LAF4. Clinical Genetics 74: 560–565. [DOI] [PubMed] [Google Scholar]
- 6. Metsu S, Rooms L, Rainger J, Taylor MS, Bengani H, et al. (2014) FRA2A is a CGG repeat expansion associated with silencing of AFF3 . PLoS Genet 10: e1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Domer PH, Fakharzadeh SS, Chen CS, Jockel J, Johansen L, et al. (1993) Acute mixed-lineage leukemia t(4;11) (q21;q23) generates an MLL-AF4 fusion product. Proceedings of the National Academy of Sciences 90: 7884–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taki T, Kano H, Taniwaki M, Sako M, Yanagisawa M, et al. (1999) AF5q31, a newly identified AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia with ins(5;11) (q31;q13q23). Proceedings of the National Academy of Sciences 96: 14535–14540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gecz J, Mulley JC (1999) Characterisation and expression of a large, 13.7 kb FMR2 isoform. Eur J Hum Genet 7: 157–162. [DOI] [PubMed] [Google Scholar]
- 10. Gu Y, Shen Y, Gibbs RA, Nelson DL (1996) Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat Genet 13: 109–113. [DOI] [PubMed] [Google Scholar]
- 11. Gu Y, McIlwain KL, Weeber EJ, Yamagata T, Xu B, et al. (2002) Impaired conditioned fear and enhanced long-term potentiation in Fmr2 knock-out mice. The Journal of Neuroscience 22: 2753–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Isnard P, Coré N, Naquet P, Djabali M (2000) Altered lymphoid development in mice deficient for the mAF4 proto-oncogene. Blood 96: 705–710. [PubMed] [Google Scholar]
- 13. Urano A, Endoh M, Wada T, Morikawa Y, Itoh M, et al. (2005) Infertility with defective spermiogenesis in mice lacking AF5q31, the target of chromosomal translocation in human infant leukemia. Molecular and cellular biology 25: 6834–6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Isaacs AM, Oliver PL, Jones EL, Jeans A, Potter A, et al. (2003) A mutation in Af4 is predicted to cause cerebellar ataxia and cataracts in the robotic mouse. The Journal of Neuroscience 23: 1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oliver PL, Bitoun E, Clark J, Jones EL, Davies KE (2004) Mediation of Af4 protein function in the cerebellum by Siah proteins. Proceedings of the National Academy of Sciences of the United States of America 101: 14901–14906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma C, Staudt LM (1996) LAF-4 encodes a lymphoid nuclear protein with transactivation leukemias potential that is homologous to AF-4, the gene fused to MLL in t(4;11). Blood 87: 734–745. [PubMed] [Google Scholar]
- 17. Bitoun E, Oliver PL, Davies KE (2007) The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Human Molecular Genetics 16: 92–106. [DOI] [PubMed] [Google Scholar]
- 18. Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, et al. (2008) H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14: 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mueller D, García-Cuéllar M-P, Bach C, Buhl S, Maethner E, et al. (2009) Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol 7: e1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulze-Gahmen U, Upton H, Birnberg A, Bao K, Chou S, et al.. (2013) The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. eLife 2. [DOI] [PMC free article] [PubMed]
- 21. Melko M, Douguet D, Bensaid M, Zongaro S, Verheggen C, et al. (2011) Functional characterization of the AFF (AF4/FMR2) family of RNA-binding proteins: insights into the molecular pathology of FRAXE intellectual disability. Human Molecular Genetics 20: 1873–1885. [DOI] [PubMed] [Google Scholar]
- 22. Bitoun E, Finelli MJ, Oliver PL, Lee S, Davies KE (2009) AF4 is a critical regulator of the IGF-1 signaling pathway during Purkinje cell development. The Journal of Neuroscience 29: 15366–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Melko M, Nguyen LS, Shaw M, Jolly L, Bardoni B, et al. (2013) Loss of FMR2 further emphasizes the link between deregulation of immediate early response genes FOS and JUN and intellectual disability. Human Molecular Genetics 22: 2984–2991. [DOI] [PubMed] [Google Scholar]
- 24. Britanova O, Lukyanov S, Gruss P, Tarabykin V (2002) The mouse Laf4 gene: exon/intron organization, cDNA sequence, alternative splicing, and expression during central nervous system development. Genomics 80: 31–37. [DOI] [PubMed] [Google Scholar]
- 25. Hiwatari M, Taki T, Taketani T, Taniwaki M, Sugita K, et al. (2003) Fusion of an AF4-related gene, LAF4, to MLL in childhood acute lymphoblastic leukemia with t(2;11) (q11;q23). Oncogene 22: 2851–2855. [DOI] [PubMed] [Google Scholar]
- 26. Fedtsova N, Quina LA, Wang S, Turner EE (2008) Regulation of the development of tectal neurons and their projections by transcription factors Brn3a and Pax7. Developmental Biology 316: 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lickiss T, Cheung AFP, Hutchinson CE, Taylor JSH, Molnár Z (2012) Examining the relationship between early axon growth and transcription factor expression in the developing cerebral cortex. Journal of Anatomy 220: 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stancik EK, Navarro-Quiroga I, Sellke R, Haydar TF (2010) Heterogeneity in ventricular zone neural precursors contributes to neuronal fate diversity in the postnatal neocortex. The Journal of Neuroscience 30: 7028–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Molyneaux BJ, Arlotta P, Menezes JRL, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nature reviews neuroscience 8: 427–437. [DOI] [PubMed] [Google Scholar]
- 30. Bi W, Sapir T, Shchelochkov OA, Zhang F, Withers MA, et al. (2009) Increased LIS1 expression affects human and mouse brain development. Nat Genet 41: 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Joset P, Wacker A, Babey R, Ingold E, Andermatt I, et al. (2011) Rostral growth of commissural axons requires the cell adhesion molecule MDGA2. Neural Development 6: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chakrabarti L, Bristulf J, Foss GS, Davies KE (1998) Expression of the murine homologue of Fmr2 in mouse brain and during development. Human Molecular Genetics 7: 441–448. [DOI] [PubMed] [Google Scholar]
- 33. Gu Y, Nelson DL (2003) FMR2 function: insight from a mouse knockout model. Cytogenet Genome Res 100: 129–139. [DOI] [PubMed] [Google Scholar]
- 34. Vogel T, Gruss P (2009) Expression of Leukaemia associated transcription factor Af9/Mllt3 in the cerebral cortex of the mouse. Gene Expression Patterns 9: 83–93. [DOI] [PubMed] [Google Scholar]
- 35. Bitoun E, Davies K (2005) The robotic mouse: Unravelling the function of AF4 in the cerebellum. The Cerebellum 4: 250–260. [DOI] [PubMed] [Google Scholar]
- 36. Knight SJ, Voelckel MA, Hirst MC, Flannery AV, Moncla A, et al. (1994) Triplet repeat expansion at the FRAXE locus and X-linked mild mental handicap. Am J Hum Genet 55: 81–86. [PMC free article] [PubMed] [Google Scholar]
- 37. Abrams MT, Doheny KF, Mazzocco MMM, Knight SJL, Baumgardner TL, et al. (1997) Cognitive, behavioral, and neuroanatomical assessment of two unrelated male children expressing FRAXE. American Journal of Medical Genetics 74: 73–81. [DOI] [PubMed] [Google Scholar]
- 38. Gecz J, Bielby S, Sutherland GR, Mulley JC (1997) Gene structure and subcellular localization of FMR2, a member of a new family of putative transcription activators. Genomics 44: 201–213. [DOI] [PubMed] [Google Scholar]
- 39. Björkblom B, Padzik A, Mohammad H, Westerlund N, Komulainen E, et al. (2012) c-Jun N-terminal kinase phosphorylation of MARCKSL1 determines actin stability and migration in neurons and in cancer cells. Molecular and cellular biology 32: 3513–3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KSJ, et al. (2011) Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proceedings of the National Academy of Sciences 108: 15751–15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mueller D, Bach C, Zeisig D, Garcia-Cuellar M-P, Monroe S, et al. (2007) A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110: 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scully R, Anderson SF, Chao DM, Wei W, Ye L, et al. (1997) BRCA1 is a component of the RNA polymerase II holoenzyme. Proceedings of the National Academy of Sciences 94: 5605–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, et al. (2005) Physical association and coordinate function of the H3K4 methyltransferase MLL1 and the H4K16 acetyltransferase MOF. Cell 121: 873–885. [DOI] [PubMed] [Google Scholar]
- 44. Takeuchi A, O‚ O’Leary DDM (2006) Radial migration of superficial layer cortical neurons controlled by novel Ig cell adhesion molecule MDGA1. The Journal of Neuroscience 26: 4460–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishikawa T, Gotoh N, Murayama C, Abe T, Iwashita M, et al. (2011) IgSF molecule MDGA1 is involved in radial migration and positioning of a subset of cortical upper-layer neurons. Developmental Dynamics 240: 96–107. [DOI] [PubMed] [Google Scholar]
- 46. Pettem KL, Yokomaku D, Takahashi H, Ge Y, Craig AM (2013) Interaction between autism-linked MDGAs and neuroligins suppresses inhibitory synapse development. The Journal of Cell Biology 200: 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K, Kim Y, Lee S-J, Qiang Y, Lee D, et al.. (2012) MDGAs interact selectively with neuroligin-2 but not other neuroligins to regulate inhibitory synapse development. Proceedings of the National Academy of Sciences. [DOI] [PMC free article] [PubMed]
- 48. Caspi M, Atlas R, Kantor A, Sapir T, Reiner O (2000) Interaction between LIS1 and doublecortin, two lissencephaly gene products. Human Molecular Genetics 9: 2205–2213. [DOI] [PubMed] [Google Scholar]
- 49. Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH (1993) Lissencephaly: A human brain malformation associated with deletion of the lis1 gene located at chromosome 17p13. JAMA 270: 2838–2842. [DOI] [PubMed] [Google Scholar]
- 50. Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, et al. (2003) RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci 6: 1277–1283. [DOI] [PubMed] [Google Scholar]
- 51. Cardoso C, Leventer RJ, Dowling JJ, Ward HL, Chung J, et al. (2002) Clinical and molecular basis of classical lissencephaly: Mutations in the LIS1 gene (PAFAH1B1). Human Mutation 19: 4–15. [DOI] [PubMed] [Google Scholar]
- 52. Tsai J-W, Bremner KH, Vallee RB (2007) Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci 10: 970–979. [DOI] [PubMed] [Google Scholar]
- 53. Kähler AK, Djurovic S, Kulle B, Jönsson EG, Agartz I, et al. (2008) Association analysis of schizophrenia on 18 genes involved in neuronal migration: MDGA1 as a new susceptibility gene. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 147B: 1089–1100. [DOI] [PubMed] [Google Scholar]
- 54. Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, et al. (2009) Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet 5: e1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li J, Liu J, Feng G, Li T, Zhao Q, et al. (2011) The MDGA1 gene confers risk to schizophrenia and bipolar disorder. Schizophrenia research 125: 194–200. [DOI] [PubMed] [Google Scholar]
- 56. Van Den Oord EG, Kuo PH, Hartmann AM, Webb BT, Möller HJ, et al. (2008) Genomewide association analysis followed by a replication study implicates a novel candidate gene for neuroticism. Archives of General Psychiatry 65: 1062–1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Modulation of Laf4 levels in cortical cells does not affect survival or axon growth. (A–C) Primary cortical cells were electroporated with the constructs indicated and cultured for 60 hours. (A) Cell death as assessed by TUNEL staining showed no significant difference in the number of apoptotic cells detected across all conditions. (B–C) Axons were visualised by NF-200 immunostaining (examples shown in (B)) and quantified, showing that axon length is not affected by levels of Laf4 or Mdga2. Scale bar: 50 µm.
(TIF)
Levels of Mdga2 are correlated with levels of Laf4. (A–B) N2a cells were transfected for 48 hours with one of five additional shRNA constructs against Laf4 (#8–#12) and levels of Laf4 (A) and Mdga2 (B) were quantified by qRT-PCR. Three out of the five (#11 and #12) additional shRNA constructs led to significant decrease of Laf4 endogenous levels (A) and this was correlated with a significant decrease in Mdga2 level, as also observed using the shRNA construct used for the slice culture experiments (shRNA-Laf4) (B). *p<0.05, 2-way ANOVA Bonferroni’s multiple comparison test.
(TIF)
Primers used in this study.
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All data are included within the manuscript.