

Abstract
While it is well known that proteins are only marginally stable in their folded states, it is often less well appreciated that most proteins are inherently aggregation-prone in their unfolded or partially unfolded states, and the resulting aggregates can be extremely stable and long-lived. For therapeutic proteins, aggregates are a significant risk factor for deleterious immune responses in patients, and can form via a variety of mechanisms. Controlling aggregation using a mechanistic approach may allow improved design of therapeutic protein stability, as a complement to existing design strategies that target desired protein structures and function. Recent results highlight the importance of balancing protein environment with the inherent aggregation propensities of polypeptide chains.
Keywords: protein aggregation, protein stability, protein interactions, computational design
Controlling aggregation is a key to successful biopharmaceutical products
Protein-based pharmaceuticals are among the fastest growing categories of therapeutic agents in the clinic and as commercial products, and typically target high-impact areas such as various cancers, auto-immune diseases, and metabolic disorders [1,2]. Although commercial scale purification processes typically result in high purity, “monomeric” protein when products are first manufactured, most if not all therapeutic proteins will form net irreversible aggregates over time as products are stored, transported, and/or administered to patients [3,4]. At a minimum, aggregates are an impurity that must stay within product specifications so as to meet the requirements of regulatory agencies – typically, these are small amounts of aggregate on a mass percentage basis (e.g., only a few percent of the total protein material, with minimal changes over the time scale of a year or more). However, more recently there has been growing concern and evidence that the presence of aggregated proteins (even humanized or fully human proteins) can significantly increase the risk that a patient will develop an immune response to the active protein monomer. This can result in the patient becoming “immune” to the drug – i.e., the drug no longer is effective for the patient – and in rare cases it may also cause serious safety issues [5]. Becoming drug-tolerant is a serious problem, as many of these drugs treat chronic, life-altering or life-threating illnesses, and there are not typically many or any competitor products available for patients that are resistant to a given drug. In extreme and rare cases, patients have developed an immunity to their own endogenous proteins, as in the case of pure red cell aplasia [6].
It remains unclear and somewhat controversial whether all aggregate types are potentially immunogenic, and how additional clinical factors can affect immunogenicity [5,7–9]. As a result, it is not known a priori which aggregate species are most important to control. Therefore, regulatory agencies must err on the side of caution and require that the properties and amount of all detectable aggregate species be well controlled, reproducible, and monitored experimentally in therapeutic protein products [8–10]. A large amount of time, effort, and financial resources can be expended to address these factors during development of protein-based therapeutics for clinical trials and commercial manufacturing. A better understanding of the molecular nature of how different aggregates form, which aspects of the protein molecules and their sample environments mitigate this process, and whether or how aggregation can be prevented altogether, could significantly reduce the time to market, cost to patient or insurer, and potential immunogenicity of future therapeutic products.
Protein aggregation (sometimes referred to as non-native aggregation) denotes the process(es) by which protein molecules assemble into stable complexes composed of two or more proteins, with the individual proteins denoted as the monomer. The monomer could be composed of a single folded chain, multiple protein chains that are disulfide bonded to one another – such as with monoclonal antibodies (MAb), or a natively multimeric complex. Aggregates are often held together by strong non-covalent contacts, and require some degree of conformational distortion (unfolding or misfolding) in order to present key stretches of amino acids that form the strong contacts between monomers (Figure 1). This type of aggregation is very difficult to reverse (e.g., the aggregates do not dissociate to a significant degree upon dilution or shifts in pH). Aggregation of this kind is distinct from protein assembly in the context of protein crystallization [11] and protein-protein binding that involves stoichiometric complexes of folded proteins[12].
Figure 1.
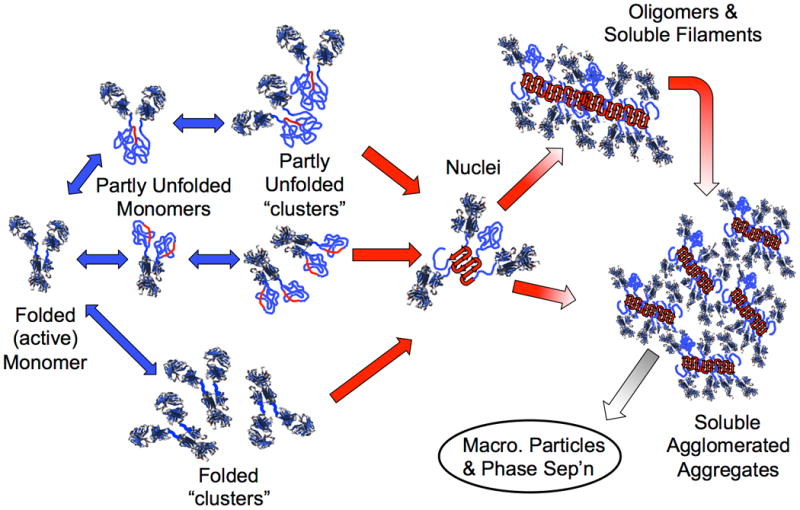
Schematic diagram illustrating multiple non-native aggregation pathways for a multi-domain protein such as a monoclonal antibody composed of a single Fc fragment and two identical Fab fragments. Red strands denote “hot spot” sequences that are prone to form strong, effectively irreversible inter-protein contacts that stabilize aggregates, but are primarily hidden or buried in fully folded monomers. Double-arrows denote effectively reversible steps. Single arrows denote irreversible steps. Nuclei are defined as the smallest net-irreversible aggregate size; growth from nuclei to form soluble aggregates spanning length scales on the order of 10 to 102 nm occurs primarily via addition of other partly unfolded monomers (upper right) or by the agglomeration of existing aggregates (lower right) [19,25,39,40]. Aggregates can become visible to the naked eye if they are sufficiently large and/or undergo phase separation [87–89]. If unfolding / aggregation is mediated by protein adsorption to bulk interfaces [90–92], and/or chemical changes such as deamidation [93–95], oxidation and other reactions [96,97], or fragmentation [98,99], then additional steps may also be kinetically important in the possible aggregation mechanism(s).
From an immunogenicity perspective, the key features of protien aggregates are: that they do not easily dissociate in vivo (aggregation is effectively irreversible), and that they retain some fraction of their original folded secondary and/or tertiary structure. The combination of these features contributes to making aggregates more prone to elicit an immune response when compared to the parent monomer [7,13,14], although it is anticipated that even the parent monomer could be immunogenic for some choices of human sequences [15]. Protein-based vaccines offer a useful example of these principles when immunogenicity is desirable: they are much more effective at eliciting an immune response when they are presented as many proteins assembled on an adjuvant particle that is similar in size to protein aggregates; conversely, the free monomer protein elicits little or no immune response [16].
A mechanistic view of protein aggregation can help to motivate current and recently developed strategies to control the aggregation processes, and provide context for next-generation approaches based on engineering new proteins or redesigning existing proteins to imbue them with greater aggregation resistance. The aggregation propensity and behavior of a given protein is also dependent on its environment, i.e. the solution pH, ionic strength, concentration of co-solutes, and exposure to different bulk liquid-fluid and liquid-solid interfaces. There are inherent difficulties in achieving active and properly folded protein therapeutics, while also balancing their inherent propensities to form aggregates. This review presents how and why a mechanistic approach to design and control of aggregation can be valuable, as well as highlighting areas for improvement. It also outlines some principles that can be useful for improving protein stability in a final product, and that aid in selecting design metrics during discovery stages when different protein candidates and/or classes are being evaluated.
Mechanisms dictate formation rates and key characteristics of aggregates
Therapeutic proteins, such as MAbs, can form different types of aggregates (Figure 1). A folded MAb contains two identical Fab (antigen binding) regions or domains, and one Fc (conserved) region or domain. The Fab domains contain the amino acid sequences that determine the specificity and affinity of a given MAb to a given epitope. Some degree of conformational distortion or “misfolding” is typically needed to allow highly aggregation-prone stretches of amino acids (so called “hot spots”) to become exposed and available to form strong inter-protein contacts between the proteins that make up an aggregate.
Aggregation-prone sequences may exist in more than one place within the same protein [17]. These may become exposed transiently via local or partial unfolding (structural distortion) of an initially folded monomer, or if one first forms small, reversibly folded oligomers or “clusters” [18]. Under conditions in which the folded state is favored over the unfolded states, monomers can initially self-associate reversibly (Figure 1, left) either as folded or partially unfolded species. These early stages are reversible, at least putatively, because there are kinetic bottlenecks that allow the hot-spot sequences to sample conformations that enable strong interactions between chains of adjacent proteins [19]. The forces that drive folding of an isolated protein molecule are also present when two or more protein chains interact with one another. The same forces that drive folding also drive aggregation, and as a result, “hot spot” sequences tend to be stretches of amino acids that are highly hydrophobic, lack charges, and are prone to form beta sheets when paired with adjacent strands (Box 1) [20].
Box 1. Folding and aggregation are driven by the same forces.
The molecular scale interactions that dictate the folded structure and stability for a given sequence of amino acids include: (i) geometric constraints due to chemical bonds and avoidance of steric overlaps, (ii) loss or gain of chain entropy due to folding or unfolding, (iii) electrostatic repulsions between amino acids with the same charge (positive or negative), (iv) electrostatic attractions between amino acids with opposite charges, (v) van der Waals interactions (relative to those with solvent) between backbone and side chain atoms, (vi) hydrogen bonding (mostly between backbone atoms), (vii) hydrophobic attractions between nonpolar side chains. Contributions (i) to (iii) resist folding, whereas contributions (iv) to (vii) promote folding. The delicate balance of these large, competing contributions leads to a relatively small number of possible molecular configurations that will provide a structured, folded state that is significantly lower in free energy than the ensemble of significantly unfolded structures. This is a fundamental basis for why protein structures are dynamic, and there is constant sampling between folded and (partially) unfolded structures, even under conditions where the folded state is predominant [84].
However, many of the same forces that must be in balance to ultimately allow folding of a single, effectively isolated, protein chain must also be present between two or more protein chains once one considers finite protein concentrations. The balance of interactions (i) to (vii) is difficult to achieve, and only a small number of different secondary structure motifs are observed to be significantly stable in folded proteins. In rough terms, these can be grouped into helices and different beta-sheet motifs. Helices are stabilized by contacts that require amino acids to be near each other in the polypeptide sequence (e.g., hydrogen bonding occurs between the backbone atoms on residue n and n ± 4, for the amino acids in an α-helix). Sheets form from strands of amino acids that are distant from one another in the amino acid sequence, and are stabilized by (iv) to (vii). For an unfolded protein, strands from different proteins can be stabilized by forming sheets between proteins, presumably while paying a smaller entropic penalty than forming sheets within the same protein, and are empirically observed to be the predominant secondary structural motif that occurs in stable aggregates. A common example for polypeptides and natively unfolded (or intrinsically disordered) proteins is that of amyloid.
Experimentally, the rate-limiting steps for aggregation typically occur after unfolding, except in the case of extreme temperatures, in which unfolding is rate-limiting. As such, experimental measurements often cannot qualitatively distinguish between unfolding before association and association before unfolding [19]. The former is supported by indirect assessments in which monomer conformational stability explains the temperature dependence of aggregation quantitatively or semi-quantitiatively [21–24]. The latter is supported by indirect assessments such as correlations of aggregation rates with the solubility (or chemical potential) of folded monomer [18].
The smallest irreversible aggregate species are sometimes termed “nuclei”, by analogy with nucleated polymerization or nucleated phase transitions. Such analogies to nucleation-and-growth processes imply that subsequent growth stages (Figure 1, right) are much faster than the creation of new nuclei [19]. However, in practice, aggregates do not always grow to macroscopic length scales (> 10 to 102 microns), and instead remain as soluble oligomers and multimers [18,25,26]. In either case, irreversible aggregation involves multiple events, each of which are thermodynamically and/or kinetically unfavorable under most solution conditions that are selected for manufacturing, storage, and administration of therapeutic proteins. As a result, the net rates of aggregate creation are rather low, with target product shelf lives on the order of two or more years in most cases [4,27].
The overall process of aggregation for proteins in this context is not under thermodynamic control, and is therefore path-dependent. Steps that are “upstream” of the rate-limiting steps will pre-equilibrate, and the relevant parameters for those upstream steps can be determined thermodynamically [21,28–30]. However, the identities of the species that are ultimately formed “downstream” depend on the irreversible path or paths that dominate for a given sample condition, as well as history and lifetime of the sample. Ultimately, a combination of the sample conditions and the protein sequence and structure determines what types of aggregates are formed [25,31,32].
Aggregated proteins are expected to be more immunogenic than their parent monomers. It is currently speculated or inferred from animal studies that the severity of immune responses also depends on the native-like residual structure in aggregates and the size of the aggregates, which indicates the number of repeating epitopes that are presented [13,33]. As such, aggregates should not be conceptually “lumped” into a composite pool, but need to be scrutinized for their differences in size, local or residual folded structure in the constituent monomers, and their “morphology.”. The term morphology refers to the three dimensional geometry of how the monomers are “packed”, such as whether the aggregates are linear or branched polymers, rodlike or globular assemblies, etc. Portions of a given aggregate species could be more or less “ordered” in terms of local packing or secondary structure. Therefore, unlike for aggregates of small peptides or intrinsically disordered polypeptides, denoting aggregates of initially folded proteins as simply “ordered” or “amorphous” would neglect their molecular-scale structural features [34].
Which mechanisms prevail?
Multiple aggregation pathways exist and can even occur in parallel: growth can occur by monomer addition, aggregate agglomeration, or phase separation, each leading to different types of aggregates (Box 2). Unfortunately, it is not yet possible to predict which pathways will be most prevalent for a given protein and choice of sample conditions. One could argue that the “intrinsic” aggregation propensity (IAP) of protein would be that of its fully unfolded amino acid sequence. Statistical algorithms exist to rank stretches of amino acids by similarity to aggregation prone sequences in polypeptide databases. However, it is very difficult to predict or detect whether such “hot spot” sequences are actually exposed in real proteins unless they exist in solvent-exposed loops or on the surface of folded proteins.
Box 2. Colloidal electrostatic interactions influence on unfolding and aggregation.
Electrostatic interactions between two charged molecules or side chains is fundamentally given by Coulomb's law, Eij ∼ qiqj / rij, with Eij denoting the magnitude and sign of the interaction between moieties i and j (Eij > for repulsive interactions, and vice versa), qi and qj are the respective charges on i and j, and rij is the center-to-center distance between them. In a solution containing dissolved salt(s) and/or buffer species, Eij is modified due to net electric field provided by all of the positively and negatively charged species in solution that surround charges i and j. To a first approximation, Eij is proportional to qiqj exp(-κrij) / rij. There is a “screening length”, 1/κ, that dictates the length scale over which charges i and j can feel electrostatic attractions or repulsions (more precisely, how quickly the magnitude of Eij decays as the separation between i and j increases). For a given choice of salt (e.g., NaCl or KCl), the magnitude of κ increases as salt concentration increases [85,86].
Changing pH changes the net charge and charge distribution on a protein and this changes the net inter-protein and intra-molecular repulsions or attractions. To a first approximation, this does not alter the non-electrostatic attractions between proteins or within proteins due to favorable van der Waals contacts and hydrophobic attractions. Therefore, decreasing the net charge on the proteins ultimately can result in strong attractions. Greater charge-charge repulsions within a protein can increase the unfolded population(s) because the charges are able to maintain larger rij values in the unfolded state(s). The aggregation “hot spots” are so strongly attractive (e.g. due to hydrophobic interactions) that they are still able to bind and form stable nuclei, albeit perhaps more slowly because of the higher charge-charge repulsions between proteins
Compared to monomer-monomer interactions, the monomer-aggregate and aggregate-aggregate repulsions are even greater because a protein in an aggregate α feels the repulsive charges from multiple proteins in its neighboring aggregate β, and to a lesser extent a monomer trying to add onto an existing aggregate α or β feels those additional repulsive forces. The magnitude of this effect depends on both net charge and κ, and this causes aggregate growth to be suppressed at sufficiently high net charge and κ. It also causes growth by monomer addition, followed by growth by aggregate-aggregate coalescence, to become prevalent as net charge or κ decrease. The figure here schematically illustrates the effects described above.
Box 2 – Figure I.
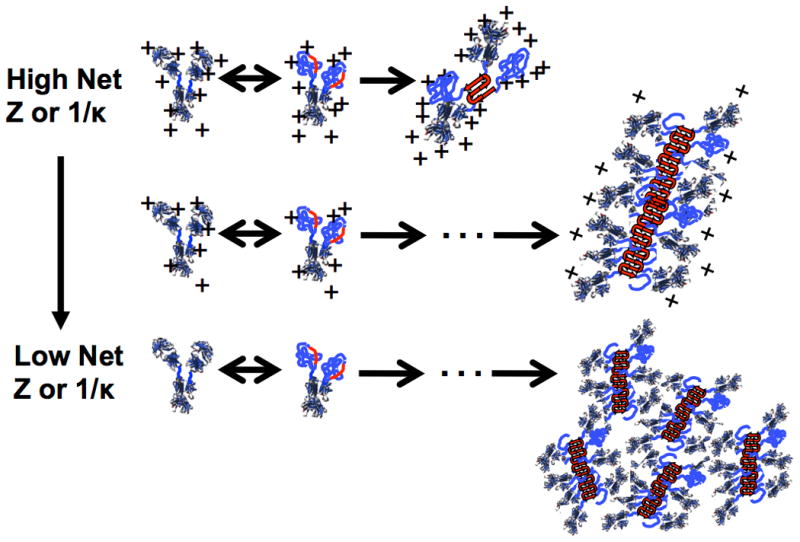
Aggregation propensity based on net charge and screening length.
If one focuses on the interactions between proteins with exposed “hot spots”, then changes in sequence that alter those “hot spots”, or other amino acids (e.g. charged residues) that cause inter-protein repulsions, could alter IAP [19,35]. As such, the IAP of a protein is an inherent function of its primary structure and the residual secondary and tertiary structure in its aggregation-prone intermediates [19]. The interactions between “hot spots” on neighboring proteins may be mitigated or eliminated altogether by altering the sequence if the hot spots can be identified correctly [35–37]. In general, there can be multiple “hot spots” within the same protein, and the structural identity of the key partially-unfolding monomer intermediates that participate in aggregation may change with solution conditions (e.g. pH, ionic strength, temperature, pressure, dissolved co-solutes, etc.) [17,38].
The solution environment, adsorption to bulk interfaces, or chemical degradation can alter the concentrations of intermediates and the interactions between them. There is therefore no sole mechanism for aggregation. Although the range of possible aggregation pathways has not been elucidated experimentally for many proteins, it has been mapped out to a reasonable extent for at least one MAb [25], and a globular protein under analogous solution conditions [31]. Based on these examples, as one increases the pH far above the pI of a protein, and/or reduces the ionic strength of the solution, protein aggregates do not grow as easily, and vice-versa (Figure 2), consistent with results from a number of MAb systems [25,32,39–44], an eye lens crystallin [36,45], an aggregation-prone cytokine [46,22], and model globular proteins [34,31].
Figure 2.
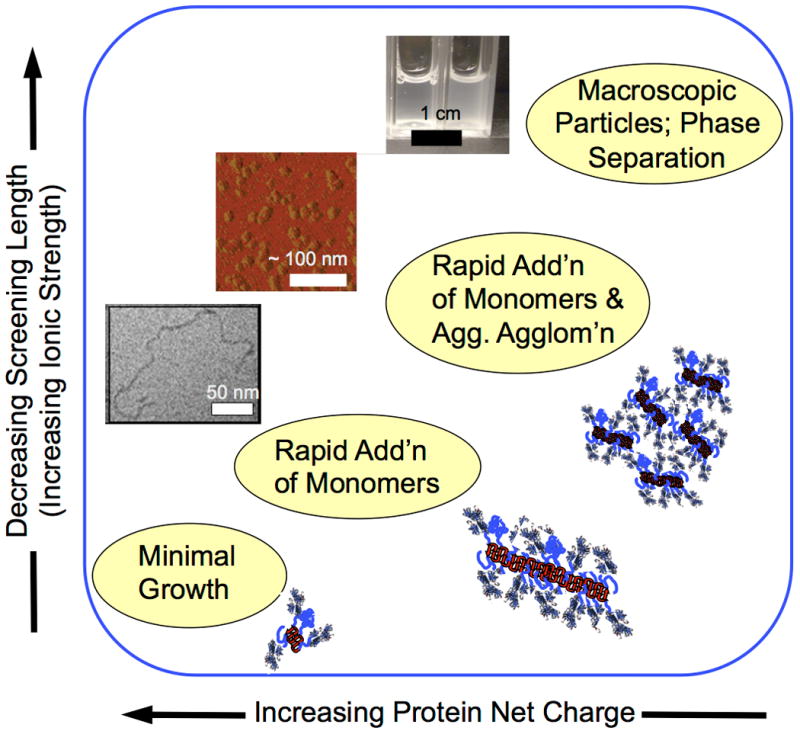
Schematic illustrating shifts in aggregation mechanisms and examples of aggregates that do not grow easily (see also Box 2), to those grown by monomer addition (lower left image, from [46]), aggregate agglomeration to form globular aggregates (middle image, from [44]), and aggregate phase separation to form macroscopic particles (upper image, from [87]].
Aggregation still occurs upon unfolding. The electrostatic repulsions are not sufficient to completely stop the “hot spot” regions from forming strong contacts, because those “hot spots” are often highly hydrophobic stretches of amino acids. This highlights a balance between the portions of the protein that drive aggregation and those that resist it (or at least slow its progression). In some cases, aggregation does not proceed beyond small oligomers, possibly because there is a monomer-oligomer equilibrium that is mediated by electrostatic repulsions, and specific salt effects [25,40,42,43]. Moving from the lower left to the upper right in Figure 2, electrostatic repulsions become weaker and short-ranged. Although growth occurs initially via addition of aggregation-prone monomers, subsequent growth becomes dominated by aggregate-aggregate coalescence and even phase separation of the previously formed aggregates [25,31,32,40,43].
Phase-separation or precipitation of proteins can also occur without a significant conformational change (e.g., “salted out” native aggregates). Such aggregates are distinct from the species of interest here because phase-separation and precipitation can be reversible upon dilution or shifts in solution conditions. Phase-separated or precipitated proteins are better considered in the context of a reversible amorphous solid-liquid or liquid-liquid transition [47]. Experimentally, precipitation of folded monomers is often accelerated by cooling. This is opposite to what is expected for non-native aggregation, although cooling does lead to higher aggregation rates or greater precipitation for non-native aggregation in some cases [47–49]. More generally, when solution conditions change, the chemical properties of unfolded species may change, leading to aggregation by different mechanisms than in previous conditions. Additionally, the overall aggregation rates at a given temperature can change dramatically as the net charge of the proteins decrease (Figure 2). This occurs because of the altered conformational stability of the parent monomer (Box 2). As a result, the net effect on aggregation rates can be a complex function of pH and ionic strength [17,25,31,41].
Designing aggregation resistance
Multiple factors must be accounted for when attempting to prevent (through protein design or controlling solution conditions) certain or all types of aggregates from forming on practical time scales. The choice of solution conditions and the material for the sample container are part of how a given protein is “formulated” to optimize its stability, along with other drug-product properties, and have been reviewed extensively elsewhere [3,4,10,50]. This section focuses on protein design or engineering through the alteration of protein sequence and structure by mutagenesis. Protein design is much more complex than simply eliminating aggregation-prone sequences (Box 1). For example, simply eliminating large stretches of hydrophobic amino acids from a folded will likely also disrupt folding. As such, there must be a balance between ameliorating aggregation and ensuring proper folding.
Engineering or alteration of “wild-type” protein sequences is a long-standing strategy when considering design targets such as improved or altered protein-ligand binding, enzyme folding, stability, and activity, and in vivo folding and expression levels for recombinant proteins [51–58]. Introducing changes in the amino acid sequence to influence key steps for in vitro aggregate formation of therapeutic proteins has received more attention only in recent years [36,59–68]. Most approaches implement one of a few main strategies: (i) experimental screening techniques that can empirically test large protein libraries to identify candidate proteins that are less prone to misfold or aggregate (e.g., when exposed to heat stress), (ii) knowledge-based approaches such as comparing structures and sequences of analogous proteins from different organisms that have evolved to tolerate stresses that promote aggregation, (iii) applying mechanistic knowledge of which relevant physical and chemical factors may predispose a given protein to aggregate. The former two strategies are informatic in nature, and do not rely on understanding the underlying physical mechanisms that mediate aggregation. As such, they also cannot easily determine “why” a given protein variant is more or less aggregation-prone. In contrast, the third strategy offers a means to address key aspects of protein design to control both aggregation rates and the properties of aggregates when they do form.
There are many different experimental and computational methods for searching through the extremely large number of possible mutations one could select for a given starting protein sequence. Consistent with the stages involved in “nucleating” new aggregates, approaches for searching the protein “design space” can be categorized as those for identifying sequences that either (i) lower the free energy of the folded state to decrease the populations of unfolded monomers that have exposed “hot spot” regions for aggregation, (ii) make relatively non-specific interactions between folded proteins or unfolded proteins more repulsive, and interactions between proteins and solvent more favorable, or (iii) reduce or eliminate “hot spots” that are inferred to have highly specific, strong interactions between polypeptide chains.
Each of the different approaches has met with some degree of success. Examples include: cellular libraries that have identified proteins that are more resistant to aggregation upon heat stress [66,69], screening of protein expression levels as a metric for aggregation propensity [70], systematic enumeration of variable-domain sequences in antibody fragments to improve heat-stress stability [53,61], computational algorithms to “supercharge” a protein surface without sacrificing folding stability [60], structure-based approaches to improve domain-domain interfaces of multi-domain proteins so as to drive (re)folding over aggregation [71–73], and a priori computational design to improve folding and/or binding [36,74,75] or reduce “hot spots” [36,62,76]. Some of these examples are qualitatively or quantitatively mechanistic in nature, whereas others are phenomenological or empirical screens.
Given that this field is much less developed than those that focus on protein function or folding, this review does not advocate for any particular mechanism-guided strategy, but rather stresses the importance of adopting such strategies in the future. Using approaches that only focus on elimination of insoluble or precipitated aggregates, or “amyloid” aggregates, are of limited utility in the context of pharmaceutical proteins. Insoluble or amyloid aggregation are extreme conditions that belie the importance of smaller aggregates and so-called sub-visible particles that current evidence indicates are of greater concern for immunogenicity in animal models [5]. This is perhaps more a question of how one implements mechanistic design, because such implementation requires more than a reliance on simple or “lumped” assays such as turbidity or dye-binding [77]. If one does not test experimentally how changes in protein sequence or environment affect different mechanistic stages of aggregation, then it is not possible to critique current or new design criteria that target a given step or steps in the aggregation mechanism. There is therefore room for improvements in both protein design and subsequent experimental characterization.
In the large majority of cases where mechanism-inspired or mechanism-guided design has been tried, the implicit or explicit assumption is that the mutations in question primarily affect only one of the steps involved in aggregation. While this may be the case, few reported studies have actually experimentally confirmed which stages in the aggregation process were qualitatively or quantitatively altered by a given mutation. Common practice is to create a given protein variant or set of variants, and then perform a “stress test” on them to check if experimental estimates of aggregation rate or propensity are different than wild-type. Some of the most common stress tests include: thermal ramping to determine the onset temperature of visible precipitation, monitoring dye-binding or turbidity, and measuring shifts in calorimetric responses or spectroscopic signals during heating scans [71].
These simple sorts of tests must be used with caution, as the net process of aggregate formation and growth involves multiple steps, and no single assay can presently distinguish the different contributions [77]. This may be acceptable if one is only interested in selecting protein variants that do not form the most easily detectable aggregate species. However, the common stress tests need to be supplemented with additional experimental characterization if mechanism-based algorithms are to be improved. In cases where, for example, changes in conformational stability are compared to changes in aggregation rates for variants that were designed to lower free energy of the folded state, there often are outliers that clearly indicate other factors affecting aggregation have changed as a result of mutations that were intended to alter only monomer unfolding [36,78,79]. In at least one case, a mutation that was introduced to reduce aggregation by eliminating hot spots was found to inadvertently harm conformational stability. However, the change in protein-protein interactions was sufficient to offset this detriment, and overall it resulted in a net aggregation-resistant variant [36]. Ultimately, if one seeks to gain or claim mechanistic insight regarding aggregation from mechanism-guided protein design, then a posteriori experimental characterization of stages in the overall aggregation process should be tested orthogonally. This will also allow for better assessments of which design strategies are quantitatively more effective than others.
Concluding remarks and future perspectives
Many of the examples above and elsewhere in the literature focus primarily on aggregation from the perspective of creation of the initial aggregate species, or implicitly lump all aggregation processes together. For example, designs that eliminate hot spots are often tested with data based upon the “growth” phase in polypeptide aggregation [35,63,80], but the experimental rates are actually a convolution of nucleation and growth [19,77]. Existing computational tools focus predominantly on predicting one of the following: changes in conformational stability (folding or unfolding) for isolated protein chains, changes in monomer-monomer interactions as folded proteins, or changes in strong “binding” propensities of hot-spot peptides as if they were fully solvent exposed and able to aggregate as excised polypeptides. Available algorithms do not account for different growth mechanisms of aggregates, or for additional pathways such as adsorption to bulk solid-liquid, liquid-liquid, or liquid-vapor interfaces.
Furthermore, most computational algorithms are narrowly focused on physiological conditions that are of questionable relevance for many pharmaceutical proteins. Very few protein drugs are processed or formulated and stored as final products at neutral pH or salt concentrations that are isotonic with blood plasma, for a variety of practical reasons [81]. It is promising that recent efforts highlight the importance of charged amino acids in protein conformational stability [54,55], as pH is one the most important adjustable factors in protein formulation [75]. In addition, the favorability of solvent exposure of different amino acids depends on the presence of neutral and charged co-solutes such as sugars, salts, and buffer species that are common additives in pharmaceutical products [33,75,76,77]. Although there is clear experimental evidence that these additives differentially affect the exposure of different amino acids, there is little or no acknowledgement by most available algorithms that the “solvent” is not just water, and near-neutral pH and isotonic salt concentrations are far from the relevant conditions for many technological or commercial applications of proteins.
In general, one must bear in mind that aggregation and different aggregation pathways and rates are a function of both the protein and its environment. There is a clear need for improved design algorithms that account for many or all of these factors when using protein engineering to control aggregation in the context of many biotechnology applications, not just for therapeutic proteins (Box 3).
Box 3. Outstanding questions.
Can proteins be designed to completely eliminate aggregates without sacrificing folding or function, or is aggregation unavoidable for some proteins for practical concentrations and time scales?
Can a priori prediction of how mutations alter aggregation rates of natively folded proteins be done quantitatively or even semi-quantitatively, without a need to fit or statistically optimize against large quantitative databases?
Will future design approaches acknowledge on the importance of the solution environment and shift away from being predominantly focused on physiological conditions that are of little relevance to aggregation and stability of proteins in biopharmaceuticals and biotechnology products?
Will it be possible to predict and design which types of aggregates form, so as to avoid aggregated forms that are more immunogenic?
Highlights.
Therapeutic proteins form a variety of aggregate types
Aggregates are risk factors for patient immune responses
Aggregation mechanisms depend on protein sequence and environment
Opportunities exist for predictive design & control of aggregation rates & mechanism
Acknowledgments
Support of the National Science Foundation and National Institutes of Health is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mullard A. 2011 FDA drug approvals. Nat Rev Drug Discov. 2012;11:91–94. doi: 10.1038/nrd3657. [DOI] [PubMed] [Google Scholar]
- 2.Rob Aggarwal S. What's fueling the biotech engine-2012 to 2013. Nat Biotechnol. 2014;32:32–39. doi: 10.1038/nbt.2794. [DOI] [PubMed] [Google Scholar]
- 3.Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm. 2005;289:1–30. doi: 10.1016/j.ijpharm.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 4.Wang W, et al. Aggregation of Therapeutic Proteins. John Wiley & Sons, Inc.; 2010. [Google Scholar]
- 5.Jiskoot W, et al. Protein instability and immunogenicity: Roadblocks to clinical application of injectable protein delivery systems for sustained release. J Pharm Sci. 2012;101:946–954. doi: 10.1002/jps.23018. [DOI] [PubMed] [Google Scholar]
- 6.Macdougall IC. Antibody-mediated pure red cell aplasia (PRCA): epidemiology, immunogenicity and risks. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc -Eur Ren Assoc. 2005;20(Suppl 4):iv9–15. doi: 10.1093/ndt/gfh1087. [DOI] [PubMed] [Google Scholar]
- 7.Hermeling S, et al. Structure-Immunogenicity Relationships of Therapeutic Proteins. Pharm Res. 2004;21:897–903. doi: 10.1023/b:pham.0000029275.41323.a6. [DOI] [PubMed] [Google Scholar]
- 8.Carpenter JF, et al. Overlooking subvisible particles in therapeutic protein products: Gaps that may compromise product quality. J Pharm Sci. 2009;98:1201–1205. doi: 10.1002/jps.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, et al. An industry perspective on the monitoring of subvisible particles as a quality attribute for protein therapeutics. J Pharm Sci. 2010;99:3302–3321. doi: 10.1002/jps.22097. [DOI] [PubMed] [Google Scholar]
- 10.Struble EB, et al. Higher-Order Structure and Protein Aggregate Characterization of Protein Therapeutics: Perspectives from Good Manufacturing Practices and Regulatory Guidance. In: Narhi LO, editor. Biophysics for Therapeutic Protein Development. Springer; New York: 2013. pp. 261–281. [Google Scholar]
- 11.McPherson A, Gavira JA. Introduction to protein crystallization. Acta Crystallogr Sect F Struct Biol Commun. 2014;70:2–20. doi: 10.1107/S2053230X13033141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bou-Abdallah F, Terpstra TR. The thermodynamic and binding properties of the transferrins as studied by isothermal titration calorimetry. Biochim Biophys Acta. 2012;1820:318–325. doi: 10.1016/j.bbagen.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006;8:E501–507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joubert MK, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem. 2012;287:25266–25279. doi: 10.1074/jbc.M111.330902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harding FA, et al. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs. 2010;2:256. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dey AK, et al. Physicochemical and functional characterization of vaccine antigens and adjuvants. Expert Rev Vaccines. 2014;13:671–685. doi: 10.1586/14760584.2014.907528. [DOI] [PubMed] [Google Scholar]
- 17.Wu H, et al. Competing Aggregation Pathways for Monoclonal Antibodies. FEBS Lett. doi: 10.1016/j.febslet.2014.01.051. in press. [DOI] [PubMed] [Google Scholar]
- 18.Banks DD, et al. Native-state solubility and transfer free energy as predictive tools for selecting excipients to include in protein formulation development studies. J Pharm Sci. 2012;101:2720–2732. doi: 10.1002/jps.23219. [DOI] [PubMed] [Google Scholar]
- 19.Roberts CJ. Non-native protein aggregation kinetics. Biotechnol Bioeng. 2007;98:927–938. doi: 10.1002/bit.21627. [DOI] [PubMed] [Google Scholar]
- 20.Caflisch A. Computational models for the prediction of polypeptide aggregation propensity. Curr Opin Chem Biol. 2006;10:437–444. doi: 10.1016/j.cbpa.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Roberts CJ. Kinetics of Irreversible Protein Aggregation: Analysis of Extended Lumry-Eyring Models and Implications for Predicting Protein Shelf Life. J Phys Chem B. 2003;107:1194–1207. [Google Scholar]
- 22.Roberts CJ, et al. Irreversible aggregation of recombinant bovine granulocyte-colony stimulating factor (bG-CSF) and implications for predicting protein shelf life. J Pharm Sci. 2003;92:1095–1111. doi: 10.1002/jps.10377. [DOI] [PubMed] [Google Scholar]
- 23.Rosa M, et al. Measuring and Modeling Hemoglobin Aggregation below the Freezing Temperature. J Phys Chem B. 2013;117:8939–8946. doi: 10.1021/jp4035369. [DOI] [PubMed] [Google Scholar]
- 24.Saluja A, et al. Significance of Unfolding Thermodynamics for Predicting Aggregation Kinetics: A Case Study on High Concentration Solutions of a Multi-Domain Protein. Pharm Res. 2014 doi: 10.1007/s11095-013-1263-5. [DOI] [PubMed] [Google Scholar]
- 25.Kim N, et al. Aggregation of anti-streptavidin immunoglobulin gamma-1 involves Fab unfolding and competing growth pathways mediated by pH and salt concentration. Biophys Chem. 2013;172:26–36. doi: 10.1016/j.bpc.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Hari SB, et al. Acid-Induced Aggregation of Human Monoclonal IgG1 and IgG2: Molecular Mechanism and the Effect of Solution Composition. Biochemistry (Mosc) 2010;49:9328–9338. doi: 10.1021/bi100841u. [DOI] [PubMed] [Google Scholar]
- 27.Weiss WF, 4th, et al. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J Pharm Sci. 2009;98:1246–1277. doi: 10.1002/jps.21521. [DOI] [PubMed] [Google Scholar]
- 28.Lomakin A, et al. Kinetic theory of fibrillogenesis of amyloid beta-protein. Proc Natl Acad Sci U S A. 1997;94:7942–7947. doi: 10.1073/pnas.94.15.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 30.Powers ET, Powers DL. The kinetics of nucleated polymerizations at high concentrations: amyloid fibril formation near and above the supercritical concentration. Biophys J. 2006;91:122–132. doi: 10.1529/biophysj.105.073767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, et al. Multi-variate approach to global protein aggregation behavior and kinetics: Effects of pH, NaCl, and temperature for α-chymotrypsinogen A. J Pharm Sci. 2010;99:645–662. doi: 10.1002/jps.21869. [DOI] [PubMed] [Google Scholar]
- 32.Sahin E, et al. Comparative effects of pH and ionic strength on protein-protein interactions, unfolding, and aggregation for IgG1 antibodies. J Pharm Sci. 2010;99:4830–4848. doi: 10.1002/jps.22198. [DOI] [PubMed] [Google Scholar]
- 33.Filipe V, et al. Aggregation and immunogenicity of therapeutic proteins. 2010:403–433. 1plate. [Google Scholar]
- 34.Krebs MRH, et al. Common motifs in protein self-assembly. Faraday Discuss. 2008;139:265–265. doi: 10.1039/b715879c. [DOI] [PubMed] [Google Scholar]
- 35.Chiti F, et al. Rationalization of the effects of mutations on peptide andprotein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 36.Sahin E, et al. Computational design and biophysical characterization of aggregation-resistant point mutations for γD crystallin illustrate a balance of conformational stability and intrinsic aggregation propensity. Biochemistry (Mosc) 2011;50:628–639. doi: 10.1021/bi100978r. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, et al. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. mAbs. 2009;1:254–267. doi: 10.4161/mabs.1.3.8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Latypov RF, et al. Elucidation of Acid-induced Unfolding and Aggregation of Human Immunoglobulin IgG1 and IgG2 Fc. J Biol Chem. 2011;287:1381–1396. doi: 10.1074/jbc.M111.297697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brummitt RK, et al. Nonnative aggregation of an IgG1 antibody in acidic conditions: part 1. Unfolding, colloidal interactions, and formation of high-molecular-weight aggregates. J Pharm Sci. 2011;100:2087–2103. doi: 10.1002/jps.22448. [DOI] [PubMed] [Google Scholar]
- 40.Brummitt RK, et al. Nonnative aggregation of an IgG1 antibody in acidic conditions, part 2: nucleation and growth kinetics with competing growth mechanisms. J Pharm Sci. 2011;100:2104–2119. doi: 10.1002/jps.22447. [DOI] [PubMed] [Google Scholar]
- 41.Arosio P, et al. Population balance modeling of antibodies aggregation kinetics. J Phys Chem B. 2012;116:7066–7075. doi: 10.1021/jp301091n. [DOI] [PubMed] [Google Scholar]
- 42.Arosio P, et al. On the role of salt type and concentration on the stability behavior of a monoclonal antibody solution. Biophys Chem. 2012:168–169. 19–27. doi: 10.1016/j.bpc.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Arosio P, et al. Aggregation Mechanism of an IgG2 and two IgG1 Monoclonal Antibodies at low pH: From Oligomers to Larger Aggregates. Pharm Res. 2013;30:641–654. doi: 10.1007/s11095-012-0885-3. [DOI] [PubMed] [Google Scholar]
- 44.Lee H, et al. Monoclonal antibody aggregation intermediates visualized by atomic force microscopy. J Pharm Sci. 2011;100:416–423. doi: 10.1002/jps.22279. [DOI] [PubMed] [Google Scholar]
- 45.Papanikolopoulou K, et al. Formation of amyloid fibrils in vitro by human γD-crystallin and its isolated domains. Mol Vis. 2008;14:81–89. [PMC free article] [PubMed] [Google Scholar]
- 46.Weiss WF, IV, et al. Nonnative protein polymers: structure, morphology, and relation to nucleation and growth. Biophys J. 2007;93:4392–4403. doi: 10.1529/biophysj.107.112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 48.Andrews JM, et al. Nucleation, Growth, and Activation Energies for Seeded and Unseeded Aggregation of α-Chymotrypsinogen A†. Biochemistry (Mosc) 2008;47:2397–2403. doi: 10.1021/bi7019244. [DOI] [PubMed] [Google Scholar]
- 49.Sahin E, et al. Aggregation and pH-temperature phase behavior for aggregates of an IgG2 antibody. J Pharm Sci. 2012 doi: 10.1002/jps.23056. [DOI] [PubMed] [Google Scholar]
- 50.Kiese S, et al. Shaken, not stirred: mechanical stress testing of an IgG1 antibody. J Pharm Sci. 2008;97:4347–4366. doi: 10.1002/jps.21328. [DOI] [PubMed] [Google Scholar]
- 51.Wang JC, Lin JH. Scoring functions for prediction of protein-ligand interactions. Curr Pharm Des. 2013;19:2174–2182. doi: 10.2174/1381612811319120005. [DOI] [PubMed] [Google Scholar]
- 52.Boder ET, et al. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc Natl Acad Sci U A. 2000;97:10701–5. doi: 10.1073/pnas.170297297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Worn A, Pluckthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- 54.Loladze VV, Makhatadze GI. Energetics of charge-charge interactions between residues adjacent in sequence. Proteins. 2011;79:3494–3499. doi: 10.1002/prot.23132. [DOI] [PubMed] [Google Scholar]
- 55.Chan CH, et al. Electrostatic contribution of surface charge residues to the stability of a thermophilic protein: benchmarking experimental and predicted pKa values. PloS One. 2012;7:e30296. doi: 10.1371/journal.pone.0030296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rowley MJ, et al. Phage display for epitope determination: a paradigm for identifying receptor-ligand interactions. Biotechnol Annu Rev. 2004;10:151–188. doi: 10.1016/S1387-2656(04)10006-9. [DOI] [PubMed] [Google Scholar]
- 57.Woodley JM. Protein engineering of enzymes for process applications. Curr Opin Chem Biol. 2013;17:310–316. doi: 10.1016/j.cbpa.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 58.Adams PD, et al. Advances, interactions, and future developments in the CNS, Phenix, and Rosetta structural biology software systems. Annu Rev Biophys. 2013;42:265–287. doi: 10.1146/annurev-biophys-083012-130253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ricci MS, et al. Mutational approach to improve physical stability of protein therapeutics susceptible to aggregation: Role of altered conformation in irreversible preciitation. In: Murphy RM, Tsai AM, editors. Misbehaving Proteins: Protein (Mis)Folding, Aggregation, and Stability. Springer; 2006. [Google Scholar]
- 60.Miklos AE, et al. Structure-Based Design of Supercharged, Highly Thermoresistant Antibodies. Chem Biol Oxf U K. 2012;19:449–455. doi: 10.1016/j.chembiol.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perchiacca JM, et al. Mutational analysis of domain antibodies reveals aggregation hotspots within and near the complementarity determining regions. Proteins. 2011;79:2637–2647. doi: 10.1002/prot.23085. [DOI] [PubMed] [Google Scholar]
- 62.Chennamsetty N, et al. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci. 2009;106:11937–11942. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fowler SB, et al. Rational design of aggregation-resistant bioactive peptides: reengineering human calcitonin. Proc Natl Acad Sci U S A. 2005;102:10105–10110. doi: 10.1073/pnas.0501215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee CC, et al. Toward aggregation-resistant antibodies by design. Trends Biotechnol. 2013;31:612–620. doi: 10.1016/j.tibtech.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 65.Schaefer JV, Plückthun A. Engineering aggregation resistance in IgG by two independent mechanisms: lessons from comparison of Pichia pastoris and mammalian cell expression. J Mol Biol. 2012;417:309–335. doi: 10.1016/j.jmb.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 66.Famm K, et al. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol. 2008;376:926–931. doi: 10.1016/j.jmb.2007.10.075. [DOI] [PubMed] [Google Scholar]
- 67.Tiller T, et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. mAbs. 2013;5 doi: 10.4161/mabs.24218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dudgeon K, et al. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A. 2012;109:10879–10884. doi: 10.1073/pnas.1202866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Orr BA, et al. Rapid method for measuring ScFv thermal stability by yeast surface display. Biotechnol Prog. 2003;19:631–638. doi: 10.1021/bp0200797. [DOI] [PubMed] [Google Scholar]
- 70.Gámez A, et al. Expression analysis of phenylketonuria mutations. Effect on folding and stability of the phenylalanine hydroxylase protein. J Biol Chem. 2000;275:29737–29742. doi: 10.1074/jbc.M003231200. [DOI] [PubMed] [Google Scholar]
- 71.Jager M, Pluckthun A. Domain interactions in antibody Fv and scFv fragments: effects on unfolding kinetics and equilibria. FEBS Lett. 1999;462:307–12. doi: 10.1016/s0014-5793(99)01532-x. [DOI] [PubMed] [Google Scholar]
- 72.Flaugh SL, et al. Contributions of hydrophobic domain interface interactions to the folding and stability of human gammaD-crystallin. Protein Sci Publ Protein Soc. 2005;14:569–581. doi: 10.1110/ps.041111405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Flaugh SL, et al. Interdomain side-chain interactions in human gammaD crystallin influencing folding and stability. Protein Sci Publ Protein Soc. 2005;14:2030–2043. doi: 10.1110/ps.051460505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gribenko AV, Makhatadze GI. Role of the charge-charge interactions in defining stability and halophilicity of the CspB proteins. J Mol Biol. 2007;366:842–856. doi: 10.1016/j.jmb.2006.11.061. [DOI] [PubMed] [Google Scholar]
- 75.Jha RK, et al. Computational Design of a PAK1 Binding Protein. J Mol Biol. 2010;400:257–270. doi: 10.1016/j.jmb.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gsponer J, Vendruscolo M. Theoretical approaches to protein aggregation. Protein Pept Lett. 2006;13:287–293. doi: 10.2174/092986606775338407. [DOI] [PubMed] [Google Scholar]
- 77.Murphy RM, Roberts CJ. Protein misfolding and aggregation research: Some thoughts on improving quality and utility. Biotechnol Prog. 2013 doi: 10.1002/btpr.1812. [DOI] [PubMed] [Google Scholar]
- 78.Chrunyk BA, Wetzel R. Breakdown in the relationship between thermal and thermodynamic stability in an interleukin-1 beta point mutant modified in a surface loop. Protein Eng. 1993;6:733–738. doi: 10.1093/protein/6.7.733. [DOI] [PubMed] [Google Scholar]
- 79.Worn A, Pluckthun A. Different equilibrium stability behavior of ScFv fragments: identification, classification, and improvement by protein engineering. Biochemistry (Mosc) 1999;38:8739–50. doi: 10.1021/bi9902079. [DOI] [PubMed] [Google Scholar]
- 80.Morris AM, et al. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochim Biophys Acta Proteins Proteomics. 2009;1794:375–397. doi: 10.1016/j.bbapap.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 81.Wang W, et al. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96:1–26. doi: 10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 82.Auton M, Bolen DW. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc Natl Acad Sci U S A. 2005;102:15065–15068. doi: 10.1073/pnas.0507053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Auton M, et al. Structural thermodynamics of protein preferential solvation: osmolyte solvation of proteins, aminoacids, and peptides. Proteins. 2008;73:802–813. doi: 10.1002/prot.22103. [DOI] [PubMed] [Google Scholar]
- 84.Creighton TE. Protein folding. W.H. Freeman and Co.; 1992. [Google Scholar]
- 85.Israelachvili JN. Intermolecular and Surface Forces. Elsevier Academic Press; 2011. [Google Scholar]
- 86.Russel WB, et al. Colloidal dispersions. Cambridge University Press; 1992. [Google Scholar]
- 87.Kroetsch AM, et al. Relating particle formation to salt- and pH-dependent phase separation of non-native aggregates of alpha-chymotrypsinogen a. J Pharm Sci. 2012;101:3651–60. doi: 10.1002/jps.23264. [DOI] [PubMed] [Google Scholar]
- 88.Philo JS. Is any measurement method optimal for all aggregate sizes and types? AAPS J. 2006;8:E564–E571. doi: 10.1208/aapsj080365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bee JS, et al. The future of protein particle characterization and understanding its potential to diminish the immunogenicity of biopharmaceuticals: a shared perspective. J Pharm Sci. 2012;101:3580–3585. doi: 10.1002/jps.23247. [DOI] [PubMed] [Google Scholar]
- 90.Hoehne M, et al. Adsorption of monoclonal antibodies to glass microparticles. J Pharm Sci. 2011;100:123–132. doi: 10.1002/jps.22275. [DOI] [PubMed] [Google Scholar]
- 91.Bee JS, et al. Aggregation of a monoclonal antibody induced by adsorption to stainless steel. Biotechnol Bioeng. 2009;105:121–129. doi: 10.1002/bit.22525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krielgaard L, et al. Effect of tween 20 on freeze-thawing- and agitation-induced aggregation of recombinant human factor XIII. J Pharm Sci. 1998;87:1593–1603. doi: 10.1021/js980126i. [DOI] [PubMed] [Google Scholar]
- 93.Flaugh SL, et al. Glutamine deamidation destabilizes human gammaD-crystallin and lowers the kinetic barrier to unfolding. J Biol Chem. 2006;281:30782–30793. doi: 10.1074/jbc.M603882200. [DOI] [PubMed] [Google Scholar]
- 94.Shi Y, et al. Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J Am Chem Soc. 2013;135:15897–15908. doi: 10.1021/ja407801x. [DOI] [PubMed] [Google Scholar]
- 95.Mukherjee R, et al. Probing deamidation in therapeutic immunoglobulin gamma (IgG1) by “bottom-up” mass spectrometry with electron transfer dissociation. Rapid Commun Mass Spectrom RCM. 2010;24:879–884. doi: 10.1002/rcm.4464. [DOI] [PubMed] [Google Scholar]
- 96.Luo Q, et al. Chemical modifications in therapeutic protein aggregates generated under different stress conditions. J Biol Chem. 2011;286:25134–25144. doi: 10.1074/jbc.M110.160440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cleland JL, et al. The development of stable protein formulations: a close look at protein aggregation, deamidation, and oxidation. Crit Rev Ther Drug Carrier Syst. 1993;10:307–377. [PubMed] [Google Scholar]
- 98.Vlasak J, Ionescu R. Fragmentation of monoclonal antibodies. MAbs. 2011;3:253–263. doi: 10.4161/mabs.3.3.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roberts CJ, et al. Effects of temperature and osmolytes on competing degradation routes for an IgG1 antibody. J Pharm Sci. 2013;102:3556–3566. doi: 10.1002/jps.23668. [DOI] [PubMed] [Google Scholar]