

Abstract
Severe blood loss in victims of trauma creates an exaggerated inflammatory background which contributes to the development of intravascular coagulopathy and multiple organ dysfunction syndrome. We hypothesized that treatment with an NF-κB inhibitor, diphenyldifluoroketone EF24, will have salutary effects in hemorrhagic shock. The objective of this study was to investigate the effect of EF24 on the expression of interleukin (IL)-1 receptor superfamily in a rat model of hypovolemic shock. Hypovolemia was induced by gradually withdrawing approximately 50% of circulating blood, and EF24 was administered intraperitoneally (0.2 mg/Kg) in 50 μl volume. After 6 h of shock, lung tissue was probed immunohistochemically and by immunoblotting for studying expression of TLR4, Il-1R, ST2, and SIGIRR. Tissue-associated pro-inflammatory cytokines, TNF-α and IL-6, were measured by ELISA. We observed a reduction in immunoreactive TLR4 and IL-1R1 in lung tissue of rats treated with EF24. Simultaneously, the pulmonary expression of ST2 and SIGIRR (the putative negative regulators of pro-inflammatory IL-1R pathway) was increased in EF24-treated hemorrhaged rats. The concentration of hemorrhage-induced TNF-α and IL-6 in lung tissue homogenates was also reduced by EF24 treatment. These results confirm our previous in vitro observations in LPS-stimulated dendritic cells that EF24 beneficially modulates IL-1R pathway and suggest that it could be investigated as an adjunct therapeutic in managing inflammation associated with hemorrhagic shock.
Keywords: Hemorrhagic shock, EF24, Inflammation, Toll-like receptor 4, Interleukin-1 receptor
Inflammation is an innate and protective immune response to a tissue injury in normal circumstances. However, unresolved, generalized and prolonged inflammation is detrimental to the well-being of the host. Exaggerated inflammation has been identified as a culprit in the pathogenesis of multiple organ dysfunction syndrome (MODS) in the victims of hemorrhagic shock and ischemia/reperfusion injury [1–3]. At cellular level, the response to inflammation is manifested by recruitment of immune cells, such as neutrophils, monocytes and dendritic cells (DCs). These cells are equipped with cell surface surveillance sensors, mainly belonging to the members of interleukin-1 receptor (IL-1R) superfamily. Upon stimulation, they recruit adaptor proteins to initiate a molecular cascade culminating into the secretion of effector cytokines, such as TNF-α and IL-6 [4, 5]. IL-1R members play a significant role in hemorrhagic shock because of their ability to interact with damage–associated molecular patterns (DAMPs) released during trauma and blood loss[6].
IL-1R superfamily comprises of receptors for IL-1 and IL-18, TLRs, single immunoglobulin IL-1R-related (SIGIRR), and an orphan receptor suppression of tumorigenicity 2 (ST2) [7–9]. They are characterized by a conserved cytosolic domain called the Toll–IL-1R (TIR) domain which activates some of the common signaling pathways. These members of IL-1R superfamily could also be classified depending on their pro- and anti-inflammatory role in the entire signaling process. Whereas IL-1R2, soluble ST2 (sST2), and SIGIRR are considered as negative regulators of inflammation, all other members of the IL-1R superfamily are pro-inflammatory. The downstream pro-inflammatory effects of IL-1R signaling are mediated by a transcription element nuclear factor-kappa B (NF-κB) [10]. Considering the diversity in theIL-1 superfamily members and their significance in perpetuating inflammation in trauma and hemorrhage, they are attractive adjunct targets in resuscitation approaches [11–13].
The objective of the presented work was to investigate the anti-inflammatory action of 3,5-bis(2-fluorobenzylidine)-4-piperidone (also known as EF24, Fig. 1a) in a hemorrhagic shock model. Originally synthesized as a synthetic curcuminoid [14], EF24 is a synthetic Michael’s acceptor which inhibits the catalytic activity of IkappaB kinase (IKK), an enzyme responsible for the phosphorylation and degradation of NF-κB inhibitory protein IkappaB[15, 16]. We recently reported that EF24 inhibits lipopolysaccharide (LPS)-induced maturation, NF-κB, and production of pro-inflammatory cytokines in DCs[15]. Here, we studied the effect of EF24 on the expression of IL-1R members, namely IL-1R1, TLR4, ST-2, and SIGIRR, in lung tissue of rats subjected to 40% blood loss.
Figure 1.
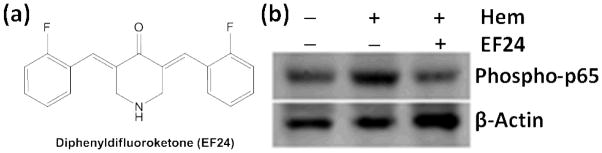
EF24 suppresses hemorrhage-induced phospho-NF-κB p65 expression in lung tissues of rats subjected to sham surgery (Ctrl), hemorrhage (Hem), and hemorrhage with EF24 treatment (Hem+EF24). To demonstrate equal loading, the membranes was stripped and re-probed with anti-β-actin antibody. The immunoblot is a representative set from three similar groups.
MATERIALS AND METHODS
The chemical synthesis and characterization of EF24 was performed in-house by the procedure published elsewhere [17]. The aqueous solution of EF24 was prepared in sterile normal saline. The animal experiments were performed according to the NIH Animal Use and Care Guidelines and were approved by the Institutional Animal Care Committee of the University of Oklahoma Health Sciences Center. Various antibodies were obtained from Cell Signaling Technology (CST, Danvers, MA), Santa Cruz Biotech (SCBT, Dallas, TX), Abcam (Cambridge, MA), and Sigma-Aldrich (Sigma, St. Louis, MO).
Rat model of fixed-volume hemorrhage
The rat hypovolemic exchange transfusion model has been described elsewhere[18]. Briefly, left femoral artery of male Sprague-Dawley rats (250–300 g) was cannulated with a catheter. The experiments were performed after allowing 2 days of recovery period. The rats were heparinized with 100 units of heparin to prevent clotting. The catheterized rats were randomized in three groups- hemorrhage without any treatment (Hem), hemorrhage with EF24 treatment (Hem + EF24), and sham control group consisting of rats undergoing surgical procedure and cannulation only. Hemorrhage was induced by withdrawing approximately 40% of circulating blood at the rate of 0.75 ml/min. During blood withdrawal, the rats were under 2% isoflurane anesthesia supplied in medical air (2 L/min). The total volume of blood was estimated as 5.7% of the total body weight. EF24 (100 μg, ~0.4 mg/Kg) was administered intraperitoneally in 50 μl of saline. After appropriate treatment, the hypovolemic rats were allowed to wake up and transferred to a fresh cage. During the entire protocol, no attempt was made to artificially maintain the body temperature of the rats and rats were allowed to inhale room air. At 6 h of hemorrhage, the rats were euthanized and lung was harvested for immunobloting and immunohistochemistry experiments. One-half of the lung was snap-frozen, whereas the other half was fixed in buffered-formalin.
Immunobloting
The samples for immunoblotting were prepared by mincing the frozen tissues (75–100 mg) and incubating them on ice for 30 min in 0.5 mL of ice-cold whole-cell lysate buffer consisting of 10% NP-40, 5 M NaCl, 1 M HEPES, 0.1 M ethyleneglycoltetraacetic acid, 0.5 M ethylenediaminetetraacetic acid, 0.1 M phenylmethylsulfonyl fluoride, 0.2 M sodium orthovanadate, 1 M NaF, 2 μg/mL aprotinin, and 2 μg/mL leupeptin. The protein was extracted by homogenization using a dounce homogenizer and centrifugation at 14,000 rpm at 4°C for 10 min. After protein estimation by bicinchoninic acid assay (Pierce-Thermo Fisher Scientific, Rockford, IL), about 25 μg of total protein was fractionated on Novex 4–20% Tris–glycine gradient SDS-PAGE gels (Invitrogen, Carlsbad, CA). The separated proteins were electro-transferred onto nitrocellulose membranes using iBlot gel transfer device (Invitrogen, Carlsbad, CA). The non-specific sites were blocked by incubating the membranes with 5% skimmed milk in Tris-buffered saline with 0.4% Tween-20 (TBST). The blocked membrane was incubated overnight at 4°C with appropriate primary antibodies in TBST. The primary antibodies against phospho-NFkB-p65 (CST), TLR4 (Abcam), Il-1R1 (Abcam), SIGIRR (SCBT), and ST2 (Abcam) were used at dilutions of 1:2,500, 1:1,000, 1:1,500, 1:1,000, and 1:1,000, respectively. After probing, the membranes were washed and incubated with HRP-conjugated-anti-rabbit-IgG antibody diluted at 1:5,000 in TBST. The immunoreactive bands were detected by SuperSignal West Femto detection reagent (Thermo Fischer Scientific, Rockford, IL). In order to ensure equal protein loading in the wells, the membranes were stripped using a stripping solution containing 10% SDS, 0.5 M Tris and β-mercaptoethanol (35 μl/ml) at 60 °C for 45 min, and re-probed with anti-β-actin antibody (Sigma; 1:5,000 in TBST). The blots were imaged using Ultraquant image acquisition machine (Claremont, CA) and the densitometric readings for the proteins were normalized with those of β-actin.
Immunohistochemistry and histopathology
The protein expression levels of TLR4, IL-1R1, ST2, and SIGIRR were assessed in rat lung tissues by IHC using a kit from DakoCytomation (Carpinteria, CA). In brief, the tissue samples were fixed with paraformaldehyde and embedded in paraffin. After washing in PBS, the slides were blocked with a protein-block solution for 20 min and incubated overnight with primary antibodies against TLR4 (Abcam, ab13867), IL-1R1 (Abcam, ab40774), ST2 (SCBT, sc18687), and SIGIRR (SCBT, sc98976) at dilutions of 1:200, 1:200, 1:100, and 1:100 in blocking solution, respectively. The slides were washed and incubated with biotinylated link universal antiserum, followed by horseradish peroxidase-streptavidin conjugate. The slides were rinsed and the color was developed using 3,3-diaminobenzidine hydrochloride as a chromogen. Finally, the sections were rinsed in distilled water, counterstained with Mayer’s hematoxylin solution and mounted with DPX mounting medium for evaluation.
For histopathological examination, the paraffin-embedded tissues were stained with hematoxylin & eosin (H & E) stain. The slides were evaluated for morphologic changes and pathological signatures of ischemia, inflammation, and any other abnormalities. The sections were observed with Olympus microscope IX701, and digital computer images were recorded using an Olympus DP70 camera.
Cytokines assay
The levels of TNF-α and IL-6 cytokines in lung tissue were estimated by rat Inflammation ELISA Strip for profiling TNF-α and IL-6 (Signosis, Sunnyvale, CA). Tissue samples were prepared by mincing the frozen lung tissues (75–100 mg) and incubating on ice for 30 min in 0.5 mL of ice-cold whole-cell lysate buffer consisting of 10% NP-40, 5 M NaCl, 1 M HEPES, 0.1 M ethyleneglycoltetraacetic acid, 0.5 M ethylenediaminetetraacetic acid, 0.1 M phenylmethylsulfonyl fluoride, 0.2 M sodium orthovanadate, 1 M NaF, 2 μg/mL aprotinin, and 2 μg/mL leupeptin. The mixture was homogenized using a dounce homogenizer and centrifuged at 14,000 rpm at 4°C for 10 min to obtain test supernatants. The ELISA was performed following manufacturer’s instructions. Briefly, 100 μl of standard, control or sample was added to the designated wells and incubated for 1 h at room temperature with gentle shaking. After washing the wells three times, 100 μl of biotin-conjugated antibody (1:1,000) was added for 1 h. The unreacted secondary antibody was washed off and the wells were probed with streptavidin-HRP conjugate for 45 min. The wells were washed again and 100 μl of 3,3′,5,5′-tetramethylbenzidine (Sigma, St. Louis, MO) substrate solution was added to generate the color. The reaction was stopped after 30 min and the optical density was read at 450 nm.
Data analysis
Unless otherwise mentioned all the results were analyzed by one-way analysis of variance (ANOVA) applying the Bonferroni post-test using Prism software (GraphPad, San Diego, CA, USA). p < 0.05 was considered statistically significant. The densitometry of immunoreactive bands was performed from three replicates using Image J 1.46r freeware (NIH, USA).
RESULTS
In our previous in vitro work we found that EF24 very potently suppresses markers of inflammation in LPS-stimulated DCs[15]. Here, we expanded our inquiry to an in vivo model of fixed-volume hemorrhage. We focused on lung because hemorrhagic shock often causes morbid pulmonary dysfunction, the severity of which determines the outcome. Secondly, lung is especially prone to injury in reduced perfusion after severe hemorrhage, and it remain sensitive to damage even after volume and oxygen deficits are corrected by resuscitation. As shown in Fig. 1, EF24 treatment markedly suppressed the hemorrhage-induced accumulation of phosphorylated p65 subunit of NF-κB. The transcriptional activity of NF-κB is regulated by the Ser536 phosphorylation of p65 subunit by IKKs [19, 20]. Therefore, tissue expression of phosopho-p65 subunit of NF-κB is a marker of NF-κB activation.
More than any other cytokine family, the IL-1 family of ligands and receptors play a major role in progression of inflammatory response [12]. Therefore, we investigated the expression of various members of IL-1R superfamily in pulmonary tissue by immunohistochemistry and immunoblotting. TLR4 and IL-1R1 are the two most important members of IL-1R family. Whereas TLR4 is a putative receptor for bacterial LPS, IL-1R1 is a glycoprotein receptor for pro-inflammatory IL-1α and IL-1β cytokines. We found that hypovolemia resulted in a significant increase in the expression of TLR4. EF24 treatment significantly (p < 0.05) reduced the hemorrhage-induced TLR4 expression (Fig. 2). Like TLR4, hemorrhage also increased the pulmonary expression of IL-1R1, which was suppressed by EF24 administration (Fig. 3).
Figure 2.

(a) IHC of TLR4 expression in lung tissues of rats subjected to sham surgery (Ctrl), hemorrhage (Hem), and hemorrhage with EF24 administration (Hem+EF24). The pictures are a representative set from three similar groups. (b) Quantitation of IHC values (* p < 0.05 vs control). (c) Immunobloting of TLR4 protein in lung tissue homogenates. To demonstrate equal loading, the membranes were stripped and re-probed with anti-β-actin antibody. (d) Actin-normalized densitometry of TLR4 immunoblots.
Figure 3.

(a) IHC of IL-1R1 expression in lung tissues of rats subjected to sham surgery (Ctrl), hemorrhage (Hem), and hemorrhage with EF24 administration (Hem+EF24). The pictures are a representative set from three similar groups. (b) Quantitation of IHC values (* p < 0.05 vs control). (c) Immunobloting of Il-1R1 protein in lung tissue homogenates. To demonstrate equal loading, the membranes were stripped and re-probed with anti-β-actin antibody. (d) Actin-normalized densitometry of Il-1R1 immunoblots.
Next, we investigated two negative regulators of IL-1R pathway, viz., ST2 and SIGIRR. Both ST2 and SIGIRR belong to the IL-1R family because of the presence of TIR domain. The membrane-bound ST2 is a receptor for cytokine IL-33. Like most other IL-1R family members, ST2 recruits MyD88 as an intracellular adaptor molecule for its downstream signaling. According to one theory, ST2 sequesters MyD88, making latter unavailable for MyD88-dependent other pro-inflammatory IL-1R pathways [7]. SIGIRR is another intracellular mediator that has been shown to negatively regulate pro-inflammatory IL-1R1 signaling [21]. We found that hemorrhage reduced the expression of both ST2 (Fig. 4) and SIGIRR (Fig. 5), whereas theEF24 administration tended to reverse the effect of hemorrhage on the expression of ST2 and SIGIRR.
Figure 4.

(a) IHC of ST2 expression in lung tissues of rats subjected to sham surgery (Ctrl), hemorrhage (Hem), and hemorrhage with EF24 administration (Hem+EF24). The pictures are a representative set from three similar groups. (b) Quantitation of IHC values (* p < 0.05 vs control). (c) Immunobloting of ST2 protein in lung tissue homogenates. To demonstrate equal loading, the membranes were stripped and re-probed with anti-β-actin antibody. (d) Actin-normalized densitometry of ST2 immunoblots.
Figure 5.

(a) IHC of SIGIRR expression in lung tissues of rats subjected to sham surgery (Ctrl), hemorrhage (Hem), and hemorrhage with EF24 administration (Hem+EF24). The pictures are a representative set from three similar groups. (b) Quantitation of IHC values (* p < 0.05 vs control). (c) Immunobloting of SIGIRR protein in lung tissue homogenates. To demonstrate equal loading, the membranes were stripped and re-probed with anti-β-actin antibody. (d) Actin-normalized densitometry of SIGIRR immunoblots.
The local pro-inflammatory cytokine content in lung increases after severe blood loss[22, 23]. We estimated TNF-α and IL-6 in pulmonary tissue homogenates of hemorrhaged rats. As shown in Fig. 6a, hemorrhage resulted in a significant increase in TNF-α. Hemorrhage also increased the levels of IL-6, but the increase was not statistically significant. The post-hemorrhage increase in the levels of TNF-α and IL-6 was subdued by EF24 treatment (p < 0.05). Pro-inflammatory cytokines, such as TNF-α and IL-6, play a major role as effector molecules in the inflammatory process that manifests itself in acute histopathologic consequences (Fig. 6b). These changes are ascribed to the inflammatory process and hemorrhage-induced cytokine storm, collectively described as generalized interstitial pneumonitis. The blood loss resulted in significant morphological changes in lung, characterized by severe pulmonary congestion and loss of alveolar integrity. There was marked thickening of alveolar epithelium and interstitial exudation. Staining for inflammatory cell infiltration was also observed in hemorrhaged rats. Treatment with EF24 markedly ameliorated the post-hemorrhage morphological changes and inflammatory cell infiltration (Fig. 6b).
Figure 6.

(a) Effect of EF24 administration on the levels of inflammatory cytokines, TNF-α and IL-6, in lung tissues of rats subjected to sham treatment (Sham Ctrl), hemorrhage (H), and hemorrhage with EF24 administration (H+EF24). (# p < 0.05 vs. Ctrl and * p < 0.05 vs. Hem). (b) Hematoxylin & eosin staining of lung tissues from a representative set of rats, showing EF24-assisted recovery from hemorrhage-induced damages (600X magnification).
Overall, the results of this study indicate that EF24 suppresses pro-inflammatory arm of IL-1R signaling while inducing the anti-inflammatory mediators in the same pathway. The salutary effect of EF24 was translated in the improvement in the lung-cytokine levels and pulmonary histology in hypovolemic rats.
DISCUSSION
In hemorrhagic shock, the cardiac and cerebral perfusion is compensated at the expense of perfusion to the peripheral organs. The ischemia in peripheral organs results in tissue injury and release of damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1). The DAMPs originating from injured tissue and other putative ligands produced during the early phase of inflammatory process interact with pattern recognition receptors (PRRs) expressed on the immune cells [24]. IL-1R superfamily constitutes the most important PRRs which assist inflammation to attain a vicious and self-damaging status. The activation of inflammatory pathways underlines the pathophysiological basis of MODS following hemorrhagic shock and resuscitation [25]. As such, timely management of the inflammatory processes and participating molecules could prevent significant morbidity and mortality associated with trauma and shock. In our previous reports, we described that EF24 potently inhibited NF-κB in an LPS-stimulated DC model of sterile inflammation [15]. Concurrently, we also found that EF24 suppresses LPS-induced expression and signaling of IL-1R1 and TLR4 in DCs. Here, we show that the in vitro actions of EF24 are effectively translated in an in vivo model of hypovolemic shock.
EF24 is known to inhibit the transcription element NF-κB, perhaps by negatively influencing the kinase activity of IkB kinase [16]. The expression of both TLR4 and IL-1R1 is transcriptionally regulated by NF-κB. NF-κB recruits E2F1 as a transcription partner to activate its genome-wide LPS-responsive genes that include TLR4 [26]. In hemorrhage-induced sterile inflammation, the inflammatory TLR4 signaling is initiated by its interaction with HMGB1 [27]. HMGB1 is a critical DAMP released by the injured tissues after hemorrhage and ischemia-reperfusion injury [27]. HMGB1 inhibitor glycyrrhizin has been shown to prevent tissue injury in animal models of ischemia-reperfusion injury[28, 29]. Therefore, it is reasonable to expect that lowered TLR4 expression will suppress inflammatory signaling originating from TLR4-HMGB1 interaction. The acute lung injury associated with hemorrhagic shock is dependent on the expression of TLR4 [30]. In our recent work, we have demonstrated that EF24 also reduces the levels of HMGB1 in lung tissue[31].
IL-1R1 is a receptor for IL-1α and IL-1β, and IL-1R signaling is the major pro-inflammatory pathway in hemorrhagic shock and ischemia-reperfusion injury. Tagawa et al, have shown that soluble IL-1R1 competitively inhibits IL-1/IL-1R1 signaling and ameliorates ischemia-reperfusion injury in orthotopic left lung isograft transplant model by improving graft oxygenation and reducing lung edema and neutrophil sequestration [32]. The expression of IL-1R1 is controlled by NF-κB, conceivably through the latter’s influence on pro-inflammatory cytokines such as IL-1β and IL-6 [33, 34]. These proinflammatory cytokine are mainly produced by macrophages, but secretion of IL-6 has also been shown from lung epithelial cells [35]. On the other hand, the main source of IL-1β appears to be the neutrophil infiltrate in inflamed lung [36], which induces IL-6 secretion from lung epithelium [37, 38]. We hypothesize that by inhibiting NF-κB, EF24 reduces the levels of these cytokines, which in turn attenuates the signaling through IL-1R1.
Contrary to IL-1R1 and TLR4, ST2 and SIGIRR are purported to be the negative regulators of inflammation. They are not known to bind IL-1α or IL-1β. Although the role of ST2 is not entirely clear, it has been speculated that ST2 sequesters MyD88, making latter unavailable for other MyD88-dependent pro-inflammatory IL-1R pathways [7]. We found that EF24 treatment increased soluble ST2 expression in LPS-stimulated DCs (unpublished). Soluble ST2 has also been reported to attenuate inflammation and lethality after intestinal ischemia and reperfusion by inducing IL-10 production in lung and intestines [39]. Unlike ST2, the SIGIRR-mediated negative regulation of inflammation follows a different mechanism. The Ig domain of SIGIRR interferes with the heterodimerization of IL-1R to its accessory proteins, whereas its intracellular TIR domain inhibits IRAK-TRAF6 interaction, thus inhibiting the activation of NF-κB[21]. The mice lacking SIGIRR show hyper-responsiveness to endotoxin challenge and IL-1 administration, accompanied by enhanced activation of TLR/IL-1 pathways [9]. Also, the loss of function mutation in the SIGIRR gene has been found to predispose ischemic kidney to acute failure, whereas wild type SIGIRR prevented tissue injury by suppressing the post-ischemic activation of intra-renal myeloid DCs [40]. Looking at these anti-inflammatory influences of ST2 and SIGIRR, we consider that any increase in their expression would have beneficial effect in controlling inflammation.
CONCLUSIONS
The results in the rat model of fixed-volume hemorrhage suggest that EF24 attenuates hemorrhage-induced inflammation at various levels of intracellular inflammatory signaling and proinflammatory mediators. It is indicated that EF24 not only reduces pro-inflammatory mediators, but also potentiates negative regulatory arm in the same pathway. These observations corroborated with the recently published anti-inflammatory effects of EF24 in the in vivo model of hemorrhage-induced inflammation[31]. The use of conventional anti-inflammatory drugs to combat trauma-associated inflammation is crippled by some serious side-effects. For instance, steroids are immunosuppressive, whereas COX-1 and selective COX-2 inhibitors are associated with bleeding and thromboembolic cum cardiovascular side-effects, respectively [41, 42]. In order to address inflammation more comprehensively, drugs modulating transcriptional control of inflammatory processes have been suggested [43]. On a cautionary note, it is important to acknowledge the role of other transcriptional elements, such as activator protein-1 (AP-1), in initiation and propagation of inflammation and much of cross-talk among their signaling pathways creates a complicated scenario. However, NF-κB is the most important transcription regulator in inflammation, regulating more than 60 pro-inflammatory genes [44, 45]. Its role has also been linked to the hemodynamic changes observed during shock [46]. Considering the potent NF-κB inhibition by EF24, as reported by us[15, 31], we speculate that EF24 is worth investigating as an adjunct salutary anti-inflammatory agent in resuscitation strategies. The reported maximum tolerated dose of EF24 is 400 mg/Kg body weight (i.p.) and 200 mg/Kg body weight (i.v.) [14], which provides a large therapeutic window for its reported usage in the hemorrhagic pathology. At the same time, it is also important to investigate the generalized immunosuppressive and coagulopathic effects of NF-κB inhibition.
Acknowledgments
The work was supported by a grant from National Heart, Lung & Blood Institute [R01HL104286].
Footnotes
Presented at the 4th International Symposium on Artificial Oxygen Carriers, September 28, 2013 Yokohama, Japan
References
- 1.McGhan LJ, Jaroszewski DE. The role of toll-like receptor-4 in the development of multi-organ failure following traumatic haemorrhagic shock and resuscitation. Injury. 2012;43:129–36. doi: 10.1016/j.injury.2011.05.032. [DOI] [PubMed] [Google Scholar]
- 2.Gruen RL, Brohi K, Schreiber M, Balogh ZJ, Pitt V, Narayan M, et al. Haemorrhage control in severely injured patients. Lancet. 2012;380:1099–108. doi: 10.1016/S0140-6736(12)61224-0. [DOI] [PubMed] [Google Scholar]
- 3.Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury multiple organ failure. Injury. 2009;40:912–8. doi: 10.1016/j.injury.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 4.Kobbe P, Lichte P, Schreiber H, Reiss LK, Uhlig S, Pape HC, et al. Inhalative IL-10 attenuates pulmonary inflammation following hemorrhagic shock without major alterations of the systemic inflammatory response. Mediators Inflamm. 2012;2012:512974. doi: 10.1155/2012/512974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavaillon JM. Pro- versus anti-inflammatory cytokines: myth or reality. Cell Mol Biol (Noisy-le-grand) 2001;47:695–702. [PubMed] [Google Scholar]
- 6.Namas R, Ghuma A, Hermus L, Zamora R, Okonkwo DO, Billiar TR, et al. The acute inflammatory response in trauma/hemorrhage and traumatic brain injury: current state and emerging prospects. Libyan J Med. 2009;4:97–103. doi: 10.4176/090325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brint EK, Xu D, Liu H, Dunne A, McKenzie AN, O’Neill LA, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–9. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 8.Dunne A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 9.Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–7. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 10.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274:10689–92. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 11.O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–8. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 13.Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling--a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004;24:1288–304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- 14.Adams BK, Ferstl EM, Davis MC, Herold M, Kurtkaya S, Camalier RF, et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg Med Chem. 2004;12:3871–83. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Vilekar P, Awasthi S, Natarajan A, Anant S, Awasthi V. EF24 suppresses maturation and inflammatory response in dendritic cells. Int Immunol. 2012;24:455–64. doi: 10.1093/intimm/dxr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasinski AL, Du Y, Thomas SL, Zhao J, Sun SY, Khuri FR, et al. Inhibition of IkappaB kinase-nuclear factor-kappaB signaling pathway by 3,5-bis(2-flurobenzylidene)piperidin-4-one (EF24), a novel monoketone analog of curcumin. Mol Pharmacol. 2008;74:654–61. doi: 10.1124/mol.108.046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagisetty P, Powell DR, Awasthi V. Synthesis and structural determination of 3 3,5-bis(2-fluorobenzylidene)-4-piperidone analogs of curcumin. J Mol Str. 2009;936:23–8. [Google Scholar]
- 18.Awasthi V, Yee SH, Jerabek P, Goins B, Phillips WT. Cerebral oxygen delivery by liposome-encapsulated hemoglobin: a positron-emission tomographic evaluation in a rat model of hemorrhagic shock. J Appl Physiol. 2007;103:28–38. doi: 10.1152/japplphysiol.00136.2006. [DOI] [PubMed] [Google Scholar]
- 19.Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, et al. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–23. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- 20.Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J Biol Chem. 2002;277:3863–9. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- 21.Qin J, Qian Y, Yao J, Grace C, Li X. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J Biol Chem. 2005;280:25233–41. doi: 10.1074/jbc.M501363200. [DOI] [PubMed] [Google Scholar]
- 22.Molina PE, Malek S, Lang CH, Qian L, Naukam R, Abumrad NN. Early organ-specific hemorrhage-induced increases in tissue cytokine content: associated neurohormonal and opioid alterations. Neuroimmunomodulation. 1997;4:28–36. doi: 10.1159/000097312. [DOI] [PubMed] [Google Scholar]
- 23.Song Y, Ao L, Raeburn CD, Calkins CM, Abraham E, Harken AH, et al. A low level of TNF-alpha mediates hemorrhage-induced acute lung injury via p55 TNF receptor. Am J Physiol Lung Cell Mol Physiol. 2001;281:L677–84. doi: 10.1152/ajplung.2001.281.3.L677. [DOI] [PubMed] [Google Scholar]
- 24.Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR. Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–7. doi: 10.1097/01.shk.0000228797.41044.08. [DOI] [PubMed] [Google Scholar]
- 25.Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS) Ann Intern Med. 1996;125:680–7. doi: 10.7326/0003-4819-125-8-199610150-00009. [DOI] [PubMed] [Google Scholar]
- 26.Lim CA, Yao F, Wong JJ, George J, Xu H, Chiu KP, et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–35. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 27.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogiku M, Kono H, Hara M, Tsuchiya M, Fujii H. Glycyrrhizin prevents liver injury by inhibition of high-mobility group box 1 production by Kupffer cells after ischemia-reperfusion in rats. J Pharmacol Exp Ther. 2011;339:93–8. doi: 10.1124/jpet.111.182592. [DOI] [PubMed] [Google Scholar]
- 29.Mabuchi A, Wake K, Marlini M, Watanabe H, Wheatley AM. Protection by glycyrrhizin against warm ischemia-reperfusion-induced cellular injury and derangement of the microcirculatory blood flow in the rat liver. Microcirculation. 2009;16:364–76. doi: 10.1080/10739680902796917. [DOI] [PubMed] [Google Scholar]
- 30.Lv T, Shen X, Shi Y, Song Y. TLR4 is essential in acute lung injury induced by unresuscitated hemorrhagic shock. J Trauma. 2009;66:124–31. doi: 10.1097/TA.0b013e318181e555. [DOI] [PubMed] [Google Scholar]
- 31.Yadav VR, Sahoo K, Roberts PR, Awasthi V. Pharmacologic suppression of inflammation by a dipheyldifluoroketone, EF24, in a rat model of fixed-volume hemorrhage improves survival. J Pharmacol Exp Ther. 2013 doi: 10.1124/jpet.113.208009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tagawa T, Dharmarajan S, Hayama M, Ishiyama T, Suda T, Itano H, et al. Endobronchial gene transfer of soluble type I interleukin-1 receptor ameliorates lung graft ischemia-reperfusion injury. Ann Thorac Surg. 2004;78:1932–9. doi: 10.1016/j.athoracsur.2004.06.008. discussion 9. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature. 2004;430:218–22. doi: 10.1038/nature02738. [DOI] [PubMed] [Google Scholar]
- 34.Ito A, Takii T, Matsumura T, Onozaki K. Augmentation of type I IL-1 receptor expression and IL-1 signaling by IL-6 and glucocorticoid in murine hepatocytes. J Immunol. 1999;162:4260–5. [PubMed] [Google Scholar]
- 35.Cromwell O, Hamid Q, Corrigan CJ, Barkans J, Meng Q, Collins PD, et al. Expression and generation of interleukin-8, IL-6 and granulocyte-macrophage colony-stimulating factor by bronchial epithelial cells and enhancement by IL-1 beta and tumour necrosis factor-alpha. Immunology. 1992;77:330–7. [PMC free article] [PubMed] [Google Scholar]
- 36.Coulter KR, Wewers MD, Lowe MP, Knoell DL. Extracellular regulation of interleukin (IL)-1beta through lung epithelial cells and defective IL-1 type II receptor expression. Am J Respir Cell Mol Biol. 1999;20:964–75. doi: 10.1165/ajrcmb.20.5.3458. [DOI] [PubMed] [Google Scholar]
- 37.Crestani B, Cornillet P, Dehoux M, Rolland C, Guenounou M, Aubier M. Alveolar type II epithelial cells produce interleukin-6 in vitro and in vivo. Regulation by alveolar macrophage secretory products. J Clin Invest. 1994;94:731–40. doi: 10.1172/JCI117392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manzer R, Wang J, Nishina K, McConville G, Mason RJ. Alveolar epithelial cells secrete chemokines in response to IL-1beta and lipopolysaccharide but not to ozone. Am J Respir Cell Mol Biol. 2006;34:158–66. doi: 10.1165/rcmb.2005-0205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fagundes CT, Amaral FA, Souza AL, Vieira AT, Xu D, Liew FY, et al. ST2, an IL-1R family member, attenuates inflammation and lethality after intestinal ischemia and reperfusion. J Leukoc Biol. 2007;81:492–9. doi: 10.1189/jlb.0606422. [DOI] [PubMed] [Google Scholar]
- 40.Lech M, Avila-Ferrufino A, Allam R, Segerer S, Khandoga A, Krombach F, et al. Resident dendritic cells prevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J Immunol. 2009;183:4109–18. doi: 10.4049/jimmunol.0900118. [DOI] [PubMed] [Google Scholar]
- 41.Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P. Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 2007;82:85–94. doi: 10.1016/j.prostaglandins.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 42.Marwali MR, Mehta JL. COX-2 inhibitors and cardiovascular risk. Inferences based on biology and clinical studies. Thromb Haemost. 2006;96:401–6. [PubMed] [Google Scholar]
- 43.Wu KK. Transcription-based COX-2 inhibition: a therapeutic strategy. Thromb Haemost. 2006;96:417–22. [PubMed] [Google Scholar]
- 44.DiDonato JA, Mercurio F, Karin M. NF-kappa B and the link between inflammation and cancer. Immunological Reviews. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 45.Villavicencio RT, Billiar TR. The role of nitric oxide in the initiation of inflammation in shock. In: Marshall JC, Cohen JC, Vincent J-L, editors. Immune response in the Critically Ill. Berlin Heidelberg: Springer-Verlag; 1999. pp. 182–9. [Google Scholar]
- 46.Doursout M-FJ, Liang YY, Uray KS, Pati S, Matijevic N, Holcomb JB. Role of the NF-kB pathway in rats subjected to moderate hemorrhage. FASEB J. 2009;23:LB380. Meeting Abstract Supplement. [Google Scholar]