

Abstract
This retrospective review on discoveries of the roles of oxidative stress in brain of subjects with Alzheimer disease (AD) and animal models thereof as well as brain from animal models of chemotherapy induced cognitive impairment (CICI) results from the author receiving the 2013 Discovery Award from the Society for Free Radical Biology and Medicine. The paper reviews our laboratory's discovery of: protein oxidation and lipid peroxidation in AD brain regions rich in amyloid β-peptide (Aβ) but not in Aβ-poor cerebellum; redox proteomics as a means to identify oxidatively modified brain proteins in AD and its earlier forms that are consistent with the pathology, biochemistry, and clinical presentation of these disorders; how Aβ in in vivo, ex vivo, and in vitro studies can lead to oxidative modification of key proteins that also are oxidatively modified in AD brain; the role of the single methionine residue of Aβ(1-42) in these processes; and some of the potential mechanisms in the pathogenesis and progression of AD.
CICI affects a significant fraction of the 14 million American cancer survivors, and due to diminished cognitive function, reduced quality of life of the persons with CICI (called “chemobrain” by patients) often results. A proposed mechanism for CICI employed the prototypical ROS-generating and non-blood brain barrier (BBB)-penetrating chemotherapeutic agent doxorubicin (Dox, also called adriamycin, ADR). Because of the quinone moiety within the structure of Dox, this agent undergoes redox cycling to produce superoxide free radical peripherally. This, in turn, leads to oxidative modification of the key plasma protein, Apolipoprotein A1 (ApoA1). Oxidized ApoA1 leads to elevated peripheral TNFα, a pro-inflammatory cytokine that crosses the BBB to induce oxidative stress in brain parenchyma that affects negatively brain mitochondria. This subsequently leads to apoptotic cell death resulting in CICI. This review outlines aspects of CICI consistent with the clinical presentation, biochemistry, and pathology of this disorder. To the author's knowledge this is the only plausible and self-consistent mechanism to explain CICI.
These two different disorders of the CNS affect millions of persons worldwide. Both AD and CICI share free radical-mediated oxidative stress in brain, but the source of oxidative stress is not the same.
Continued research is necessary to better understand both AD and CICI. The discoveries about these disorders from the Butterfield laboratory that led to the 2013 Discovery Award from the Society of Free Radical and Medicine provides a significant foundation from which this future research can be launched.
Keywords: 2013 SFRBM Discovery Award, Alzheimer disease (AD) and its earlier forms (amnestic MCI and preclinical AD), Aβ(1-42) associated oxidative stress, redox proteomics, chemotherapy induced cognitive impairment (“chemobrain”), plasma derived elevated TNFα and its sequela in brain
1. Introduction
Being presented the 2013 Discovery Award from the Society for Free Radical Biology & Medicine at its annual national meeting in San Antonio, TX in November 2013 ranks as one of the significant highlights of my academic career along with my being awarded the Presidential Award for Excellence in Science, Mathematics, and Engineering Mentoring by President Clinton in the White House in 1998, being elected a Fellow of SFRBM in 2012, and receiving the Alkmeon International Prize for Progress in Science from the European Brain Research Institute in 2014 in Rome, Italy.
Counting my three years as a Ph.D. student and two years as a NIH Postdoctoral Fellow at Duke University or Duke University School of Medicine in Physical Chemistry and Neuroscience, respectively, I have been involved in academic biomedical research for nearly 44 years, with most of this time at the University of Kentucky. Highly talented graduate students, postdoctoral scholars, visiting scientists, and undergraduate students, along with outstanding University of Kentucky collaborators and collaborators worldwide, have contributed greatly to the research that led to the Discovery Award and that has led to over 580 refereed scientific publications from my laboratory. I trained more than 60 Ph.D. and M.S. students, over 150 undergraduates, 30 postdoctoral scholars and visiting scientists, and all have been critical to the success of the laboratory in making discoveries about oxidative stress in brain in subjects with neurodegenerative disorders and in patients following certain chemotherapy regimens. So, to this outstanding group of scientists, including University of Kentucky and worldwide collaborators, this grateful scientist is highly appreciative and dedicates this review to each of you.
Our laboratory has been involved in the beginning with discovering oxidative stress in brains from subjects with aged related neurodegenerative disorders, especially Alzheimer disease (AD) through all its stages, but also in model systems of AD, Parkinson disease (PD), Huntington disease (HD), and amyotrophic lateral sclerosis (ALS). In addition, we have investigated oxidative stress in various tissues from patients with chemotherapy induced cognitive impairment (CICI, often called “chemobrain” by patients) and models thereof. The organization of this review will highlight some significant discoveries and achievements from humans and model systems related first to AD and then to CICI.
Since the Discovery Award was presented to me based on research in our laboratory, I will focus this review on mostly our own work. Other laboratories have made highly important contributions related to oxidative stress in AD, and omission of citation of references of such work is not meant to convey any message other than this review focuses on aspects of our own laboratory's work related to AD associated with the 2013 Discovery Award from SFRBM. Therefore, with any unintended hubris, I respectfully ask the indulgence of all appropriate researchers.
2. Oxidative Stress Indices and Redox Proteomics Employed in Our Laboratory
Oxidative stress is a condition that arises when free radical production exceeds biochemical means of scavenging these free radicals, and is manifested, among other ways, by protein oxidation and lipid peroxidation [1]. For markers for protein oxidation, protein carbonyls [2] and 3-nitrotyrosine [3] are the most prominently used, and while several markers of lipid peroxidation are available [4], protein-bound HNE (4-hydroxy-2-nonenal) [5] is most often used in our laboratory (Table I). Protein carbonyls arise as a consequence of at least four processes involving free radical reactions [2]: (a) Cleavage of the peptide backbone to produce carbon-centered radicals that combine rapidly with paramagnetic oxygen; (b) Oxidation of specific protein side chains, for example, Lys, His, Thr, etc. (c) Covalent modification by reactive products of lipid peroxidation, e.g., alkenals like HNE via Michael addition; and (d) Glycoxidation of oligosaccharide chains. HNE is produced from a lipid acyl chain hydroperoxide, particularly that from arachidonic acid. The hydroperoxide is formed following free radical attack on allylic H atoms of these acyl chains and subsequent chain reaction involving oxygen reaction with carbon-centered free radicals and further attack on allylic H atoms on acyl chains of lipids [1]. 3-Nitrotyrosine is formed by reaction of tyrosine with peroxynitrite (ONOO-), the latter, in turn, formed following reaction of nitric oxide and superoxide free radical and subsequent involvement of carbon dioxide [3].
Table I. Commonly Used Indices of Protein Oxidation and Lipid Peroxidation, Two Key Markers of Oxidative Stress, Employed in the Butterfield Laboratory*.
| Index | Comments | |
|---|---|---|
| Protein Oxidation | ||
| Protein Carbonyls (PC) | 3-Nitrotyrosine (3-NT) | PC quantified by immunochemical detection of 2,4-dintrophenylhydrazone adduct to proteins. 3-NT is produced following formation of ONOO- from reaction of NO and superoxide radical anion. Subsequent involvement of CO2 and scission of nitrosoperoxylcarbonate leads to NO2. 3-NT is detected immunochemically. |
| Lipid Peroxidation | ||
| Protein-bound 4-hydroxy-2-nonenal (HNE) | HNE is formed from oxidized arachidonic acid and binds to Cys, His, and Lys on proteins via Michael addition. | |
See Butterfield and Stadtman [2] for more details
Redox proteomics, pioneered in the Butterfield laboratory [6], is that branch of the field of proteomics used to identify proteins oxidatively modified by one or more of the processes noted above. Detailed methodology for protein identification and caveats of the methods are given in recent reviews [6-8] (see Fig. 1).
Figure 1.
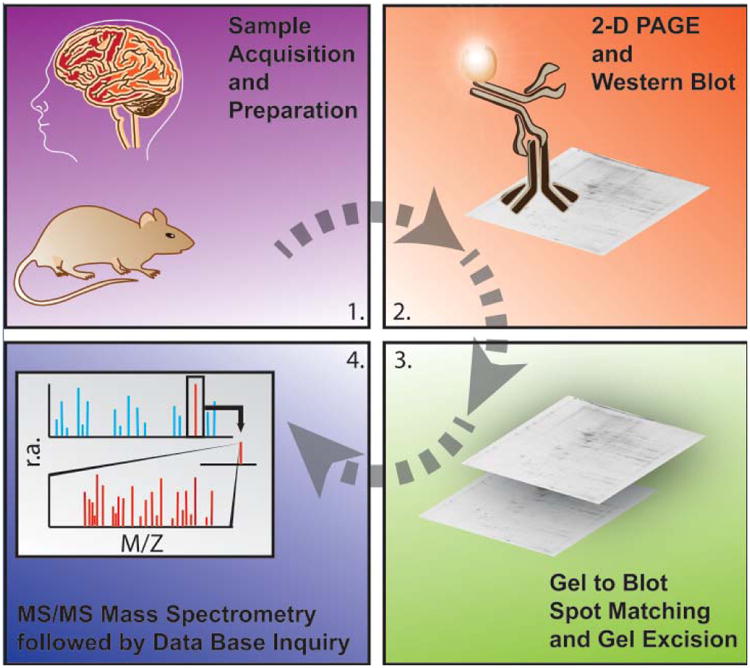
Schematic representation of the major methodological steps involved in redox proteomics. For more details see recent reviews in [6, 7]. The figure, used with permission from Elsevier Science Publishers, is taken from reference [122].
3. Alzheimer Disease
3.1 Brief Background of AD
The cause of the largest fraction (about 70-75 percent) of dementia in the USA, AD is an age-dependent progressive neurodegenerative disorder characterized by loss of cognition, including reasoning and memory. Approximately 5.5 million Americans currently are diagnosed with AD, and recent studies suggest that with proper coding of the actual cause of death of AD patients would rank AD as the 3rd or 4th leading cause of death in the USA [9]. Pathology may occur at least 20 years before the onset of symptoms, so diagnostic biomarkers are clearly needed for this devastating disease. The three major characteristic neuropathological hallmarks of AD include the presence of senile plaques (SP, composed of a core of amyloid β-peptide (Aβ) surrounded by dystrophic neuritis with several associated proteins), neurofibrillary tangles (NFT, composed of hyperphosphorylated tau protein) [10] and the loss of synapses in brain parenchyma [11]. Aβ, a 39-43 amino acid peptide (Fig. 2) is formed by proteolytic cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase (Fig. 3).
Figure 2.

Sequences of Aβ(1-42) and variants of this sequence mentioned in this retrospective paper. Note the key methionine residue at position 35 of Aβ(1-42).
Figure 3.
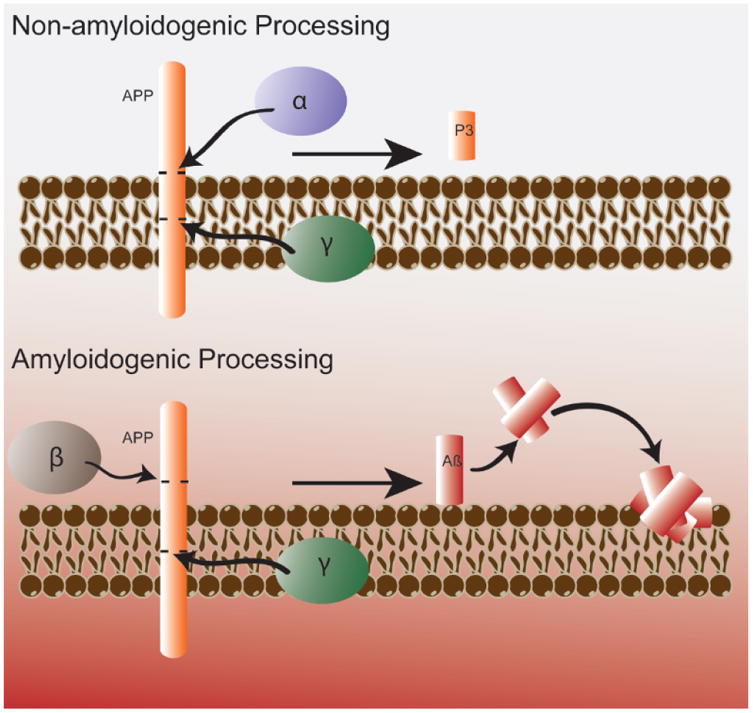
Non-amyloidogenic (Top) and amyloidogenic (Bottom) pathways of APP processing. Note the latter leads to olgimeric Aβ(1-42) that enters the lipid bilayer leading to damaging lipid peroxidation and its sequelae as discussed in the text. The figure, used with permission from Elsevier Science Publishers, is taken from reference [122].
Mild cognitive impairment [12]; [13], particularly amnestic MCI, is arguably a prodromal form of AD. Patients with amnestic MCI have memory loss but normal activities of daily living. Preclinical AD (PCAD) is gaining acceptance as the earliest form of AD [14]. Patients with PCAD have normal indices of cognition but significant AD-relevant pathology in their brains.
The amyloid cascade hypothesis, suitably updated, has provided a framework for understanding clinical presentation and pathology of AD [15]. Although SP density does not correlate with performance on instruments of cognition, Aβ(1-42) oligomer density does [15]. Oligomers of Aβ(1-42) injected into mice brain significantly diminish long-term potentiation (LTP), which is needed for learning and memory [16]. Many other detrimental biological effects of Aβ(1-42) oligomers are known, and most researchers in the AD field accept that oligomers are the toxic species of Aβ(1-42) [17].
Our laboratory along with the late William R. Markesbery developed the notion of another complementary component of AD pathogenesis, namely, the involvement of free radical oxidative stress in the pathogenesis and progression of AD [18, 19], with our laboratory emphasizing the role of Aβ(1-42) oligomers in this oxidative stress [20, 21].
This part of this invited review associated with the 2013 Discovery Award from the Society for Free Radical Biology and Medicine will highlight some discoveries from our laboratory of the role of Aβ(1-42)-associated oxidative stress in the pathogenesis and progression of AD.
3.2. Studies of Oxidative Stress in Brain of Subjects of AD and Its Earliest Stages from the Butterfield Laboratory
We demonstrated for the first time that protein oxidation indexed by elevated protein carbonyls was localized to brain regions of subjects with AD that were rich in Aβ(1-42), viz, hippocampus and inferior parietal lobule, but not in brain regions poor in Aβ, e.g., cerebellum [22]. Subsequently, we, showed that elevated protein-bound HNE was observed in AD brain [4, 23-25], complementing research by Markesbery and colleagues, who showed that free HNE was elevated in AD brain [26]. We also demonstrated that 3-NT was elevated in AD brain in the same regions in which protein carbonyls were elevated [27, 28]. Similar studies in brain of subjects with amnestic MCI demonstrated elevated levels of protein carbonyls [29, 30], protein-bound HNE [31, 32], and 3-NT [33]. Similar studies in early AD revealed elevated protein-bound HNE [34]. The inferior parietal lobule region from subjects with PCAD in our hands did not show evidence of oxidative stress [35], while others reported only HNE elevation in hippocampus from PCAD subjects [36]. Taken together, our studies suggested that oxidative stress in brain occurred early in the progression of AD.
The lipid peroxidation product HNE and Aβ(1-42) were shown by us to cause loss of phospholipid asymmetry in synaptic membranes [37, 38]. The loss of lipid asymmetry in brain membranes caused by Aβ(1-42) was prevented by several antioxidant compounds (Structures given in Fig. 4). Exposure of phosphatidylserine (PtdSer) to the outer bilayer lamella normally invokes a response by membrane-bound flippase, an ATP-dependent enzyme with a critical cysteine in its active site, to return PtdSer to its normal location, the inner lamella of the lipid bilayer. Loss of lipid asymmetry by HNE and Aβ(1-42)-produced HNE conceivably results from binding of this alkenal to the active site cysteine of flippase. Using this information, we determined that synaptic membranes from AD and amnestic MCI inferior parietal lobule had lost lipid asymmetry, i.e., greater exposure of PtdSer to the outer bilayer lamella [39], which is a marker of apoptotic cells. These results showed that loss of lipid asymmetry occurred early in the progression of AD and were correlated with levels of Aβ(1-42). Similar studies with the human double mutant APP/PS-1 knock in mouse model of AD showed loss of lipid asymmetry in brain synaptic membranes occurred as a function of age, reaching a maximum loss with the onset of Aβ(1-42) deposition [40].
Figure 4.
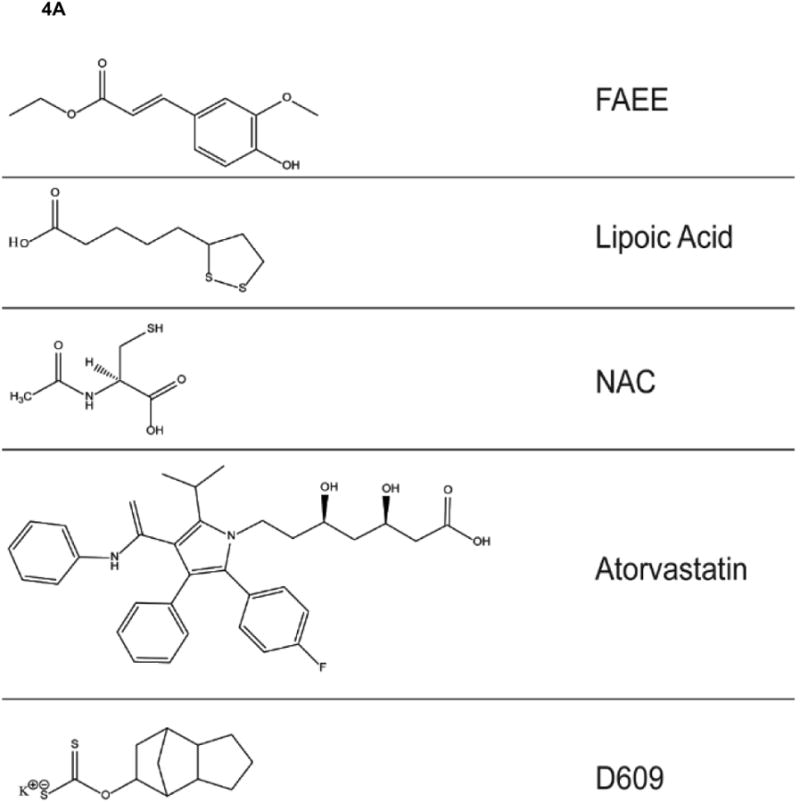
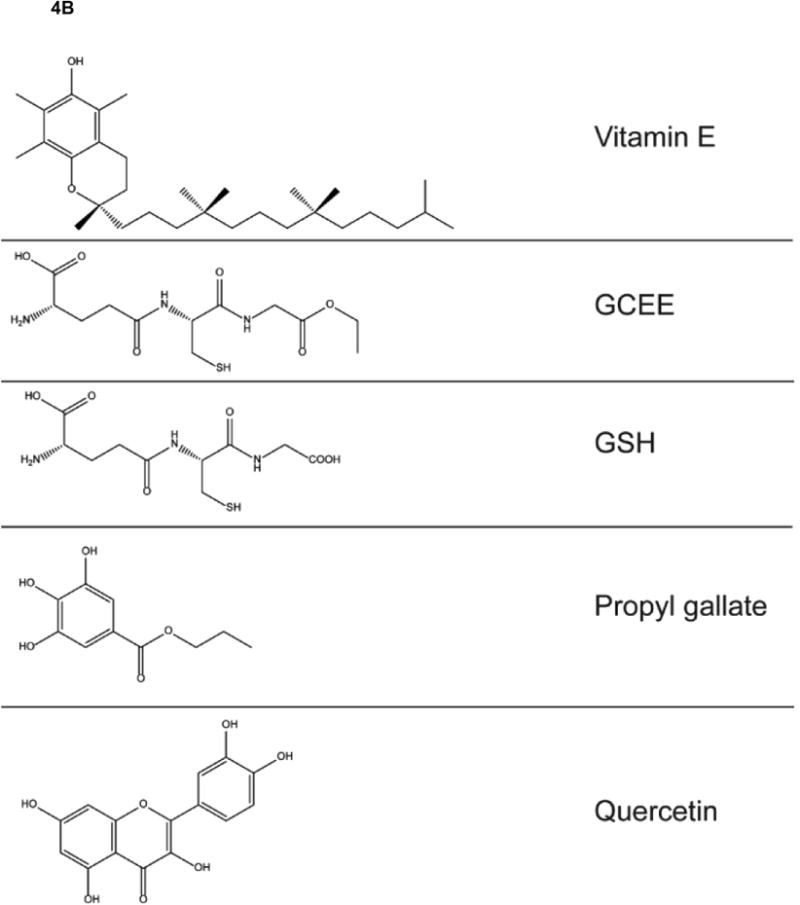
A,B. Structures of antioxidant compounds mentioned in this retrospective paper.
Redox proteomics, pioneered in our laboratory, is a subset of the field of proteomics that leads to the identification of oxidatively modified and, almost always, dysfunctional proteins [7, 8, 41, 42]. Metabolic proteins were identified by redox proteomics as oxidatively dysfunctional in AD and aMCI brain, consistent with the observation that glucose utilization is markedly depressed in both conditions, with the AD brain showing greater effects than the former [6, 25, 27, 29, 32, 43-49]. Among these proteins were various glycolytic enzymes, TCA enzymes, and components of ATP synthase. Lack of ATP is consistent with altered synaptic function in AD and induced by Aβ(1-42) oligomers [16]. Diminution of ATP also would greatly affect cellular and mitochondrial potential, glutamate neurotransmission, learning and memory, and maintenance of lipid asymmetry.
All these functions are lost in AD and aMCI, demonstrating again that redox proteomics-identified proteins often mirror the pathology and clinical presentation of this disease. One such protein that appears oxidatively modified at all stages of AD is enolase [50]. This protein is not only involved in glycolysis, but plays roles in activation of plasminogen, which in turn is activated to plasmin, a protease that, among other proteins, targets Aβ(1-42) for destruction. Moreover, enolase through c-myc activates pro-survival pathways involving ERK1/2. Thus, oxidatively dysfunctional enolase in AD would lead to elevated neurotoxic Aβ(1-42) and to loss of pro-survival processes [50], both effects consistent with observations in AD brain. Moreover, comparison of brain from subjects with PCAD, who have no cognitive loss, to those of subjects with amnestic MCI, who have only memory loss but no dementia, revealed that enolase is oxidatively modified in aMCI brain [51], consistent with the notion that this protein is involved in memory, and its oxidation may be associated with loss of memory in this early form of AD.
Other classes of proteins identified by redox proteomics or Western blot methods in our laboratory as oxidatively modified and likely dysfunctional in AD and/or aMCI brain include proteins that normally function as: chaperones [52]; metabolic proteins [53]; proteins involved neurite extension [54]; proteasomal degradation of damaged, aggregated proteins [54]; glutathionylation [55-57] proteins involved in antioxidant defense [27, 28, 45, 47, 58, 59] cell cycle proteins [60, 61] p53-mediated non-transcriptional apoptosis [62, 63] glycoproteins [64]; [65, 66] proteins associated with PSD95 [67]; LRP-1, a major clearance protein that removes Aβ(1-42) from brain to blood [68]; heme oxygenase-1 that degrades pro-oxidant heme forming biliverdin [69]; biliverdin reductase, which rapidly converts biliverdin to the antioxidant and denitrifying molecule, bilirubin, is involved in various pro-survival signaling pathways [70, 71] lipoamide dehydrogenase-mediated reduction of lipoic acid [72]; and protein synthesis [25].
These oxidatively modified and dysfunctional proteins are consonant with the clinical presentation and pathology of AD and various stages thereof. They also represent potentially important targets for therapeutic intervention in early stages of AD. And these numerous proteins, involved in many pathways, are consistent with a free radical source in AD in which any protein modified by free radicals or free radical reaction products (i.e., HNE) would by oxidatively modified, of altered structure [73], and consequently dysfunctional.
An initial redox proteomics study of brain in familial AD (FAD) was conducted by our laboratory in collaboration with Ralph Martins, now of Edith Cowan University in Australia. Brain proteins associated with energy metabolism, nitric oxide utilization, the ubiquitin-proteasome system, and the spectrin actin cytoskeletal network were found to be oxidatively modified [74]. Some of these proteins were identified as oxidatively modified and dysfunctional in brain of sporadic AD and amnestic MCI as noted above.
Down syndrome (DS) is a consequence of trisomy of chromosome 21. With improvements in medical care, persons with DS often live to the sixth decade of life or longer. At approximately 40-45 years of age, Alzheimer-like dementia appears coincident with AD-like neuropathology [75]. In collaboration with Prof. Elizabeth Head at the University of Kentucky and Prof. Marzia Perluigi of the University of Rome-La Sapienza, we determined that elevated oxidative stress occurred in frontal cortex [76] and other brain regions of DS subjects with AD-like pathology [75]. In addition, redox proteomics analyses identified brain proteins, including those associated with dysfunctional proteastasis network, as oxidatively modified and likely dysfunctional in DS subjects with AD-like pathology [77-79]. The APP gene is located on chromosome 21; consequently, there is a dose effect of Aβ(1-42) in DS that likely contributes to the oxidative stress of this disorder. Other moieties like Cu,Zn-SOD also are coded for on chromosome 21 and also may contribute to oxidative stress in this disorder. However, oxidative stress and redox proteomics-identified oxidatively modified proteins also are found early in DS: for example, amniotic fluid from mothers carrying a DS fetus had elevated indices of oxidative stress and increased oxidative modification of key proteins such as apolipoprotein A1 [80].
3.3. Potential Biomarkers of AD and Its Earlier Forms
Ideally, biomarkers of AD and its earlier forms would be found in plasma or at least cerebral spinal fluid (CSF) [81, 82]. Given that oxidative stress may be a integral aspect of the pathogenesis of AD [21, 24], oxidatively modified proteins potentially may be among such biomarkers. The Butterfield laboratory in collaboration with the Perluigi laboratory of the University of Rome-La Sapienza demonstrated decreased expression and increased oxidation of plasma haptoglobin in AD patients [83] and alterations of the HO-1/BVR-A system in plasma of probable AD patients and MCI patients [84], suggesting that these damaged proteins could be part of a panel of altered proteins to serve as a potential biomarker in AD and its earlier forms.
In addition to plasma and CSF, we proposed that oxidatively damaged mitochondria isolated from peripheral lymphocytes potentially could contribute to a biomarker for AD and MCI [85, 86]. That elevated indices of oxidative damage to mitochondria isolated from lymphocytes inversely correlated with performance on measures of cognitive function in both AD and MCI, and that proteomics analysis of these mitochondria demonstrated differential levels of key proteins involved in ATP production, protection against oxidative stress and other pathways previously identified by our proteomics studies of brains of subjects with AD and MCI noted above, support our hypothesis that mitochondria isolated from peripheral lymphocytes potentially could be part of a biomarker for AD and its earlier forms.
3.4. In Vivo Studies of Models of Alzheimer Disease
As noted above, oligomeric Aβ(1-42) is viewed by many (most) AD researchers as underlying the pathology and clinical presentation of this dementing disorder. Accordingly, we performed in vivo studies of models of AD to assess the role of Aβ(1-42) to induce oxidative stress and oxidize specific proteins. Moreover, we investigaed the role of antioxidant compounds administered in vivo on oxidative stress induced by Aβ(1-42) in vivo and ex vivo.
Using a muscle promoter, DNA corresponding to human Aβ(1-42) was inserted into C.elegans, and oxidative stress was measured [87]. The phenotype observed in this constitutive Aβ(1-42) expression system was paralysis of the worms, and elevated protein carbonyls were demonstrated. Subsequent studies using a temperature-sensitive mutant of C.elegans that allowed for controlled temporal expression of Aβ(1-42) showed that elevated protein carbonyls occurred precisely when the paralysis phenotype occurred and prior to deposition of fibrillar Aβ(1-42), suggesting that Aβ(1-42)-induced oxidized muscle proteins led to the paralysis and that oligomeric Aβ(1-42) was the causative agent [88]. Redox proteomics analysis identified worm orthologues of human brain proteins in C.elegans expressing human Aβ(1-42) that had been identified previously [89]. If the codon for Met-35 of human Aβ(1-42) were mutated to Cys, no oxidative stress was found [87].
Human double mutant APP/PS-1 knock-in mice (APPNLh/APPNLh × PS-1P264L/PS-1P264L) were studied for oxidative stress in brain [90, 91]. Use of knock in mice obviates any putative compensatory responses in the mouse, since the mouse gene is knocked out, but the mouse promoter remains. This means that human mutant APP and PS-1 genes are inserted, resulting in the correct amount and correct location of the human proteins, albeit with mutations that lead to Aβ(142) deposition beginning at 6 months, becoming more obvious at 9 months of age, and displaying frank neuritic plaques at 12 months of age [40, 92]. Elevated protein carbonyls, protein-bound HNE, and 3-NT were observed in brain at all ages of the APP/PS-1 human double mutant knock in mouse, including embryonic neurons [93], suggesting that oxidative stress is a prominent and perhaps fundamental aspect of pathology in this AD mouse model. Consistent with this notion, this mouse model was treated in vivo with N-acetylcysteine (NAC, Fig. 4) in the drinking water, which provided the rate-limiting substrate for glutathione (GSH) elevation in brain [91]. The time of treatment spanned five months beginning at either 4 months or 7 months of age, terminating at the time of Aβ deposition or at the time of frank neuritic plaque formation, respectively. Decreased protein carbonyls in brain were found in both cases, with the magnitude of the decrease greater in brain of mice whose treatment began at an earlier age (Fig. 5). These results are consistent with the idea that elevated GSH might be a good therapeutic strategy in AD and that earlier stage treatment (amnestic MCI for example) may be more beneficial. Proteomics studies of brain from APP/PS-1 mice as a function of age identified oxidatively modified proteins in the same pathways as found in AD and MCI brain noted above [92].
Figure 5.
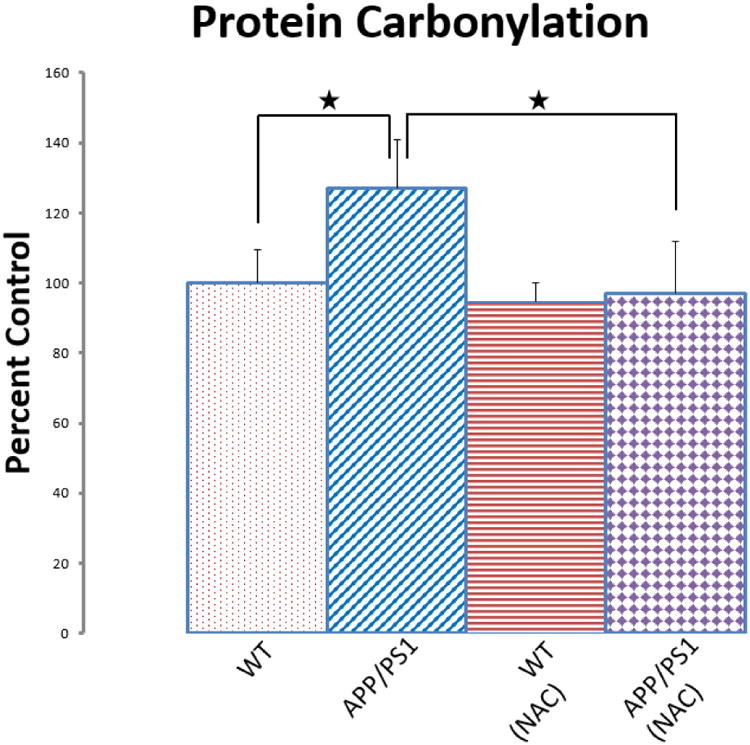
N-Acetylcysteine (NAC) given in drinking water inhibits oxidative stress in brain at 9 mos. of age of the APP/PS-1 human double mutant knock in mouse model of AD. Protein oxidation in brain assessed by protein carbonyls is shown. The figure is adapted from [91].
Similar studies of another AD mouse model, the transgenic PDAPP(Swe/Ind) mouse, also called the J20 mouse, that has two mutations in APP, showed elevated levels of human Aβ(1-42) and elevated oxidative stress in brain [94]. NAC treatment also led to decreased oxidative stress in this mouse model of AD as this antioxidant molecule did in the APP/PS-1 human double mutant knock-in mouse noted above, and proteomics identified brain proteins in this mouse that likely contributed to this neuroprotection following treatment with NAC [95].
In a different kind of in vivo study of Aβ(1-42), this neurotoxic peptide was injected into the nucleus basalis of Mynert in the forebrain of rats in collaboration with Giancarlo Pepeu of the University of Florence, his a well-known laboratory with expertise in cholinergic neurons [96]. This brain region is highly involved in AD with dramatic neuronal loss and innervated by cholinergic neurons, many axons of which project to the outer molecular layer of the hippocampus. Coupled to the known diminution of choline acetyltransferase activity in AD brain [97], the cholinergic hypothesis of AD was developed [98]. One week post injection of Aβ(1-42) to the NBM region, we determined in the hippocampus that elevated protein oxidation was present, but not in saline-injected rats. Further, redox proteomics analyses identified many oxidatively modified hippocampal proteins in Aβ(1-42)-treated rats that mirrored those in AD and MCI brain. These results are consistent with the notions that Aβ causes oxidative stress in vivo and that oxidative stress in one brain region can be directed to another connected brain region [96].
The senescence-accelerated mouse, type 8 (SAMP8), is a good model of AD as demonstrated by age-related cognitive decline and deposition of Aβ(1-42) [99, 100]. Treatment of aged SAMP8 mice with lipoic acid and/or NAC led to significantly decreased oxidative damage in brain [101]. Quantitative proteomics identified brain proteins of differential levels and oxidative modification in aged SAMP8 mice [102], and redox proteomics studies identified which brain proteins were less oxidized by lipoic acid [103]. These latter proteins were largely those found in pathways we had identified to be damaged in AD and MCI as noted above. Diminution of brain proteins thought to be involved in the pathogenesis of AD by use of antisense oligonucleotide treatment led to significantly decreased oxidative stress and decreased markers of AD pathology but increased learning and memory in aged SAMP8 mice [104-106]. In particular, antisense directed against APP [104], PS-1 [105], or glycogen synthase kinase-3β [106] led to decreased levels of Aβ(1-42) deposition and phosphorylated tau protein, respectively. These studies strongly support the notion that these proteins, known to be fundamental to pathological alterations in AD, can be reduced by appropriate treatments, and such treatments improve cognitive behavior of this AD mouse model. These results support the notion that these proteins may be potential therapeutic targets in AD and MCI.
The beagle dog deposits Aβ(1-42) of identical sequence as that of human Aβ(1-42) in the form of plaques as the animal ages. Consequently, we hypothesized that the aged beagle dog brain would show evidence of oxidative stress and that dietary intervention with a high antioxidant diet and program of behavioral enrichment would lower this oxidative damage. Drs. Carl Cotman and Elizabeth Head of the University of California at Irvine had fed 12-year old beagles over a 3-year period with a diet rich in antioxidants, including lipoic acid and polyphenols, and provided behavioral enrichments to a cohort of these dogs. These researchers demonstrated a markedly reduced error rate in learning and memory of such treated then 15 year-old dogs compared to dog-chow fed beagles of the same age [107]. Dr. Head relocated to the University of Kentucky and provided brains of these treated dogs. We found elevated protein oxidation in brain of dog-chow fed 15-year old dogs and significantly reduced protein oxidation in brain of the dogs fed a high antioxidant diet and put on a program of behavioral enrichment [108]. Both interventions were necessary to produce the decreased oxidative stress in brain, and this lower oxidative stress correlated with significantly improved learning and memory performance, to the extent that both the oxidative stress and the cognitive improvement were reminiscent of brain from a 4 year-old beagle. If this paradigm is translatable to humans, it would suggest that people strongly consider eating a healthy, high antioxidant diet and remain intellectually engaged throughout their lifespan, and, as they are able, to engage in regular exercise to increase probability of a healthy brain and lower one's risk of AD as one ages.
Statins, such as atorvastatin, do not cross the blood brain barrier in a significant amount; yet, statins may have neuroprotective effects [109]. How can this neuroprotection occur? In collaboration with Dr. Elizabeth Head's laboratory, we used the beagle dog to investigate further the neuroprotective effects of atrovastatin. Middle-aged dogs were treated over a year with atrovastatin or vehicle. Several important findings were determined: (a) There was decreased oxidative stress in brain of atrovastatin-treated dogs relative to vehicle-treated dogs, and the former had elevated levels of inducible heme oxygenase-1 that correlated with improved learning and memory [110]. (b) Since biliverdin is produced by HO-1, we examined the enzyme biliiverdin reductase-A from atovastatin- and vehicle-treated dog brains and determined that in the former treatment this enzyme is much less oxidatively modified and is phosphorylated on Ser and Thr residues, the latter required to activate the reductase activity of this enzyme [111]. The BVR-A product, bilirubin, is a good antioxidant at low levels, and protection of brain-resident BVR-A by atrovastatin therapy was correlated with improved learning and memory. (c) Inhibition of HMG-CoA reductase by statins also reduce synthesis of Co-enzyme Q10, which is an electron carrier in the electron transport chain. Hence, we treated atrovastatin-treated dogs also with CoQ10 and found improved learning and memory function [112]. Based on these studies in beagle dogs taken together, we hypothesize that statins are neuroprotective in brain of AD patients not necessarily because of cholesterol lowering, but due to decreased oxidative stress in brain associated with upregulation and decreased oxidative damage to the HO-1/BVR-A system in brain regions of relevance to AD and MCI [109, 113].
3.5. Ex Vivo Studies of Aβ-induced Oxidative Stress
Aging is the single most risk factor for AD, but another prominent risk factor for AD is inheritance of the gene, apolipoprotein E, allele 4 (ApoE). This gene has three alleles, ApoE2, ApoE3, and ApoE4. Persons who inherit two copies of ApoE4 have as much as a 70 percent chance of developing AD, while persons who inherit two copies of ApoE2 are highly unlikely to develop AD. To investigate the roles of Aβ peptide and ApoE allele status, we performed several studies. First, synaptic membranes from ApoE knockout mice were treated with Aβ(1-40), and these synaptosomes were more susceptible to oxidative stress than synaptosomes from WT mice [114]. In a second, direct test of the hypothesis that ApoE4 would confer a greater risk of oxidative stress induced by Aβ(1-42), synaptic membranes from human ApoE2, ApoE3, or ApoE4 targeted-replacement (mouse gene knocked out, human gene knocked in) mice were treated with Aβ(1-42). In all measures of oxidative stress, synaptic membranes from ApoE4 targeted-replacement mice were significantly more elevated following addition of Aβ(1-42) compared to those from ApoE2 and ApoE3 targeted-replacement mice [115]. Taken together, these results are consistent with the notion that the greater risk of developing AD by inheritance of ApoE4 may be related to the significantly more elevated oxidative stress in brain associated with Aβ(1-42).
The xanthate, tricyclodecan-9-yl-xanthogenate (D609), (Fig. 4) is a powerful antioxidant [116] that works through the thiol functionality of the molecule. Indeed, during the scavenging of radicals, the disulfide is formed, and this disulfide in turn is a substrate for glutathione reductase to recycle back to the reduced thiol [117]. Gerbils were injected i.p. with D609 or saline and synaptosomes were isolated. We demonstrated that Aβ(1-42) addition to the synaptosomes from saline-injected rodents led to significantly elevated protein oxidation and lipid peroxidation, both prevented in D609-injected rodents [118]. Also, mitochondria isolated from brain of the D609- or saline-injected rodents were treated with Aβ(1-42) and those of saline-treated rodents had significantly elevated oxidative stress that was prevented in brain mitochondria isolated from D609-treated animals [119]. It is unlikely that free D609 could be responsible for this neuroprotection, since in order to produce synaptosomes or to isolate mitochondria numerous centrifugation and washing steps are required. Rather, we hypothesize that D609 induces a cellular stress response that leads to upregulation of protective genes, likely through a Nrf-2 response. A similar hypothesis was confirmed in an analogous study involving ferulic acid ethyl ester (FAEE) (Fig. 4). Ferulic acid has the structure of essentially half of curcumin, and the ethyl ester functionality permits transfer across the BBB [118]. Synaptic membranes were obtained from rodents treated i.p. with FAEE and treated with Aβ(1-42) or saline. Elevated protein oxidation and lipid peroxidation were demonstrated in synaptic membranes from saline-treated rodents but prevented in FAEE-treated rodents [118]. In vivo treatment of D609 or FAEE protected synaptic membranes from loss of Aβ(1-42)-mediated lipid asymmetry [38]. This neuroprotection was associated with elevated levels of heat shock protein 70, heme oxygenase-1 and decreased levels of i-NOS, a result that also was observed in primary neuronal membranes treated with Aβ(1-42) and FAEE [120], suggesting a cellular stress response (sometimes called hormesis).
The unifying theme of in vivo and ex vivo studies of Aβ(1-42) is that this neurotoxic peptide is associated with free radical oxidative stress in brain. It is our laboratory's contention that this peptide contributes extensively and may be the critical aspect of the significant oxidative stress under which brain of amnestic MCI and AD patients exists [21, 24, 121, 122].
3.6. In Vitro Studies of Aβ-Mediated Oxidative Stress and Neurotoxicity
We demonstrated that incubation of Aβ or Aβ oligomers caused protein oxidation and neurotoxicity in rat embryonic neuronal cultures that was abrogated by the antioxidants propylgallate, vitamin E, quercetin, upregulation of glutathione, D609, or FAEE, [87, 120, 123-129]. Aβ addition to neuronal cultures led to altered expression of creatine kinase BB isoform and altered levels of mRNA for Cu,Zn-superoxide dismutase and Mn-superoxide dismutase [125]. Protein carbonyls were elevated on glutamine synthetase, and creatine kinase, as well as other antioxidant enzymes by Aβ peptides [130-132]. Loss of activity of these enzymes in AD brain due to oxidative modification would have profound negative effects on free radical scavenging, mitochondrial dysfunction, neurotransmission, ammonia levels and pH balance, and glucose metabolism, all of which are observed in AD brain [121]. Redox proteomics studies of synaptosomes treated with Aβ(1-42) identified a number of oxidatively modified proteins that had been identified in AD and/or MCI brain using this method [133].
Several transport systems are known to be altered in AD brain [134]. Aβ(1-42) addition to synaptic preparations or neuronal cultures was able to reproduce this oxidative modification. For example, the glutamate transporter, EAAT2 (Glt-1) was found to be oxidatively modified by the lipid peroxidation product HNE in AD brain and separately by addition of Aβ(1-42) to synaptic preparations that contain EAAT2 [23], likely accounting for the loss of activity in both AD and following addition of this peptide to astrocytic cultures [135-137]. These results are consistent with the notion that glutamate-mediated excitotoxicity is one means of neuronal death in AD.
The activities of both Na+,K+-ATPase and Ca2+-ATPase in hippocampal neuronal cultures and in human autopsy material from normal controls were inhibited following addition of Aβ(1-40) or Aβ(25-35) [138]. Dyshomeostasis of Ca2+ and cell death were observed in neuronal cultures by inhibition of these ion-motive ATPases.
Taken together, these above-cited results are consistent with Aβ-associated free radical-mediated oxidative stress and have relevance to the protein oxidation present in AD and MCI brain [24, 121].
As noted above, lipid peroxidation is a prominent feature of brain from subjects with AD and amnestic MCI [24, 25, 49, 139]. Our laboratory first demonstrated lipid peroxidation in synaptic membranes following exposure to Aβ indexed by protein-bound HNE or other means [140]; [23], and vitamin E, a lipid-soluble, chain-breaking antioxidant, prevented Aβ-mediated lipid peroxidation and ROS formation [127, 141]. Aβ-induced lipid peroxidation was subsequently demonstrated in neuronal cultures and prevented by overexpression of the anti-apoptotic protein Bcl-2 [142] or by antioxidants such as those listed above in the discussion of Aβ-induced protein oxidation. Mitochondrial lipid peroxidation also was evident following Aβ(1-42) addition [119]. Aβ(25-35) led to loss of the phosphatidylethanolamine pool of lipids [143], which also was observed in AD brain [144]. Such changes may have relevance to a recent report that altered lipid composition may be able to diagnose AD up to three years prior to onset of clinical symptoms [145]. When covalently modified by HNE, synaptic membrane proteins have altered conformation, and therefore likely altered function [73]. Such changes are consistent with the known alterations of synaptic functions in AD and MCI brain that may significantly contribute to the cognitive deficits in both disorders [16].
Taken together, these results are consistent with the notion that the known elevation of lipid peroxidation in AD and amnestic MCI has a significant contribution from free radical processes associated with Aβ(1-42) [4, 21] and that these alterations occur early in the progression of AD [4]; [49].
3.7 Role of the Single Methionine Residue of Aβ(1-42) in Oxidative Stress Associated with This Neurotoxic Peptide
Given that neurotoxic oligomers of Aβ(1-42) are associated with oxidative stress how does this occur? We hypothesized that the single methionine (Met) residue at position 35 of Aβ(1-42) is critical to the oxidative stress associated with this peptide [146-148]. Key to this hypothesis is the reasonable assertion that in order for lipid peroxidation to occur, a free radical source has to be localized to the lipid bilayer near allylic H-atoms of acyl chains of phospholipids: a reactive free radical localized outside the bilayer would simply be too reactive to diffuse into the lipid bilayer to encounter a lipid resident allylic H-atom. The free radical mediated mechanism of lipid peroxidation is shown in Fig. 6. In this case oligomeric Aβ(1-42) must be in the bilayer, consistent with the model for methionine-mediated, oxidative stress-associated neurotoxicity shown in Fig. 6.
Figure 6.
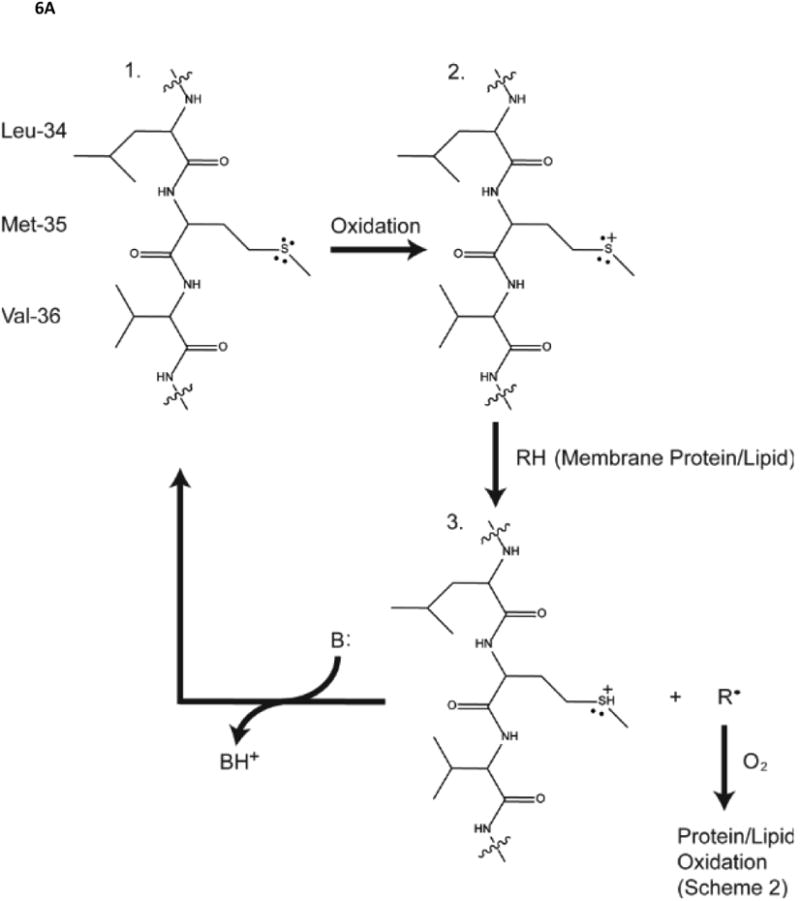
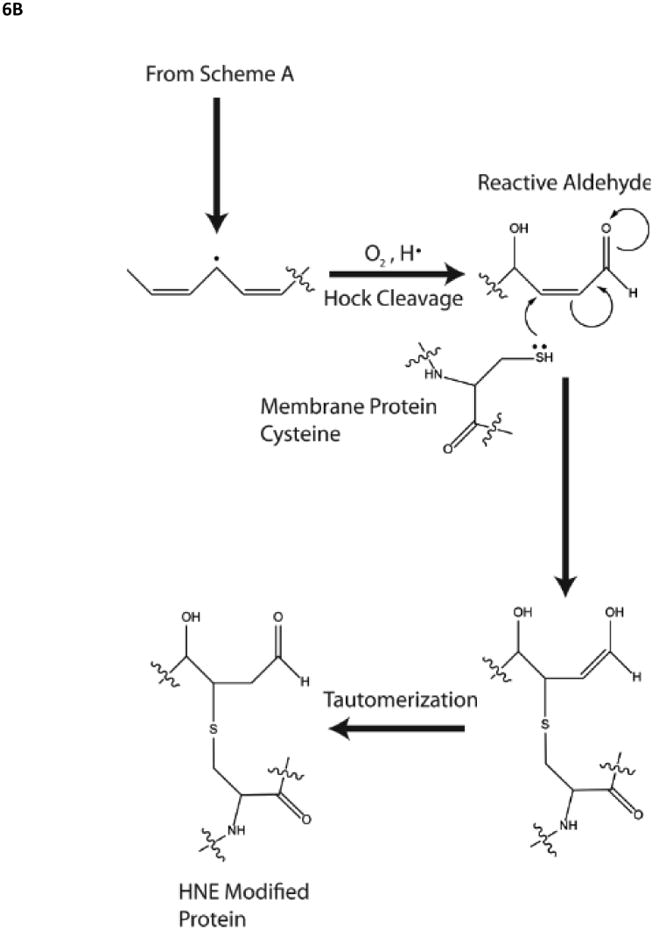
A,B. Role of the single methionine residue of Aβ(1-42) in lipid peroxidation, whose mechanism is shown. Note the HNE formed by lipid peroxidation forms Michael adducts with protein-resident Cys, His, or Lys residues, changing the conformation and decreasing function of the HNE-modified proteins.
Support for the hypothesis stated above comes from both in vitro and in vivo studies outlined below:
(A) In contrast to native Aβ(1-42), no neurotoxicity nor protein oxidation occurred in neuronal cultures treated with Aβ(1-42M35Norleucine) [20]. This latter peptide has a substitution of the Met S atom by CH2, resulting in no net change in length or dipole moment of the Met residue, but simply lacking the S atom of Met. This is a one-atom change in an approximately 4000 MW peptide. Moreover, if other amino acids of Aβ(1-42) were critical to the oxidative stress associated with this peptide, then the resulting norleucine-substituted peptide also should have elicited oxidative stress, but this did not occur [87].
(B) Substitution of Gly-37 to Asp in Aβ(1-42) has the effect of having a highly negative charge in the lipid bilayer, i.e., a medium of low dielectric constant. Consequently, this substituted Aβ peptide Met would be removed from the bilayer. That is, even though Aβ(1-42)G37D has a Met residue, localization of the Met region of the peptide to outside the lipid bilayer removed the allylic H targets for the Met-resident sulfuranyl free radical to attack and hence no lipid peroxidation [149].
(C) The great majority of proteins and peptides that reside in the lipid bilayer adopt a helical secondary structure. Given the i+4 rule of alpha-helices, this means Ile at residue 31 of Aβ(1-42) interacts with Met at residue 35. Indeed, high resolution NMR studies show that the carbonyl oxygen of Ile-31 is within a van der Waals distance of the S-atom of Met-35 [147, 150]. Since O is more electronegative than S, the lone pairs of electrons on the S atom of Met would be drawn to the O of Ile-31, loosening the influence of the positive charges on the protons in the S nucleus and making the lone pairs of electrons on the S-atom of Met-35 vulnerable to a one-electron oxidation [20, 42, 122, 151, 152]. The ultimate identity of the oxidant is unknown, but two possibilities among other conceivable moieties are molecular oxygen and Cu2+ bound to the Met-35 residue, resulting in superoxide free radical and Cu+, respectively. Paramagnetic molecular oxygen has zero dipole moment so is highly soluble in the low dielectric environment of the lipid bilayer. Cu2+ has relatively low affinity for Met, but only weak binding is necessary for electron transfer to take place. Consistent with Fig. 7 above, the attack of the resulting sulfuranyl free radical on labile allylic H atoms of acyl chains of the lipid bilayer would form an acid on the S atom of Met-35 and a C-centered free radical on the acyl chain from which the allylic H-atom originated. This C-centered free radical would immediately bind molecular oxygen in a rapid radical-radical recombination reaction forming a lipid peroxyl free radical. The latter would attack another acyl chain resident allylic H-atom to form a lipid hydroperoxide, from which HNE and other reactive alkenals, as well is isoprostanes, can form. This chain reaction process will continue as long as there are allylic H-atoms available, and this process is consistent with the observation that a “chain-breaking” antioxidant like vitamin E can prevent lipid peroxidation as noted above. The acid formed on the S atom of Met of Aβ(1-42) following allylic H-atom abstraction has a pKa of minus 5, meaning that any base would immediately remove the H+ from the S atom in Met-35, reforming the original Met residue. That is, this whole process is catalytic and explains why even a small amount of total Aβ(1-42) in the lipid bilayer that undergoes this one-electron oxidation has its oxidative effect greatly amplified by lipid peroxidation resulting from the chain reaction process described above, yielding in significant protein and lipid damage. Moreover, this process can solve the quandary of explaining the observation that clioquinol, a divalent cation chelator previously proposed by others to remove Cu2+ from Aβ plaques and hence dissolve them from brain, can work even though Cu2+ binding to Aβ-bound Cu2+ is reportedly attomolar [153], while Cu2+ binding to clioquinol is nanomolar [154]. If instead of the Cu2+ tightly bound to the three His residues (at positions 6,13,14) of Aβ(1-42), the Cu2+ chelated by clioquinol is weakly (micromolar) bound to Met-35, then that latter Cu2+ could participate in an electron transfer reaction with one of the lone pair electrons on the S-atom of Met-35, producing the sulfuranyl free radical discussed above and Cu+ [20, 42, 151]. The latter could both participate in Fenton chemistry producing damaging free radicals and potentially impart a SOD-like characteristic for Aβ(1-42) as has been proposed [155].
Figure 7.
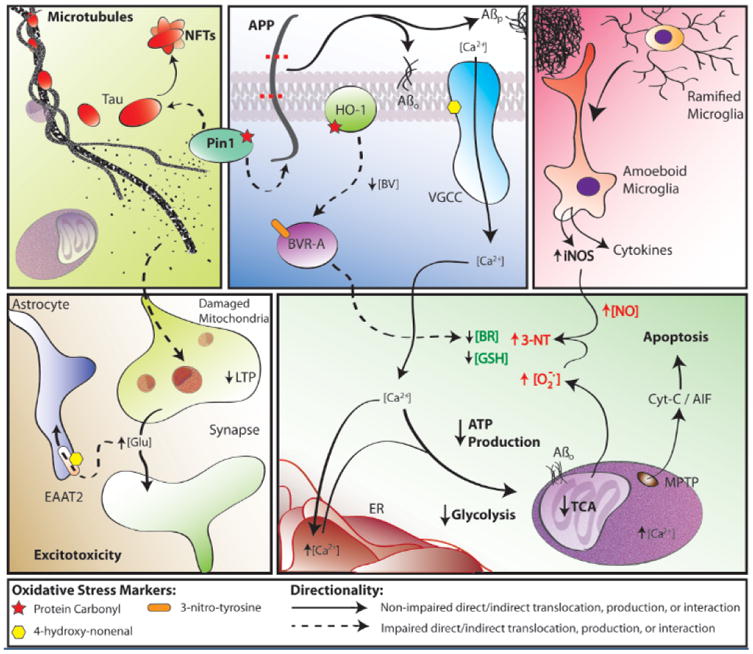
Some findings and discoveries related to Aβ(1-42) oligomer-induced oxidative stress and its sequela published from the Butterfield laboratory that contribute to a unifying oxidative stress centric hypothesis for neuronal death in AD that is consistent with the pathology, biochemical alterations, and clinical presentation in this disorder. Amyloid β-peptide, produced by β- and γ-secretase (top, middle panel), aggregates into extracellular fibrils forming the core of SP or aggregates into hydrophobic oligomers that insert into the plasma membrane. One-electron oxidation of the S-atom of Met-35 of Aβ(1-42) [see text] initates the chain reaction of lipid peroxidation (LPO), greatly amplyfing the damage of the initial free radical on the peptide. HNE, produced by LPO, binds to and causes dysfunction of key proteins in the PM and cytosol. Among the former are ion-motive ATPases, e.g., Na,K-ATPase and Ca-ATPase. The resultant loss of cell potential opens voltage gated Ca2+ channels, leading to a massive influx of Ca2+ to the cytosol (bottom, right panel), and subsequent attempts to sequester this Ca2+ in ER and mitochondria. However, this massive overload of Ca2+ causes ER to undergo unfolded protein response and damage and decreases protein synthesis. Mitochondrial Ca2+ overload causes swelling of mitochondria and opening of the MPTP with release of cytochrome c to initiate the intrinsic pathway for apoptosis (bottom, right panel). The intracellular Ca2+ also activates numerous degradative enzymes such as calpains, PLA2, endonucleases, etc. causing necrotic mechanisms to engage (see text). The oxidative stress in cytosol and mitochondria lead to damage to many glycolytic, TCA, and ETC enzymes or complexes, resulting in dramatic loss of ATP production (bottom, right panel), which leads to loss of many important neuronal functions, ranging from axonal transport to neurotransmission (bottom, left panel), as well as maintaining the cellular and organelle potentials. This oxidative stress also damages Pin1, which regulates both APP and Tau (top, left/middle panels), but also a key tau kinase (GSK-3β) and a tau phosphatase (PP2A). The resultant hyperphosphorylated tau, falls off microtubules (MT) [top, left panel], leading to cessation of anterograde and retrograde transport. Among other determimental consequences of this, synaptic-resident mitochondria are no longer able to produce the required ATP to maintain presynaptic function, including loss of LTP, needed for learning and memory (bottom, left panel). Note that Pin1 is involved in three major neuropathological hallmarks of AD: SP, NFT, and synapse loss. The intracellular detritus resulting from all these aberrant processes is not removed, since the proteostasis network of the ubiquitin-proteasome system (UPS), ER, and autophagy are all damaged by oxidative stress (see text). The Aβ(1-42)-initiated oxidative stress also leads to damage to heme oxygenase -1 (HO-1) and biliverdin reductase-A (BVR-A), which decreases intracellular antioxidant bilirubin (top, middle panel; bottom, right panel). Moreover, glutathione (GSH) is decreased because of elevated oxidative stress and since its rate limiting synthetic enzyme, γ-Glu-Cys ligase, is damaged by oxidative stress. There are many other processes associated with oxidative stress on which we and others have published. However, this schematic diagram outlines some of the key processes involved in neuronal death in AD that fit the pathology, biochemical alterations, and clinical presentations observed in AD that were published from our laboratory based on neurochemical, oxidative stress, and proteomics studies of AD and its earlier forms.
(D) Spin trapping studies using ultrapure [repeatedly recrystallized] spin trap phenyl-t-butyl nitrone (PBN) in buffers made with deionized water that had been treated overnight with chelex-100 beads and containing desferal to chelate as many adventitious metal ions as possible led to EPR spectra of a trapped radical when treated with Aβ(1-42) but no EPR spectrum of either the PBN alone or with Aβ(1-42)M35Norleucine in which the S-atom of Met-35 was replaced by a CH2 group [20]. This latter peptide, as noted above, was non-reactive and non-neurotoxic. Moreover, this norleucine substituted peptide was produced by the same company as native Aβ(1-42), presumably having the same amount of any metal ion bound to it and suggesting that it is the Met-35 S-atom that contributes to the oxidative stress associated with this peptide. Absence of molecular oxygen from the buffer solution prevented formation of an EPR spectrum [156], suggesting in this in vitro system O2 could be an oxidant to oxidize the S-atom of Met-35.
(E) As noted above, Aβ(1-42) oligomers in the lipid bilayer of neuronal plasma membranes adopt a helical structure, and this secondary structure contributes to the reactivity of the peptide [157]. Also noted above, alpha helices obey the i+4 rule of amino acid interaction. Thus, we substituted the alpha-helix breaking amino acid proline for Ile at position 31. Addition of Aβ(1- 42)I31P to neuronal cultures no longer led to protein oxidation nor to neuronal cell death [157].
(F) Substitution of the codon of Cys for Met-35 in the DNA of Aβ(1-42) in C.elegans led to a peptide that deposited to the same extent as the native peptide within the nematode, but resulted in no elevated protein oxidation, in marked contrast to native Aβ(1-42) [87]. Using the J20 mouse described above, addition of a third mutation to APP to form PDAPPM631L led to a mouse containing Leu in place of the Met residue at position 631 of APP, a position that corresponds to residue 35 of Aβ(1-42). Comparison of oxidative stress parameters in brain of the J20 mouse with those of the J20M631L mouse demonstrated significant oxidative stress in the former but no oxidative stress of the latter mouse at 9 months of age [158]. Proteomics studies identified differential levels and decreased oxidation of brain proteins in the J20M631L mouse compared to the J20 mouse [94, 159].
Taken together, these in vitro and in vivo studies strongly support the hypothesis that the S-atom of the single Met residue of Aβ(1-42) is key to the oxidative stress associated with this neurotoxic peptide.
3.8. Conclusions Related to Discoveries in AD from the Butterfield Laboratory
Two major interrelated discoveries from our laboratory relevant to Alzheimer disease are that the vast majority of evidence outlined above supports the notion that Aβ(1-42) is associated with oxidative stress and neurotoxicity, and that such oxidative stress is abundant in the brain of subjects with AD and amnestic MCI. These discoveries have contributed to changing the paradigm of AD pathogenesis to include oxidative stress as a fundamental factor in the pathology and progression of this disorder (Fig. 7) [18, 19, 21, 122]. Lipid peroxidation and its sequelae, e.g., HNE for example, bind to and damage key proteins that contribute to Ca2+ dyshomeostasis and subsequent neuronal death. Redox proteomics, pioneered by our laboratory, identified oxidatively modified proteins in brain from AD and its earlier forms, whose dysfunction likely contributes to the pathology, biochemistry, and clinical presentation of the disease. Moreover, new potential therapeutic targets to slow progression of AD have emerged from these studies. Future investigations from the Butterfield laboratory are aimed at utilizing these discoveries to engage in disease-modifying approaches that slow onset and progression of this devastating dementing disorder.
4. Chemotherapy Induced Cognitive Impairment (CICI)
4.1. Brief Background of CICI
With 14 million cancer survivors in the United States alone, chemotherapy associated cognitive dysfunction is an increasing concern. A significant number experience Chemotherapy Induced Cognitive Impairment (CICI), called “chemobrain” by patients [160]. CICI reduces their quality of life with serious, even devastating, symptoms such as diminished executive function and intellectual impairment. There is an urgent, important need for treatment interventions that lead to a high quality of life for cancer survivors—projected to number 18 million within 10 years. Depending on the study, CICI affects up to 70% of cancer survivors, whose chemotherapy induced neurological deficits result in loss of executive function, inability to multi-task, and slowness in intellectual reasoning [160]. Furthermore, symptoms can be relatively long lasting, up to 10 years post-chemotherapy. Associated with these symptoms are significant MRI-determined volumetric diminution of the hippocampus [161], altered white [162-164] and gray [165] matter and decreased glucose utilization assessed by PET scanning [166].
A now well-recognized condition, CICI seriously limits the quality of life of cancer survivors [167]. Currently, no evidence-based treatment or preventative intervention for CICI exists, as the underlying mechanisms remain poorly understood. Particularly perplexing is that CICI often is associated with ROS generating chemotherapeutic agents that do not cross the BBB.
ROS-mediated mitochondrial injury is an important mechanism of chemotherapy-induced injury in tissues with high-energy demand, such as heart and brain. Doxorubicin (Dox), often used in therapy for solid tumors and leukemia, undergoes redox cycling involving its quinone structure to produce superoxide free radicals (Fig. 8), which are thought to underlie Dox-induced cardiomyopathy and limit its lifetime therapeutic dosage [1]. Dox, one of approximately 50% of FDA-approved chemotherapeutic agents associated with ROS, does not cross the BBB [168].
Figure 8.
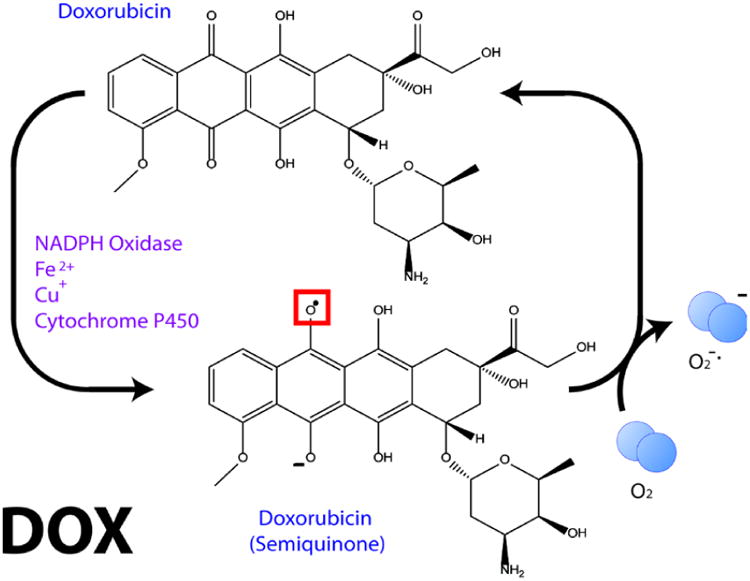
Redox cycling of Dox to produce superoxide free radical. See text for more details.
4.2. Oxidative Stress in Brain and Plasma Following ROS-associated Chemotherapy
Intraperitoneal injection of Dox led to elevated oxidative damage to brain [169, 170]. How can oxidative stress occur in brain by a drug that does not cross the BBB? To investigate this key question, we determined that i.p administration of Dox led to elevated levels of the pro-inflammatory cytokine TNFα in plasma and in brain [171]. Moreover, this treatment led to translocation of p53 to mitochondria where it bound to the anti-apoptotic protein, Bcl-2, consistent with induction of apoptosis, which we also demonstrated via, cytochrome c release, TUNEL staining and elevation of caspase 3. Mitochondria also are dysfunctional based on loss of respiration following Dox treatment [171]. If animals were injected i.p. by the glutathione precursor, gamma-glutamylcysteine ethyl ester (GCEE, Fig. 4), brain GSH levels increase and Dox-induced, TNFα mediated oxidative stress in brain is prevented [170]. Of course, such an approach could not be used therapeutically, since elevated glutathione would be conjugated to the chemotheapeuric agent by glutathione-S-transferase and exported from the cancer cell by the multidrug resistant protein, MRP-1 [172, 173], However, this study does demonstrate again that oxidative stress is induced in brain by Dox treatment in the periphery. Plasma of Dox-treated wild-type mice showed evidence of oxidative stress that was blocked by treatment with GCEE, [81].
That peripheral (plasma) TNFα is involved in this process was demonstrated by the abrogation of all these processes noted above when anti-TNFα antibody was administered along with Dox [171]. This result is consistent with the notion it is TNFα, among the many proinflammatory cytokines, that is key to Dox-induced oxidative stress in brain. This concept is strengthened by our observation using a cytokine array that only TNFα was statistically significantly elevated in plasma from persons receiving Dox therapy [174].
4.3 Role of Nitrosative Stress in Dox-induced, TNFα-Mediated Damage to Brain
Given that respiration is compromised in brain mitochondria isolated from Dox-treated mice [171], that MnSOD is fundamental to mitochondrial function [175], and that 3-NT is elevated in brain of Dox-treated WT mice [170], we performed studies of brain mitochondria isolated from Dox-treated i-NOS knock-out mice [176]. Fig. 9 shows that absence of i-NOS (and hence no NO produced from NOS2) abrogates: loss of brain mitochondrial respiration, nitration of MnSOD, and diminution of MnSOD activity. Consequently, since i-NOS is downstream from NFκ-B activation [177], which, in turn, is activated by TNFα, we hypothesize that damaged mitochondria and apoptotic cell death in brain are secondary to TNFα accumulation in brain following Dox administration and in the sequence:
Figure 9.
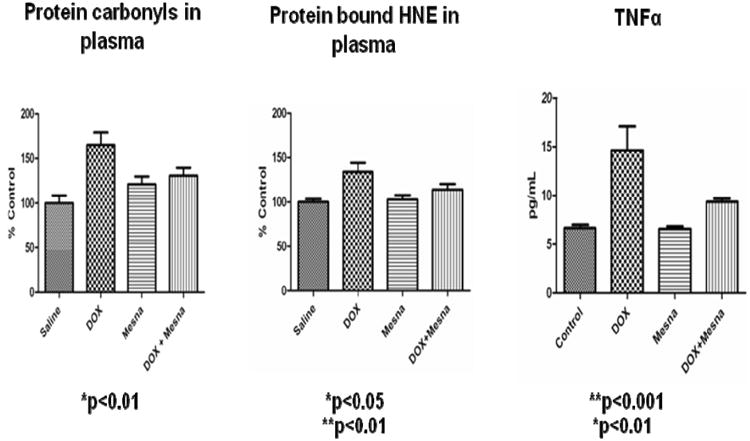
Dox administration to WT mice (i.p.) leads in plasma to protein oxidation and lipid peroxidation (assessed by protein carbonyls and protein-bound HNE, respectively), and to elevated TNFα. Concomitant administration of Mesna significantly depresses these markers to essentially control levels. The figure, used with permission from Elsevier Science Publishers, is taken from reference [174].
Nitric oxide reacts with superoxide radical anion to form peroxynitrte that in the presence of CO2 leads to 3-NT formation [2].
4.4 Plasma ApoA1 Involvement in Dox-induced, TNFα-mediated Oxidative Stress in Brain
Using redox proteomics, Apolipoprotein A1 (ApoA1) was identified as an oxidized protein in plasma of children with lymphoma treated with Dox [174]. ApoA1 is a major component of the high density lipoprotein particle and is involved with cholesterol transport away from tissues. Importantly, ApoA1 normally keeps TNFα levels low in plasma by both preventing monocyte-T cell interactions and by interaction with ABCA1, the cholesterol transporter [178]. Going from beside to bench, we confirmed in plasma of Dox-treated mice that ApoA1 was oxidatively modified (assessed by protein carbonyls). Further, addition of native ApoA1 to macrophage culture did not lead to TNFα elevation, but addition of oxidized ApoA1 to macrophage culture did result in significant TNFα release [174]. We speculate that, due to a changed conformation secondary to oxidation [2], oxidized ApoA1 interacts with ABCA1 differently and does not suppress TNFα elevation.
4.5 Prevention by Mesna of Biochemical Alterations in Brain Following Dox Treatment
Protein oxidation was clearly apparent in plasma from children with lymphoma treated with Dox, but, surprisingly, not so with lipid peroxidation assessed by protein-bound HNE [174]. However, examination of the medical records demonstrated that about half the patients studied had been given Mesna (2-mercaptoethane-1 sulfonate, Na+ salt). Segregation of the data from the Mesna-treated group from the non-Mesna-treated group of patients showed significant suppression of lipid peroxidation in the former and significant elevation of lipid peroxidation of the latter, i.e., the sum of changes in the two groups was about zero compared to control [174].
Mesna is often given in therapeutic regimens containing ifosphamide and/or cyclophosphamide to prevent hemorrhagic cystitis in the bladder secondary to acrolein, formed as a metabolic byproduct of these two drugs [179]. An important feature of Mesna is its negative charge that prevents this molecule from entering cancer cells to interfere with cancer chemotherapy. This is an important distinction with other thiol compounds, for example NAC, which as described above if given to patients would have the effect of leading to export of the chemotherapeutic agent from cancer cells, thereby harming the cancer patient. Mesna does not suffer this fate, as it does not penetrate cancer cells and does not interfere with cancer chemotherapy [180].
Again pursuing a bedside to bench strategy, we treated WT mice with both Mesna and Dox, and found plasma levels of protein oxidation, lipid peroxidation, and, importantly, TNFα levels were suppressed to levels similar to those in plasma from saline-treated mice and in marked contrast to Dox-treated mice in the absence of Mesna [174] (Fig. 10). Based on these exciting results, and this time going from bench to bedside, our team of basic scientists and clinicians recently have conducted a NCI-sponsored pilot study of oxidative biomarkers and TNFα signature in plasma from patients with breast cancer or lymphoma randomized in the first cycle to Mesna or saline and the second cycle to the opposite treatment. The results of this latter study will be reported in a separate publication after statistical analyses are completed, but initial results are promising.
Figure 10.
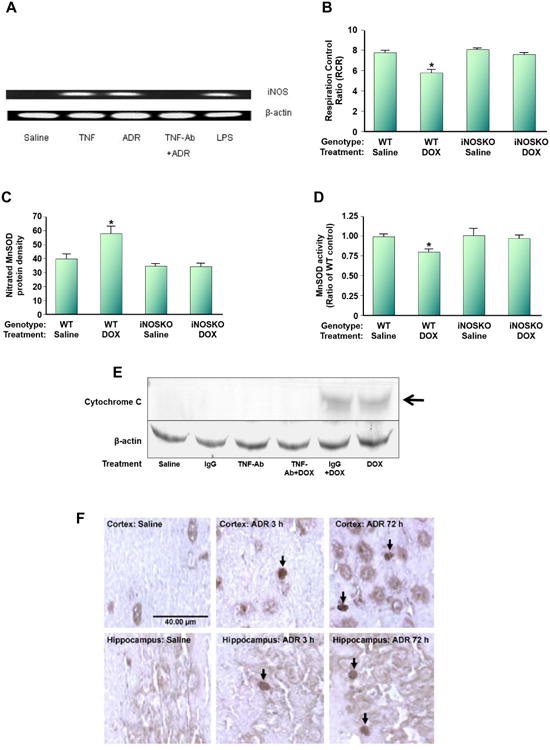
Dox (also called adriamycin, ADR) induces nitrosative stress in brain mitochondria. WT mice were injected i.p. with Dox. (A). i-NOS message was induced, but anti-TNFα antibody prevents i-NOS induction following ADR treatment. (B). The RCR, a measure of oxygen consumption, in brain mitochondria isolated from ADR-treated WT mice is significantly depressed, but not in i-NOS knock-out mice. (C). Mitochondrial-resident MnSOD is nitrated following ADR addition to WT mice, but not in i-NOS knock-out mice. (D). MnSOD activity is significantly depressed in mitochondria isolated from brain of ADR-treated WT mice, but not in i-NOS knock-out mice. (E). Mitochondria isolated from brain of ADR-treated WT mice lead to cytochrome c release, but not in mice also treated with anti-TNFα antibody. Note that ADR-treated WT mice also treated with IgG still lead to cytochrome c release from mitochondria isolated from brain, suggesting specificity of the anti-TNFα treatment. (F). Consistent with the results of (E), apoptosis occurs in brain of ADR-treated WT mice as assessed by TUNEL staining even at 3h post-ADR treatment, and pronounced apoptosis at 72h post-ADR treatment occurred. This latter time is the time at which oxidative stress in brain is maximal. The figure, used with permission from Elsevier Science Publishers, was modified from reference [171].
4.6 Conclusions and Discussion of CICI, with Overall Model
Based on our studies in humans and mice treated with Dox, we have developed the following model that for the first time provides a rational molecular mechanism for CICI. Fig. 11 shows the model. Redox cycling of Dox leads to superoxide free radicals to cause plasma protein oxidation and lipid peroxidation that, in turn, lead to oxidation of the key plasma lipoprotein, ApoA1. The proinflammatory cytokine TNFα is elevated in plasma as a consequence of ApoA1 oxidation. These processes are blocked by co-administration of Mesna. The plasma resident elevated TNFα is transported to the brain parenchyma via receptor-mediated endocytosis. This process is blocked by co-administration of anti-TNFα antibody. Once in brain, TNFα through a series of biochemical events, including activation of i-NOS, leads to mitochondrial dysfunction, cytochrome c release, and apoptosis. When enough neurons have died, symptoms of CICI appear.
Figure 11.
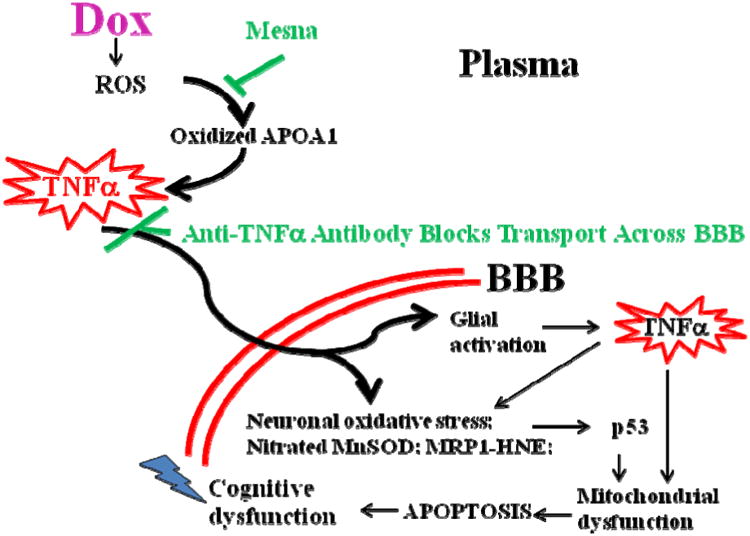
Model for CICI (“chemobrain”) based on demonstrated results from our laboratory. Dox (ADR), as a prototypical ROS-generating agent that does not cross the BBB, causes oxidation of ApoA1 in plasma, which leads to increased levels of TNFα. This cytokine crosses the BBB to enter glia and neurons, generating even more TNFα. Subsequent biochemical events lead to brain mitochondrial dysfunction and resultant apoptosis as explained more in the text. Mesna, which does not interfere with cancer chemotherapy, blocks Dox-mediated oxidation of ApoA1 and elevation of TNFα. Anti- TNFα antibody blocks elevated TNFα from crossing the BBB. Both treatments inhibit the biochemical events that otherwise cause neuronal death. See text and [169-171, 174, 176, 181, 182] for more details.
5.0 Overall Summary
Alzheimer disease and chemotherapy induced cognitive impairment each affect millions of persons worldwide. Both disorders are united by cognitive issues likely deriving from, or at least associated with, free radical events. But each disorder is unique: AD is associated with Aβ-associated oxidative stress within the brain parenchyma, leading to lipid peroxidation and protein oxidation (among other oxidative stress indices), which damages mitochondria, leads to oxidative dysfunction of key proteins involved in numerous pathways, including glucose metabolic proteins and death of neurons; in contrast, CICI involves oxidative stress events originating in the plasma that lead to TNFα elevation and subsequent transfer to brain glia and neurons, which in turn leads to more oxidative stress, mitochondrial dysfunction, and cell death. Neither condition is fully understood, and while pharmacological suppression of CICI shows initial promise, this still remains elusive in the case of AD. Continued studies of molecular mechanisms involved in AD and CICI and translational investigations in models of both disorders are ongoing in our laboratory.
Acknowledgments
The results mentioned in this paper were obtained with support by NIH grants and by funds from the UK Markey Cancer Center. The 2013 Discovery Award from the Society for Free Radical Biology and Medicine was presented to me for discoveries that emanated from my laboratory related to fundamental free radical processes in both AD and CICI. However, I am first to state that this recognition is in large part due to the great fortune and opportunity I have had to train immensely talented and motivated Ph.D., M.S., and undergraduate students, postdoctoral scholars, and visiting scientists, as well as collaborate with generous and supportive faculty at the University of Kentucky and elsewhere. It is to this entire group of wonderful people that I am grateful and appreciative. The 2013 Discovery Award from the Society for Free Radical Biology and Medicine is a highlight of my career. I would like to mention a few University of Kentucky collaborators for special thanks. First I wish to acknowledge the late William R. Markesbery, M.D., a consummate gentleman and outstanding neuropathologist, who made a number of seminal contributions to the field of AD and provided me with important guidance throughout my career. Others to thank for their assistance and collaboration with my laboratory in the field of AD include Mark Mattson, Ph.D., James Geddes, Ph.D., Stephen Scheff, Ph.D., Steven Estus, Ph.D., M. Paul Murphy, Ph.D., Elizabeth Head, Ph.D., Peter Nelson, M.D. and Linda Van Eldik, Ph.D. In our studies of CICI, I thank Daret K. St. Clair, Ph.D., Jeffrey A. Moscow, M.D., Mary Vore Iwamoto, Ph.D., John Hayslip, M.D., Heidi Weiss, Ph.D., Subbaro Bondada, Ph.D., and B. Mark Evers, M.D., each a highly respected scientist or physician-scientist. Together we formed a team, without which our progress in CICI could not have occurred. This group provided enormous assistance and friendship for which I will always be grateful.
I thank Ms. Mollie Fraim for secretarial assistance with this manuscript and many others throughout my career.
Lastly, I want to give special thanks to my wife of 46 years, Marcia T. Butterfield, B.A., R.N. Marcia has always been supportive of my career that often involved long days and nights, and extensive travel away from Lexington. Moreover, she always understood the concentration and effort required during grant proposal writing periods. Marcia always has been my inspiration and I am truly grateful to have her in my life.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. Oxford University Press; 2007. p. 704. [Google Scholar]
- 2.Butterfield DA, Stadman ER. Protein oxidation processes in aging brain. Advances in Cell Aging and Gerontology. 1997;2:161–191. [Google Scholar]
- 3.Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods in enzymology. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer's disease. Biochim Biophys Acta. 2010;1801:924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 6.Butterfield DA, Perluigi M, Reed T, Muharib T, Hughes CP, Robinson RA, Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal. 2012;17:1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butterfield DA, Di Domenico F, Robinson RAS. Mass Spectrometry and Redox Proteomics: Applications in Disease. Mass Spectrometry Reviews. 2014 doi: 10.1002/mas.21374. in press. [DOI] [PubMed] [Google Scholar]
- 8.Butterfield DA, Dalle-Donne I. Redox proteomics: from protein modifications to cellular dysfunction and disease. Mass spectrometry reviews. 2014;33:1–6. doi: 10.1002/mas.21404. [DOI] [PubMed] [Google Scholar]
- 9.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. Journal of neuropathology and experimental neurology. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selkoe DJ. Alzheimer's disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 11.Scheff SW, Price DA. Alzheimer's disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–115. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- 12.Petersen RC, Caracciolo B, Brayne C, Gauthier S, Jelic V, Fratiglioni L. Mild cognitive impairment: a concept in evolution. Journal of internal medicine. 2014;275:214–228. doi: 10.1111/joim.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markesbery WR. Neuropathologic alterations in mild cognitive impairment: a review. J Alzheimers Dis. 2010;19:221–228. doi: 10.3233/JAD-2010-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vos SJ, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, Cairns NJ, Morris JC, Holtzman DM, Fagan AM. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957–965. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 16.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 17.Klein WL. Synaptotoxic amyloid-beta oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer's disease? J Alzheimers Dis. 2013;33 Suppl 1:S49–65. doi: 10.3233/JAD-2012-129039. [DOI] [PubMed] [Google Scholar]
- 18.Butterfield DA. beta-Amyloid-associated free radical oxidative stress and neurotoxicity: implications for Alzheimer's disease. Chemical research in toxicology. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 19.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 20.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. Journal of structural biology. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 21.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 22.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, Butterfield DA. Brain regional correspondence between Alzheimer's disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 23.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 24.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 25.Perluigi M, Coccia R, Butterfield DA. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxid Redox Signal. 2012;17:1590–1609. doi: 10.1089/ars.2011.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller JN, Hanni KB, Markesbery WR. 4-hydroxynonenal increases neuronal susceptibility to oxidative stress. J Neurosci Res. 1999;58:823–830. [PubMed] [Google Scholar]
- 27.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 28.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 31.Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2006;397:170–173. doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 32.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer's disease. Brain Res. 2007;1148:243–248. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reed TT, Pierce WM, Markesbery WR, Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 35.Aluise CD, Robinson RA, Beckett TL, Murphy MP, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Preclinical Alzheimer disease: brain oxidative stress, Abeta peptide and proteomics. Neurobiol Dis. 2010;39:221–228. doi: 10.1016/j.nbd.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradley MA, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med. 2010;48:1570–1576. doi: 10.1016/j.freeradbiomed.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castegna A, Lauderback CM, Mohmmad-Abdul H, Butterfield DA. Modulation of phospholipid asymmetry in synaptosomal membranes by the lipid peroxidation products, 4-hydroxynonenal and acrolein: implications for Alzheimer's disease. Brain Res. 2004;1004:193–197. doi: 10.1016/j.brainres.2004.01.036. [DOI] [PubMed] [Google Scholar]
- 38.Mohmmad Abdul H, Butterfield DA. Protection against amyloid beta-peptide (1-42)-induced loss of phospholipid asymmetry in synaptosomal membranes by tricyclodecan-9-xanthogenate (D609) and ferulic acid ethyl ester: implications for Alzheimer's disease. Biochim Biophys Acta. 2005;1741:140–148. doi: 10.1016/j.bbadis.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Bader Lange ML, Cenini G, Piroddi M, Abdul HM, Sultana R, Galli F, Memo M, Butterfield DA. Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer disease. Neurobiol Dis. 2008;29:456–464. doi: 10.1016/j.nbd.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bader Lange ML, St Clair D, Markesbery WR, Studzinski CM, Murphy MP, Butterfield DA. Age-related loss of phospholipid asymmetry in APP(NLh)/APP(NLh) × PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice: relevance to Alzheimer disease. Neurobiol Dis. 2010;38:104–115. doi: 10.1016/j.nbd.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sultana R, Perluigi M, Newman SF, Pierce WM, Cini C, Coccia R, Butterfield DA. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer's disease. Antioxid Redox Signal. 2010;12:327–336. doi: 10.1089/ars.2009.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butterfield DA, Swomley AM, Sultana R. Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal. 2013;19:823–835. doi: 10.1089/ars.2012.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butterfield DA. Proteomics: a new approach to investigate oxidative stress in Alzheimer's disease brain. Brain Res. 2004;1000:1–7. doi: 10.1016/j.brainres.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 44.Butterfield DA, Boyd-Kimball D, Castegna A. Proteomics in Alzheimer's disease: insights into potential mechanisms of neurodegeneration. J Neurochem. 2003;86:1313–1327. doi: 10.1046/j.1471-4159.2003.01948.x. [DOI] [PubMed] [Google Scholar]
- 45.Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer's disease brain: new insights from redox proteomics. European journal of pharmacology. 2006;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 46.Reed TT, Owen J, Pierce WM, Sebastian A, Sullivan PG, Butterfield DA. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J Neurosci Res. 2009;87:408–417. doi: 10.1002/jnr.21872. [DOI] [PubMed] [Google Scholar]
- 47.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer's disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006;27:1564–1576. doi: 10.1016/j.neurobiolaging.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 48.Sultana R, Butterfield DA. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. Journal of bioenergetics and biomembranes. 2009;41:441–446. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sultana R, Perluigi M, Allan Butterfield D. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med. 2013;62:157–169. doi: 10.1016/j.freeradbiomed.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butterfield DA, Lange ML. Multifunctional roles of enolase in Alzheimer's disease brain: beyond altered glucose metabolism. J Neurochem. 2009;111:915–933. doi: 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aluise CD, Robinson RA, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer's disease: insights into memory loss in MCI. J Alzheimers Dis. 2011;23:257–269. doi: 10.3233/JAD-2010-101083. [DOI] [PubMed] [Google Scholar]
- 52.Di Domenico F, Sultana R, Tiu GF, Scheff NN, Perluigi M, Cini C, Butterfield DA. Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: an investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res. 2010;1333:72–81. doi: 10.1016/j.brainres.2010.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 54.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 55.Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, Klein JB, Turner DM, Butterfield DA. An increase in S-glutathionylated proteins in the Alzheimer's disease inferior parietal lobule, a proteomics approach. J Neurosci Res. 2007;85:1506–1514. doi: 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]
- 56.Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 in Alzheimer's disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochemical research. 2004;29:2215–2220. doi: 10.1007/s11064-004-7028-0. [DOI] [PubMed] [Google Scholar]
- 57.Di Domenico F, Perluigi M, Foppoli C, Blarzino C, Coccia R, De Marco F, Butterfield DA, Cini C. Protective effect of ferulic acid ethyl ester against oxidative stress mediated by UVB irradiation in human epidermal melanocytes. Free Radic Res. 2009;43:365–375. doi: 10.1080/10715760902777329. [DOI] [PubMed] [Google Scholar]
- 58.Sultana R, Perluigi M, Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer's disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal. 2006;8:2021–2037. doi: 10.1089/ars.2006.8.2021. [DOI] [PubMed] [Google Scholar]
- 59.Sultana R, Piroddi M, Galli F, Butterfield DA. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochemical research. 2008;33:2540–2546. doi: 10.1007/s11064-008-9593-0. [DOI] [PubMed] [Google Scholar]
- 60.Sultana R, Butterfield DA. Regional expression of key cell cycle proteins in brain from subjects with amnestic mild cognitive impairment. Neurochemical research. 2007;32:655–662. doi: 10.1007/s11064-006-9123-x. [DOI] [PubMed] [Google Scholar]
- 61.Keeney JT, Swomley AM, Harris JL, Fiorini A, Mitov MI, Perluigi M, Sultana R, Butterfield DA. Cell cycle proteins in brain in mild cognitive impairment: insights into progression to Alzheimer disease. Neurotox Res. 2012;22:220–230. doi: 10.1007/s12640-011-9287-2. [DOI] [PubMed] [Google Scholar]
- 62.Cenini G, Sultana R, Memo M, Butterfield DA. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer's disease. J Cell Mol Med. 2008;12:987–994. doi: 10.1111/j.1582-4934.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cenini G, Sultana R, Memo M, Butterfield DA. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008;45:81–85. doi: 10.1016/j.freeradbiomed.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, Butterfield DA. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer's disease and mild cognitive impairment: implications for progression of AD. Journal of proteome research. 2009;8:471–482. doi: 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Di Domenico F, Owen JB, Sultana R, Sowell RA, Perluigi M, Cini C, Cai J, Pierce WM, Butterfield DA. The wheat germ agglutinin-fractionated proteome of subjects with Alzheimer's disease and mild cognitive impairment hippocampus and inferior parietal lobule: Implications for disease pathogenesis and progression. J Neurosci Res. 2010;88:3566–3577. doi: 10.1002/jnr.22528. [DOI] [PubMed] [Google Scholar]
- 66.Butterfield DA, Owen JB. Lectin-affinity chromatography brain glycoproteomics and Alzheimer disease: insights into protein alterations consistent with the pathology and progression of this dementing disorder. Proteomics Clin Appl. 2011;5:50–56. doi: 10.1002/prca.201000070. [DOI] [PubMed] [Google Scholar]
- 67.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Owen JB, Sultana R, Aluise CD, Erickson MA, Price TO, Bu G, Banks WA, Butterfield DA. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Abeta accumulation in AD brain. Free Radic Biol Med. 2010;49:1798–1803. doi: 10.1016/j.freeradbiomed.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barone E, Di Domenico F, Sultana R, Coccia R, Mancuso C, Perluigi M, Butterfield DA. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med. 2012;52:2292–2301. doi: 10.1016/j.freeradbiomed.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barone E, Di Domenico F, Cenini G, Sultana R, Cini C, Preziosi P, Perluigi M, Mancuso C, Butterfield DA. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim Biophys Acta. 2011;1812:480–487. doi: 10.1016/j.bbadis.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barone E, Di Domenico F, Mancuso C, Butterfield DA. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: it's time for reconciliation. Neurobiol Dis. 2014;62:144–159. doi: 10.1016/j.nbd.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hardas SS, Sultana R, Clark AM, Beckett TL, Szweda LI, Murphy MP, Butterfield DA. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox biology. 2013;1:80–85. doi: 10.1016/j.redox.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Subramaniam R, Roediger F, Jordan B, Mattson MP, Keller JN, Waeg G, Butterfield DA. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997;69:1161–1169. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- 74.Butterfield DA, Gnjec A, Poon HF, Castegna A, Pierce WM, Klein JB, Martins RN. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer's disease: an initial assessment. J Alzheimers Dis. 2006;10:391–397. doi: 10.3233/jad-2006-10407. [DOI] [PubMed] [Google Scholar]
- 75.Perluigi M, Butterfield DA. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr Gerontol Geriatr Res. 2012;2012:724904. doi: 10.1155/2012/724904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cenini G, Dowling AL, Beckett TL, Barone E, Mancuso C, Murphy MP, Levine H, 3rd, Lott IT, Schmitt FA, Butterfield DA, Head E. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta. 2012;1822:130–138. doi: 10.1016/j.bbadis.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perluigi M, Butterfield DA. The identification of protein biomarkers for oxidative stress in Down syndrome. Expert review of proteomics. 2011;8:427–429. doi: 10.1586/epr.11.36. [DOI] [PubMed] [Google Scholar]
- 78.Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C, Perluigi M. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer's disease neuropathology: Redox proteomics analysis of human brain. Biochim Biophys Acta. 2013;1832:1249–1259. doi: 10.1016/j.bbadis.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perluigi M, Di Domenico F, Buttterfield DA. Unraveling the complexity of neurodegeneration in brains of subjects with Down syndrome: insights from proteomics. Proteomics Clin Appl. 2014;8:73–85. doi: 10.1002/prca.201300066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perluigi M, di Domenico F, Fiorini A, Cocciolo A, Giorgi A, Foppoli C, Butterfield DA, Giorlandino M, Giorlandino C, Schinina ME, Coccia R. Oxidative stress occurs early in Down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteomics Clin Appl. 2011;5:167–178. doi: 10.1002/prca.201000121. [DOI] [PubMed] [Google Scholar]
- 81.Aluise CD, Sowell RA, Butterfield DA. Peptides and proteins in plasma and cerebrospinal fluid as biomarkers for the prediction, diagnosis, and monitoring of therapeutic efficacy of Alzheimer's disease. Biochim Biophys Acta. 2008;1782:549–558. doi: 10.1016/j.bbadis.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Di Domenico F, Coccia R, Butterfield DA, Perluigi M. Circulating biomarkers of protein oxidation for Alzheimer disease: expectations within limits. Biochim Biophys Acta. 2011;1814:1785–1795. doi: 10.1016/j.bbapap.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 83.Cocciolo A, Di Domenico F, Coccia R, Fiorini A, Cai J, Pierce WM, Mecocci P, Butterfield DA, Perluigi M. Decreased expression and increased oxidation of plasma haptoglobin in Alzheimer disease: Insights from redox proteomics. Free Radic Biol Med. 2012;53:1868–1876. doi: 10.1016/j.freeradbiomed.2012.08.596. [DOI] [PubMed] [Google Scholar]
- 84.Di Domenico F, Barone E, Mancuso C, Perluigi M, Cocciolo A, Mecocci P, Butterfield DA, Coccia R. HO-1/BVR-a system analysis in plasma from probable Alzheimer's disease and mild cognitive impairment subjects: a potential biochemical marker for the prediction of the disease. J Alzheimers Dis. 2012;32:277–289. doi: 10.3233/JAD-2012-121045. [DOI] [PubMed] [Google Scholar]
- 85.Sultana R, Mecocci P, Mangialasche F, Cecchetti R, Baglioni M, Butterfield DA. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer's disease: insights into the role of oxidative stress in Alzheimer's disease and initial investigations into a potential biomarker for this dementing disorder. J Alzheimers Dis. 2011;24:77–84. doi: 10.3233/JAD-2011-101425. [DOI] [PubMed] [Google Scholar]
- 86.Sultana R, Baglioni M, Cecchetti R, Cai J, Klein JB, Bastiani P, Ruggiero C, Mecocci P, Butterfield DA. Lymphocyte mitochondria: toward identification of peripheral biomarkers in the progression of Alzheimer disease. Free Radic Biol Med. 2013;65c:595–606. doi: 10.1016/j.freeradbiomed.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer's amyloid beta-peptide (1-42) Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. discussion 339-342. [DOI] [PubMed] [Google Scholar]
- 88.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer's disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 89.Boyd-Kimball D, Poon HF, Lynn BC, Cai J, Pierce WM, Jr, Klein JB, Ferguson J, Link CD, Butterfield DA. Proteomic identification of proteins specifically oxidized in Caenorhabditis elegans expressing human Abeta(1-42): implications for Alzheimer's disease. Neurobiol Aging. 2006;27:1239–1249. doi: 10.1016/j.neurobiolaging.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 90.Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang Q, Aluise CD, Joshi G, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: toward therapeutic modulation of mild cognitive impairment. J Neurosci Res. 2010;88:2618–2629. doi: 10.1002/jnr.22422. [DOI] [PubMed] [Google Scholar]
- 92.Sultana R, Robinson RA, Di Domenico F, Abdul HM, St Clair DK, Markesbery WR, Cai J, Pierce WM, Butterfield DA. Proteomic identification of specifically carbonylated brain proteins in APP(NLh)/APP(NLh) × PS-1(P264L)/PS-1(P264L) human double mutant knock-in mice model of Alzheimer disease as a function of age. Journal of proteomics. 2011;74:2430–2440. doi: 10.1016/j.jprot.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, Markesbery WR, Butterfield DA. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1-42), H2O2, and kainic acid: implications for Alzheimer's disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- 94.Robinson RA, Lange MB, Sultana R, Galvan V, Fombonne J, Gorostiza O, Zhang J, Warrier G, Cai J, Pierce WM, Bredesen DE, Butterfield DA. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: effects of the amyloid-beta peptide of amyloid precursor protein. Neuroscience. 2011;177:207–222. doi: 10.1016/j.neuroscience.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Robinson RA, Joshi G, Huang Q, Sultana R, Baker AS, Cai J, Pierce W, St Clair DK, Markesbery WR, Butterfield DA. Proteomic analysis of brain proteins in APP/PS-1 human double mutant knock-in mice with increasing amyloid beta-peptide deposition: insights into the effects of in vivo treatment with N-acetylcysteine as a potential therapeutic intervention in mild cognitive impairment and Alzheimer's disease. Proteomics. 2011;11:4243–4256. doi: 10.1002/pmic.201000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boyd-Kimball D, Sultana R, Poon HF, Lynn BC, Casamenti F, Pepeu G, Klein JB, Butterfield DA. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1-42) into rat brain: implications for Alzheimer's disease. Neuroscience. 2005;132:313–324. doi: 10.1016/j.neuroscience.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 97.Davies P. Loss of choline acetyltransferase activity in normal aging and in senile dementia. Advances in experimental medicine and biology. 1978;113:251–256. doi: 10.1007/978-1-4684-8893-7_16. [DOI] [PubMed] [Google Scholar]
- 98.Contestabile A. The history of the cholinergic hypothesis. Behavioural brain research. 2011;221:334–340. doi: 10.1016/j.bbr.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 99.Morley JE, Armbrecht HJ, Farr SA, Kumar VB. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer's disease. Biochim Biophys Acta. 2012;1822:650–656. doi: 10.1016/j.bbadis.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 100.Butterfield DA, Poon HF. The senescence-accelerated prone mouse (SAMP8): a model of age-related cognitive decline with relevance to alterations of the gene expression and protein abnormalities in Alzheimer's disease. Experimental gerontology. 2005;40:774–783. doi: 10.1016/j.exger.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 101.Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, Butterfield DA, Morley JE. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- 102.Poon HF, Castegna A, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience. 2004;126:915–926. doi: 10.1016/j.neuroscience.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 103.Poon HF, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Proteomic analysis of specific brain proteins in aged SAMP8 mice treated with alpha-lipoic acid: implications for aging and age-related neurodegenerative disorders. Neurochemistry international. 2005;46:159–168. doi: 10.1016/j.neuint.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 104.Poon HF, Farr SA, Banks WA, Pierce WM, Klein JB, Morley JE, Butterfield DA. Proteomic identification of less oxidized brain proteins in aged senescence-accelerated mice following administration of antisense oligonucleotide directed at the Abeta region of amyloid precursor protein. Brain Res Mol Brain Res. 2005;138:8–16. doi: 10.1016/j.molbrainres.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 105.Fiorini A, Sultana R, Forster S, Perluigi M, Cenini G, Cini C, Cai J, Klein JB, Farr SA, Niehoff ML, Morley JE, Kumar VB, Allan Butterfield D. Antisense directed against PS-1 gene decreases brain oxidative markers in aged senescence accelerated mice (SAMP8) and reverses learning and memory impairment: A proteomics study. Free Radic Biol Med. 2013;65c:1–14. doi: 10.1016/j.freeradbiomed.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Farr SA, Ripley JL, Sultana R, Zhang Z, Niehoff ML, Platt TL, Murphy MP, Morley JE, Kumar V, Butterfield DA. Antisense oligonucleotide against GSK-3beta in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic Biol Med. 2014;67:387–395. doi: 10.1016/j.freeradbiomed.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cotman CW, Head E, Muggenburg BA, Zicker S, Milgram NW. Brain aging in the canine: a diet enriched in antioxidants reduces cognitive dysfunction. Neurobiol Aging. 2002;23:809–818. doi: 10.1016/s0197-4580(02)00073-8. [DOI] [PubMed] [Google Scholar]
- 108.Opii WO, Joshi G, Head E, Milgram NW, Muggenburg BA, Klein JB, Pierce WM, Cotman CW, Butterfield DA. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer's disease. Neurobiol Aging. 2008;29:51–70. doi: 10.1016/j.neurobiolaging.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barone E, Di Domenico F, Butterfield DA. Statins more than cholesterol lowering agents in Alzheimer disease: Their pleiotropic functions as potential therapeutic targets. Biochemical pharmacology. 2014;88:605–616. doi: 10.1016/j.bcp.2013.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Butterfield DA, Barone E, Di Domenico F, Cenini G, Sultana R, Murphy MP, Mancuso C, Head E. Atorvastatin treatment in a dog preclinical model of Alzheimer's disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int J Neuropsychopharmacol. 2012;15:981–987. doi: 10.1017/S1461145711001118. [DOI] [PubMed] [Google Scholar]
- 111.Barone E, Mancuso C, Di Domenico F, Sultana R, Murphy MP, Head E, Butterfield DA. Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J Neurochem. 2012;120:135–146. doi: 10.1111/j.1471-4159.2011.07538.x. [DOI] [PubMed] [Google Scholar]
- 112.Martin SB, Cenini G, Barone E, Dowling AL, Mancuso C, Butterfield DA, Murphy MP, Head E. Coenzyme Q10 and cognition in atorvastatin treated dogs. Neurosci Lett. 2011;501:92–95. doi: 10.1016/j.neulet.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Butterfield DA, Barone E, Mancuso C. Cholesterol-independent neuroprotective and neurotoxic activities of statins: perspectives for statin use in Alzheimer disease and other age-related neurodegenerative disorders. Pharmacol Res. 2011;64:180–186. doi: 10.1016/j.phrs.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lauderback CM, Hackett JM, Keller JN, Varadarajan S, Szweda L, Kindy M, Markesbery WR, Butterfield DA. Vulnerability of synaptosomes from apoE knock-out mice to structural and oxidative modifications induced by A beta(1-40): implications for Alzheimer's disease. Biochemistry. 2001;40:2548–2554. doi: 10.1021/bi002312k. [DOI] [PubMed] [Google Scholar]
- 115.Lauderback CM, Kanski J, Hackett JM, Maeda N, Kindy MS, Butterfield DA. Apolipoprotein E modulates Alzheimer's Abeta(1-42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res. 2002;924:90–97. doi: 10.1016/s0006-8993(01)03228-0. [DOI] [PubMed] [Google Scholar]
- 116.Zhou D, Lauderback CM, Yu T, Brown SA, Butterfield DA, Thompson JS. D609 inhibits ionizing radiation-induced oxidative damage by acting as a potent antioxidant. The Journal of pharmacology and experimental therapeutics. 2001;298:103–109. [PubMed] [Google Scholar]
- 117.Lauderback CM, Drake J, Zhou D, Hackett JM, Castegna A, Kanski J, Tsoras M, Varadarajan S, Butterfield DA. Derivatives of xanthic acid are novel antioxidants: application to synaptosomes. Free Radic Res. 2003;37:355–365. doi: 10.1080/1071576021000040664. [DOI] [PubMed] [Google Scholar]
- 118.Perluigi M, Joshi G, Sultana R, Calabrese V, De Marco C, Coccia R, Cini C, Butterfield DA. In vivo protective effects of ferulic acid ethyl ester against amyloid-beta peptide 1-42-induced oxidative stress. J Neurosci Res. 2006;84:418–426. doi: 10.1002/jnr.20879. [DOI] [PubMed] [Google Scholar]
- 119.Ansari MA, Joshi G, Huang Q, Opii WO, Abdul HM, Sultana R, Butterfield DA. In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid beta-peptide and other oxidative stressors: relevance to Alzheimer's disease and other oxidative stress-related neurodegenerative disorders. Free Radic Biol Med. 2006;41:1694–1703. doi: 10.1016/j.freeradbiomed.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sultana R, Ravagna A, Mohmmad-Abdul H, Calabrese V, Butterfield DA. Ferulic acid ethyl ester protects neurons against amyloid beta- peptide(1-42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem. 2005;92:749–758. doi: 10.1111/j.1471-4159.2004.02899.x. [DOI] [PubMed] [Google Scholar]
- 121.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Swomley AM, Forster S, Keeney JT, Triplett J, Zhang Z, Sultana R, Butterfield DA. Abeta, Oxidative Stress in Alzheimer Disease: Evidence Based on Proteomics Studies. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbadis.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harris ME, Hensley K, Butterfield DA, Leedle RA, Carney JM. Direct evidence of oxidative injury produced by the Alzheimer's beta-amyloid peptide (1-40) in cultured hippocampal neurons. Experimental neurology. 1995;131:193–202. doi: 10.1016/0014-4886(95)90041-1. [DOI] [PubMed] [Google Scholar]
- 124.Aksenov MY, Aksenova MV, Butterfield DA, Hensley K, Vigo-Pelfrey C, Carney JM. Glutamine synthetase-induced enhancement of beta-amyloid peptide A beta (1-40) neurotoxicity accompanied by abrogation of fibril formation and A beta fragmentation. J Neurochem. 1996;66:2050–2056. doi: 10.1046/j.1471-4159.1996.66052050.x. [DOI] [PubMed] [Google Scholar]
- 125.Aksenov MY, Aksenova MV, Markesbery WR, Butterfield DA. Amyloid beta-peptide (1-40)-mediated oxidative stress in cultured hippocampal neurons. Protein carbonyl formation, CK BB expression, and the level of Cu, Zn, and Mn SOD mRNA. J Mol Neurosci. 1998;10:181–192. doi: 10.1007/BF02761773. [DOI] [PubMed] [Google Scholar]
- 126.Yatin SM, Aksenova M, Aksenov M, Markesbery WR, Aulick T, Butterfield DA. Temporal relations among amyloid beta-peptide-induced free-radical oxidative stress, neuronal toxicity, and neuronal defensive responses. J Mol Neurosci. 1998;11:183–197. doi: 10.1385/JMN:11:3:183. [DOI] [PubMed] [Google Scholar]
- 127.Yatin SM, Varadarajan S, Butterfield DA. Vitamin E Prevents Alzheimer's Amyloid beta-Peptide (1-42)-Induced Neuronal Protein Oxidation and Reactive Oxygen Species Production. J Alzheimers Dis. 2000;2:123–131. doi: 10.3233/jad-2000-2212. [DOI] [PubMed] [Google Scholar]
- 128.Boyd-Kimball D, Sultana R, Poon HF, Mohmmad-Abdul H, Lynn BC, Klein JB, Butterfield DA. Gamma-glutamylcysteine ethyl ester protection of proteins from Abeta(1-42)-mediated oxidative stress in neuronal cell culture: a proteomics approach. J Neurosci Res. 2005;79:707–713. doi: 10.1002/jnr.20393. [DOI] [PubMed] [Google Scholar]
- 129.Ansari MA, Abdul HM, Joshi G, Opii WO, Butterfield DA. Protective effect of quercetin in primary neurons against Abeta(1-42): relevance to Alzheimer's disease. The Journal of nutritional biochemistry. 2009;20:269–275. doi: 10.1016/j.jnutbio.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aksenov MY, Aksenova MV, Carney JM, Butterfield DA. Oxidative modification of glutamine synthetase by amyloid beta peptide. Free Radic Res. 1997;27:267–281. doi: 10.3109/10715769709065765. [DOI] [PubMed] [Google Scholar]
- 131.Butterfield DA, Hensley K, Cole P, Subramaniam R, Aksenov M, Aksenova M, Bummer PM, Haley BE, Carney JM. Oxidatively induced structural alteration of glutamine synthetase assessed by analysis of spin label incorporation kinetics: relevance to Alzheimer's disease. J Neurochem. 1997;68:2451–2457. doi: 10.1046/j.1471-4159.1997.68062451.x. [DOI] [PubMed] [Google Scholar]
- 132.Aksenov M, Aksenova M, Butterfield DA, Markesbery WR. Oxidative modification of creatine kinase BB in Alzheimer's disease brain. J Neurochem. 2000;74:2520–2527. doi: 10.1046/j.1471-4159.2000.0742520.x. [DOI] [PubMed] [Google Scholar]
- 133.Boyd-Kimball D, Castegna A, Sultana R, Poon HF, Petroze R, Lynn BC, Klein JB, Butterfield DA. Proteomic identification of proteins oxidized by Abeta(1-42) in synaptosomes: implications for Alzheimer's disease. Brain Res. 2005;1044:206–215. doi: 10.1016/j.brainres.2005.02.086. [DOI] [PubMed] [Google Scholar]
- 134.Sultana R, Butterfield DA. Alterations of some membrane transport proteins in Alzheimer's disease: role of amyloid beta-peptide. Molecular bioSystems. 2008;4:36–41. doi: 10.1039/b715278g. [DOI] [PubMed] [Google Scholar]
- 135.Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol. 1996;40:759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- 136.Harris ME, Carney JM, Cole PS, Hensley K, Howard BJ, Martin L, Bummer P, Wang Y, Pedigo NW, Jr, Butterfield DA. beta-Amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: implications for Alzheimer's disease. Neuroreport. 1995;6:1875–1879. doi: 10.1097/00001756-199510020-00013. [DOI] [PubMed] [Google Scholar]
- 137.Harris ME, Wang Y, Pedigo NW, Jr, Hensley K, Butterfield DA, Carney JM. Amyloid beta peptide (25-35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem. 1996;67:277–286. doi: 10.1046/j.1471-4159.1996.67010277.x. [DOI] [PubMed] [Google Scholar]
- 138.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sultana R, Butterfield DA. Identification of the oxidative stress proteome in the brain. Free Radic Biol Med. 2011;50:487–494. doi: 10.1016/j.freeradbiomed.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Butterfield DA, Hensley K, Harris M, Mattson M, Carney J. beta-Amyloid peptide free radical fragments initiate synaptosomal lipoperoxidation in a sequence-specific fashion: implications to Alzheimer's disease. Biochemical and biophysical research communications. 1994;200:710–715. doi: 10.1006/bbrc.1994.1508. [DOI] [PubMed] [Google Scholar]
- 141.Subramaniam R, Koppal T, Green M, Yatin S, Jordan B, Drake J, Butterfield DA. The free radical antioxidant vitamin E protects cortical synaptosomal membranes from amyloid beta-peptide(25-35) toxicity but not from hydroxynonenal toxicity: relevance to the free radical hypothesis of Alzheimer's disease. Neurochemical research. 1998;23:1403–1410. doi: 10.1023/a:1020754807671. [DOI] [PubMed] [Google Scholar]
- 142.Bruce-Keller AJ, Begley JG, Fu W, Butterfield DA, Bredesen DE, Hutchins JB, Hensley K, Mattson MP. Bcl-2 protects isolated plasma and mitochondrial membranes against lipid peroxidation induced by hydrogen peroxide and amyloid beta-peptide. J Neurochem. 1998;70:31–39. doi: 10.1046/j.1471-4159.1998.70010031.x. [DOI] [PubMed] [Google Scholar]
- 143.Koppal T, Subramaniam R, Drake J, Prasad MR, Dhillon H, Butterfield DA. Vitamin E protects against Alzheimer's amyloid peptide (25-35)-induced changes in neocortical synaptosomal membrane lipid structure and composition. Brain Res. 1998;786:270–273. doi: 10.1016/s0006-8993(97)01466-2. [DOI] [PubMed] [Google Scholar]
- 144.Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer's disease. Neurochemical research. 1998;23:81–88. doi: 10.1023/a:1022457605436. [DOI] [PubMed] [Google Scholar]
- 145.Mapstone M, Cheema AK, Fiandaca MS. Plasma phospholipids identify antecedent memory impairment in older adults. 2014;20:415–418. doi: 10.1038/nm.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Butterfield DA, Kanski J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer's amyloid beta-peptide 1-42. Peptides. 2002;23:1299–1309. doi: 10.1016/s0196-9781(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 147.Butterfield DA, Boyd-Kimball D. The critical role of methionine 35 in Alzheimer's amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta. 2005;1703:149–156. doi: 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 148.Butterfield DA, Sultana R. Methionine-35 of abeta(1-42): importance for oxidative stress in Alzheimer disease. Journal of amino acids. 2011;2011:198430. doi: 10.4061/2011/198430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kanski J, Aksenova M, Butterfield DA. The hydrophobic environment of Met35 of Alzheimer's Abeta(1-42) is important for the neurotoxic and oxidative properties of the peptide. Neurotox Res. 2002;4:219–223. doi: 10.1080/10298420290023945. [DOI] [PubMed] [Google Scholar]
- 150.Schoneich C. Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer's disease. Biochim Biophys Acta. 2005;1703:111–119. doi: 10.1016/j.bbapap.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 151.Varadarajan S, Kanski J, Aksenova M, Lauderback C, Butterfield DA. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer's Abeta(1--42) and Abeta(25--35) Journal of the American Chemical Society. 2001;123:5625–5631. doi: 10.1021/ja010452r. [DOI] [PubMed] [Google Scholar]
- 152.Butterfield DA, Swomley AM, Sultana R. Amyloid beta-Peptide (1-42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxid Redox Signal. 2013;19:823–835. doi: 10.1089/ars.2012.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 154.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 155.Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, Barrow CJ, Masters CL, Bush AI, Barnham KJ. Alzheimer's disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. The Journal of biological chemistry. 2001;276:20466–20473. doi: 10.1074/jbc.M100175200. [DOI] [PubMed] [Google Scholar]
- 156.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kanski J, Aksenova M, Schoneich C, Butterfield DA. Substitution of isoleucine-31 by helical-breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer's amyloid beta-peptide. Free Radic Biol Med. 2002;32:1205–1211. doi: 10.1016/s0891-5849(02)00821-3. [DOI] [PubMed] [Google Scholar]
- 158.Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R, Bredesen DE. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010;48:136–144. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sultana R, Robinson RA, Lange MB, Fiorini A, Galvan V, Fombonne J, Baker A, Gorostiza O, Zhang J, Cai J, Pierce WM, Bredesen DE, Butterfield DA. Do proteomics analyses provide insights into reduced oxidative stress in the brain of an Alzheimer disease transgenic mouse model with an M631L amyloid precursor protein substitution and thereby the importance of amyloid-beta-resident methionine 35 in Alzheimer disease pathogenesis? Antioxid Redox Signal. 2012;17:1507–1514. doi: 10.1089/ars.2011.4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Raffa RB, Tallarida RJ. Chemo-Fog. Landis Biosci. Press; Austin: 2010. [Google Scholar]
- 161.Raffa RB. Is a picture worth a thousand (forgotten) words?: neuroimaging evidence for the cognitive deficits in ‘chemo-fog’/‘chemo-brain’. Journal of clinical pharmacy and therapeutics. 2010;35:1–9. doi: 10.1111/j.1365-2710.2009.01044.x. [DOI] [PubMed] [Google Scholar]
- 162.Conroy SK, McDonald BC, Smith DJ, Moser LR, West JD, Kamendulis LM, Klaunig JE, Champion VL, Unverzagt FW, Saykin AJ. Alterations in brain structure and function in breast cancer survivors: effect of post-chemotherapy interval and relation to oxidative DNA damage. Breast cancer research and treatment. 2013;137:493–502. doi: 10.1007/s10549-012-2385-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.de Ruiter MB, Reneman L, Boogerd W, Veltman DJ, Caan M, Douaud G, Lavini C, Linn SC, Boven E, van Dam FS, Schagen SB. Late effects of high-dose adjuvant chemotherapy on white and gray matter in breast cancer survivors: converging results from multimodal magnetic resonance imaging. Human brain mapping. 2012;33:2971–2983. doi: 10.1002/hbm.21422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McDonald BC, Saykin AJ. Alterations in brain structure related to breast cancer and its treatment: chemotherapy and other considerations. Brain imaging and behavior. 2013;7:374–387. doi: 10.1007/s11682-013-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Simo M, Rifa-Ros X, Rodriguez-Fornells A, Bruna J. Chemobrain: a systematic review of structural and functional neuroimaging studies. Neuroscience and biobehavioral reviews. 2013;37:1311–1321. doi: 10.1016/j.neubiorev.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 166.Ahles TA, Saykin AJ. Candidate mechanisms for chemotherapy-induced cognitive changes. Nature reviews Cancer. 2007;7:192–201. doi: 10.1038/nrc2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ganz PA, Kwan L, Castellon SA, Oppenheim A, Bower JE, Silverman DH, Cole SW, Irwin MR, Ancoli-Israel S, Belin TR. Cognitive complaints after breast cancer treatments: examining the relationship with neuropsychological test performance. Journal of the National Cancer Institute. 2013;105:791–801. doi: 10.1093/jnci/djt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen Y, Jungsuwadee P, Vore M, Butterfield DA, St Clair DK. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Molecular interventions. 2007;7:147–156. doi: 10.1124/mi.7.3.6. [DOI] [PubMed] [Google Scholar]
- 169.Joshi G, Sultana R, Tangpong J, Cole MP, St Clair DK, Vore M, Estus S, Butterfield DA. Free radical mediated oxidative stress and toxic side effects in brain induced by the anti cancer drug adriamycin: insight into chemobrain. Free Radic Res. 2005;39:1147–1154. doi: 10.1080/10715760500143478. [DOI] [PubMed] [Google Scholar]
- 170.Joshi G, Aluise CD, Cole MP, Sultana R, Pierce WM, Vore M, St Clair DK, Butterfield DA. Alterations in brain antioxidant enzymes and redox proteomic identification of oxidized brain proteins induced by the anti-cancer drug adriamycin: implications for oxidative stress-mediated chemobrain. Neuroscience. 2010;166:796–807. doi: 10.1016/j.neuroscience.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tangpong J, Cole MP, Sultana R, Joshi G, Estus S, Vore M, St Clair W, Ratanachaiyavong S, St Clair DK, Butterfield DA. Adriamycin-induced, TNF-alpha-mediated central nervous system toxicity. Neurobiol Dis. 2006;23:127–139. doi: 10.1016/j.nbd.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 172.Jungsuwadee P, Cole MP, Sultana R, Joshi G, Tangpong J, Butterfield DA, St Clair DK, Vore M. Increase in Mrp1 expression and 4-hydroxy-2-nonenal adduction in heart tissue of Adriamycin-treated C57BL/6 mice. Molecular cancer therapeutics. 2006;5:2851–2860. doi: 10.1158/1535-7163.MCT-06-0297. [DOI] [PubMed] [Google Scholar]
- 173.Jungsuwadee P, Nithipongvanitch R, Chen Y, Oberley TD, Butterfield DA, St Clair DK, Vore M. Mrp1 localization and function in cardiac mitochondria after doxorubicin. Mol Pharmacol. 2009;75:1117–1126. doi: 10.1124/mol.108.052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Aluise CD, Miriyala S, Noel T, Sultana R, Jungsuwadee P, Taylor TJ, Cai J, Pierce WM, Vore M, Moscow JA, St Clair DK, Butterfield DA. 2-Mercaptoethane sulfonate prevents doxorubicin-induced plasma protein oxidation and TNF-alpha release: implications for the reactive oxygen species-mediated mechanisms of chemobrain. Free Radic Biol Med. 2011;50:1630–1638. doi: 10.1016/j.freeradbiomed.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 175.Dhar SK, St Clair DK. Manganese superoxide dismutase regulation and cancer. Free Radic Biol Med. 2012;52:2209–2222. doi: 10.1016/j.freeradbiomed.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 176.Tangpong J, Cole MP, Sultana R, Estus S, Vore M, St Clair W, Ratanachaiyavong S, St Clair DK, Butterfield DA. Adriamycin-mediated nitration of manganese superoxide dismutase in the central nervous system: insight into the mechanism of chemobrain. J Neurochem. 2007;100:191–201. doi: 10.1111/j.1471-4159.2006.04179.x. [DOI] [PubMed] [Google Scholar]
- 177.Keeney JT, Forster S, Sultana R, Brewer LD, Latimer CS, Cai J, Klein JB, Porter NM, Butterfield DA. Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: implications for low vitamin D-dependent age-related cognitive decline. Free Radic Biol Med. 2013;65:324–334. doi: 10.1016/j.freeradbiomed.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Keeney JT, Swomley AM, Forster S, Harris JL, Sultana R, Butterfield DA. Apolipoprotein A-I: insights from redox proteomics for its role in neurodegeneration. Proteomics Clin Appl. 2013;7:109–122. doi: 10.1002/prca.201200087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Furlanut M, Franceschi L. Pharmacology of ifosfamide. Oncology. 2003;65 Suppl 2:2–6. doi: 10.1159/000073350. [DOI] [PubMed] [Google Scholar]
- 180.Zhang Y, Kawedia JD, Myers AL, McIntyre CM, Anderson PM, Kramer MA, Culotta KS. Physical and chemical stability of high-dose ifosfamide and mesna for prolonged 14-day continuous infusion. Journal of oncology pharmacy practice : official publication of the International Society of Oncology Pharmacy Practitioners. 2014;20:51–57. doi: 10.1177/1078155213478284. [DOI] [PubMed] [Google Scholar]
- 181.Aluise CD, St Clair D, Vore M, Butterfield DA. In vivo amelioration of adriamycin induced oxidative stress in plasma by gamma-glutamylcysteine ethyl ester (GCEE) Cancer letters. 2009;282:25–29. doi: 10.1016/j.canlet.2009.02.047. [DOI] [PubMed] [Google Scholar]
- 182.Aluise CD, Sultana R, Tangpong J, Vore M, St Clair D, Moscow JA, Butterfield DA. Chemo brain (chemo fog) as a potential side effect of doxorubicin administration: role of cytokine-induced, oxidative/nitrosative stress in cognitive dysfunction. Advances in experimental medicine and biology. 2010;678:147–156. doi: 10.1007/978-1-4419-6306-2_19. [DOI] [PubMed] [Google Scholar]