

Summary
Bacterial pathogens can induce an inflammatory response from epithelial tissues due to secretion of the pro-inflammatory chemokine interleukin-8 (IL-8). Many bacterial pathogens manipulate components of the focal complex (FC) to induce signaling events in host cells. We examined the interaction of several bacterial pathogens with host cells, including Campylobacter jejuni, to determine if the FC is required for induction of chemokine signaling in response to bacterial pathogens. Our data indicate that secretion of IL-8 is triggered by C. jejuni, Helicobacter pylori, and Salmonella enterica serovar Typhimurium in response to engagement of β1 integrins. Additionally, we found that the secretion of IL-8 from C. jejuni infected epithelial cells requires FAK, Src, and paxillin, which in turn are necessary for Erk 1/2 recruitment and activation. Targeting the FC component paxillin with siRNA prevented IL-8 secretion from cells infected with several bacterial pathogens, including C. jejuni, Helicobacter pylori, Salmonella enterica serovar Typhimurium, Staphylococcus aureus, Pseudomonas aeruginosa, and Vibrio parahaemolyticus. Our findings indicate that maximal IL-8 secretion from epithelial cells in response to bacterial infection is dependent on the FC. Based on the commonality of the host response to bacterial pathogens, we propose that the FC is a signaling platform for an epithelial cell response to pathogenic organisms.
Keywords: Bacteria-host cell interactions, cell signaling, integrin, FAK, Erk1/2, focal complex
Introduction
Interleukin-8 is a chemokine that can be secreted by epithelial cells in response to stress or injury, including contact with bacterial and viral pathogens (Hoffmann et al., 2002). IL-8 diffuses locally upon secretion, acting as a chemo-attractant recruiting immune cells to the site of injury (Baggiolini et al., 1997). Thus, IL-8 plays a key role in initiating the inflammatory response against bacterial pathogens. While expression of IL-8 is partially controlled through histone modification and mRNA stabilization, both NF-κB and AP-1 activation are required for IL-8 gene CXCL8 transcription. NF-κB activation is often associated with activation of the Toll-like receptors that recognize bacterial components such as lipopolysaccharide (LPS) and flagellin (Hoffmann et al., 2002). AP-1 is localized to the nucleus where it is activated by MAP kinases. Pathogenic bacteria produce unique proteins that activate the MAP kinases via a variety of mechanisms (Mitsuno et al., 2001, Watson et al., 2005, Matlawska-Wasowska et al., 2010, Samuelson et al., 2013).
Mitogen activated protein kinases (MAP kinases) are signaling proteins that are important in many cellular processes, including regulation of IL-8 expression (Schaeffer et al., 1999, Hoffmann et al., 2002). MAP kinase activation requires the activation of a MAP kinase-kinase-kinase [or MAP triple kinase (e.g., Raf)]. The activated triple kinase then phosphorylates its target MAP kinase-kinase (e.g., MEK 1/2), leading to phosphorylation of the MAP kinase (e.g., Erk 1/2) (Schaeffer et al., 1999). A scaffold protein is required for MAP kinase activation and the prototypical MAP kinase scaffold is KSR (kinase suppressor of Ras), but other proteins, including components of the focal complex (FC), may function as MAP kinase scaffolds (Morrison, 2001, Roy et al., 2002, Ishibe et al., 2003).
Integrins are surface exposed heterodimeric cell receptors, which are common to many types of metazoan (animal) life, consisting of one α and one β subunit (Giancotti et al., 1999). The integrins function both as anchoring points that interact with specific extracellular matrix (ECM) components, as well as intracellular signaling proteins (Schwartz et al., 2002). The association of bacteria with the extracellular matrix of host cells or an associated integrin receptor is a common theme shared among pathogenic bacteria. Cellular association with the ECM results in recruitment and activation of focal complex components. The proteins of the FC are host cell structural and signaling molecules that contribute to cell motility and spreading by coordinating broad signaling cascades (Wozniak et al., 2004). Bacteria have evolved interactions with these highly conserved integrins and their associated ECM proteins, particularly fibronectin (Henderson et al., 2011).
Three proteins recruited to integrins, as part of the FC, are focal adhesion kinase (FAK), paxillin, and epidermal growth factor receptor (EGFR) (Giancotti et al., 1999, Zouq et al., 2009). The cytoplasmic tails of activated β1 integrins interact with FAK leading to its auto-phosphorylation and the association of the non-receptor tyrosine kinase Src (Chen et al., 1996, Orr et al., 2004, Parsons et al., 2004, Hall et al., 2011). Following association, Src phosphorylates multiple tyrosine residues on FAK, leading to an increase in FAK kinase activity (Orr et al., 2004). The fully activated FAK-Src complex then phosphorylates other associated proteins, including paxillin (Schaller et al., 1995, Orr et al., 2004, Hall et al., 2011). Of particular interest FAK-Src phosphorylation of paxillin can result in the recruitment of the MAP kinase Erk 1/2 (Schaller et al., 1995, Ishibe et al., 2003). Similarly, the EGFR can be activated through integrin activation utilizing the scaffold protein p130Cas as well as Src (Cabodi et al., 2004). Active Erk 1/2 translocates to the nucleus where it contributes to the transcription of IL-8 through activation of AP-1 (Hoffmann et al., 2002).
Pathogens often induce host inflammatory responses. For example, Campylobacter jejuni, which is a leading bacterial cause of gastroenteritis worldwide, causes an intense inflammatory response and diarrhea with blood and leukocytes in the stools. The latter is characteristic of a pathogen that invades the intestinal epithelium as cell invasion results in epithelial barrier disruption. C. jejuni possesses two fibronectin binding proteins termed CadF and FlpA (Konkel et al., 1997, Flanagan et al., 2009). Invasion of the cells lining the intestinal track is requires the binding of the bacteria to the host cells via the adhesins followed by the delivery of effector proteins termed the Campylobacter invasion antigens (Cia proteins) to host cells (Konkel et al., 2010). The Cia proteins are synthesized when C. jejuni are co-cultured with epithelial cells (Konkel et al., 1993). We have recently demonstrated that the CiaD effector is, in part, responsible for the activation of Erk 1/2, though the cellular components of this activation remain unknown (Samuelson et al., 2013). We hypothesize that the C. jejuni adhesins and secreted proteins act cooperatively to subvert components of the host cell focal adhesion complex to facilitate internalization, and that these virulence proteins also contribute to IL-8 secretion.
The recurring theme of bacterial interaction with components of the FC system, including the ECM components and the integrins, led us to hypothesize that these proteins are serving a critical role in bacterial pathogenesis. The goal of this work was to identify membrane associated and cytosolic signaling components required for Erk 1/2 activation as a prerequisite for IL-8 secretion in response to bacterial pathogens. We hypothesized that bacterial activation of the FC directly contributes to the activation of the MAP kinase signaling pathway in epithelial cells. We demonstrate that β1 integrins, FAK, Src, and paxillin are required for Erk 1/2 activation and IL-8 secretion in response to C. jejuni. We further show that several bacterial pathogens require paxillin for the generation of an IL-8 response, including Helicobacter pylori, Salmonella enterica serovar Typhimurium, Pseudomonas aeruginosa, Vibrio parahaemolyticus and Staphylococcus aureus. This work suggests an expanded role for the FC in the detection of pathogenic bacteria.
Results
β1 integrin is required for IL-8 secretion from multiple pathogens
We hypothesized that host epithelial cells have evolved the capacity to detect pathogenic bacteria via their interactions with the extracellular matrix (ECM). Given the prevalence of fibronectin binding proteins among these bacteria, we hypothesized that pathogen detection requires β1 integrin receptors. To test this hypothesis, we treated INT 407 human epithelial cells with siRNA specific to β1 integrin or a scrambled siRNA control, infected the cells with various pathogenic bacteria, and used an ELISA to measure the level of IL-8 in the supernatants. The cells were infected with Campylobacter jejuni, Helicobacter pylori, and Salmonella enterica Serovar Typhimurium. Uninfected cells served as a negative control. Knockdown of the β1 integrin with siRNA lead to a significant decrease in the level of IL-8 secreted following infection with all three organisms (Fig. 1A–C). The knockdown of β1 integrin in the siRNA treated cells was confirmed by immunoblot analysis (Fig. 1D). Based on these data, we concluded that the β1 integrin is required for a maximal IL-8 response.
Fig. 1. The β1 integrin in epithelial cells contributes to the IL-8 response to multiple bacterial pathogens.

Panels: (A–C) INT 407 cells were transfected with either siRNA specific for the β1 integrin or scrambled (Scrm) siRNA. The cells were then infected with C. jejuni, H. pylori, or S. Typhimurium. Secreted IL-8 levels were determined by ELISA following 24 hr (C. jejuni) or 5 hr (H. pylori and S. Typhimurium) incubations. Non-transfected cells infected with each bacteria served as a positive control and untreated uninfected cells served as a negative control. (D) The efficiency of the β1 integrin knockdown was tested by immunoblot analysis of cell lysates, with actin serving as a loading control.
C. jejuni must be metabolically active to promote IL-8 secretion from epithelial cells
We chose to use C. jejuni to dissect the role of the IL-8 response, as this pathogen activates a robust inflammatory response. To determine the role of the bacteria in inducing IL-8 secretion from epithelial cells, experiments were initially performed to determine if the bacteria must be metabolically active. IL-8 was not detected in the supernatants when C. jejuni was incubated with host cells for 24 hr in the presence of the bacterial protein synthesis inhibitor chloramphenicol (Fig. S1), indicating that the bacteria must be metabolically active to generate a host response. This result is consistent with previous findings (Samuelson et al., 2013), and demonstrates that constitutively synthesized bacterial components are not sufficient to promote IL-8 secretion.
C. jejuni activates the Raf/MEK/Erk MAPK signaling pathway
Erk 1/2 is highly activated in response to C. jejuni at every time point over a course of a 24 hr infection period (time points: 30 min, 3 hr, and 24 hr), as judged by experiments using a Map Kinase phospho-array (not shown). This finding is consistent with a previous report (Watson et al., 2005). To determine if C. jejuni activates the entire Raf/MEK/Erk MAP kinase pathway, INT 407 cells were infected with C. jejuni and lysed following a 30 min incubation. The lysates were immunoblotted with phospho-specific antibodies for Raf (S-338), MEK (S-217/221), and Erk 1/2 (T-202/Y-204), with total Erk 1/2 serving as a loading control. Uninfected cells served as a negative control. Erk 1/2, MEK 1/2 and c-Raf were all activated in response to C. jejuni (Fig. 2A and 2B). To determine if the activation of the Raf/MEK/Erk MAP kinase pathway leads to IL-8 secretion, INT 407 cells were treated with Raf Inhibitor I and Erk 1/2-activation inhibitor PD98059. Vehicle treated INT 407 cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control. A reduction in the level of IL-8 secreted from C. jejuni infected host cells was observed following treatment of the cells with Raf Inhibitor I (Fig. 2C) and Erk 1/2-activation inhibitor PD98059 (Fig. 2D). The effect of Raf inhibition was not as potent as that of the Erk 1/2 inhibition regardless of the Raf inhibitors tested (Fig. S2), suggesting that other MAP triple kinases may be acting on the pathway. Taken together, these results indicate that C. jejuni activates the Raf/MEK/Erk MAP kinase pathway and that the Raf- 1, MEK 1/2, and Erk 1/2 components are required for maximal IL-8 secretion.
Fig. 2. C. jejuni activates Erk 1/2 by the canonical MAPK activation pathway.

Panels: (A) Immunoblot analysis of INT 407 cells infected with C. jejuni following a 30 min incubation. The level of phosphorylated c-Raf, MEK 1/2 and Erk 1/2 were determined by immunoblot with phospho-specific monoclonal antibodies for each protein. Total Erk 1/2 (tErk) served as a loading control. The images shown are a composite of different sections of the same immunoblot. (B) Densitometric analysis of phosphorylated Raf/MEK/Erk from three samples. (C) INT 407 cells were treated with Raf Inhibitor I and IL-8 secretion was determined by ELISA 24 hr following infection with C. jejuni. Uninfected cells served as a negative control. (D) INT 407 cells were treated with the Erk 1/2 activation inhibitor PD98059, infected with C. jejuni, and the level of IL-8 in supernatants were measured by ELISA. Vehicle treated cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control.
β1 integrins are required for Erk 1/2 activation
We hypothesized that β1 integrin activation is linked with Erk 1/2 activation and is required for IL-8 secretion. To test this hypothesis, we used siRNA to knockdown β1 integrin expression, infected cells with C. jejuni, and examined the level of Erk 1/2 activation. Non-specific scrambled siRNA was used to control for the effect of transfection. Knockdown of the β1 integrin in host cells resulted in a significant reduction in the amount of activated (phosphorylated) Erk 1/2 when compared to untreated cells (Fig. 3A). To determine if the decrease in β1 integrin expression was effecting the level of IL-8 gene activation, transcripts were measured by RT-PCR analysis. Scrambled siRNA treated cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control. INT 407 cells treated with β1 integrin siRNA were significantly reduced in IL-8 mRNA levels as compared to controls treated with nonspecific siRNA (Fig. 3B). This finding is consistent with our hypothesis, indicating that the β1 integrin is necessary for Erk 1/2 activation and IL-8 gene expression.
Fig. 3. The β1 integrin is required for full activation of Erk 1/2 and IL-8 gene transcription in response to C. jejuni.

Panels: (A) INT 407 cells were transfected with siRNA specific for the β1 integrin or scramble (Scrm) siRNA. The cells were infected with C. jejuni and incubated for 30 min. Uninfected cells served as a negative control. The cell lysates were analyzed by immunoblot using antibody against pErk 1/2. The membranes were re-probed using antibody against actin as a loading control. Densitometric analysis was performed to quantify the level of Erk 1/2 activation. (B) INT 407 cells were treated with siRNA and infected with C. jejuni. The level of IL-8 gene expression following a 6 hr infection was measured by RT-PCR. IL-8 expression was normalized to GAPDH transcript levels with uninfected cells serving as negative control.
Src is required for IL-8 secretion
Several proteins are known to associate with the clustered β1 integrins, including the EGFR, the scaffold protein p130Cas, and the tyrosine kinase Src (Cabodi et al., 2004). Importantly, the EGFR and Src kinase are both known to be required for C. jejuni invasion of host cells (Eucker et al., 2012). Therefore, we performed studies to determine if the secretion of IL-8 from C. jejuni infected epithelial cells involves EGFR, p130Cas, and Src (Fig. 4).
Fig. 4. Src is required for full Erk 1/2 activation and IL-8 secretion.

Panels: (A) INT 407 cells were treated with PP2 and erlotinib to inhibit Src and the EGFR, respectively. Vehicle treated cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control. Secreted IL-8 was quantified by ELISA. (B) INT 407 cells were treated with PP2 and erlotinib to inhibit Src and the EGFR, respectively. Erk 1/2 phosphorylation (activation) following a 30 min infection was measured by immunoblot using antibodies against phospho-Erk 1/2 (pErk). The membranes were re-probed using antibody against total Erk 1/2 (tErk) as a loading control. Densitometric analysis was used to quantify the level of pErk activation. (C) INT 407 cells were transfected with siRNA specific to p130Cas or scrambled siRNA (control). Non-transfected cells infected with C. jejuni served as a positive control and untreated cells served as a negative control. The efficiency of p130Cas knockdown was determined by immunoblot analysis. The membranes were re-probed using antibody against actin as a loading control.
To test the contribution of the EGFR in IL-8 secretion, INT 407 cells were treated with the EGFR inhibitor erlotinib and infected with C. jejuni. Uninfected cells served as a negative control. We measured the amount of IL-8 secreted from infected cells and found that inhibition of the EGFR did not significantly reduce IL-8 secretion (Fig. 4A). Because activated EGFR can lead to Erk 1/2 activation (Roberts et al., 2007), we also examined if Erk 1/2 was activated in response to C. jejuni when EGFR activation was prevented by erlotinib. Inhibition of EGFR activation with erlotinib did not prevent Erk 1/2 activation following C. jejuni infection (Fig. 4B). This finding indicates that Erk 1/2 is activated independent of EGFR. We also infected INT 407 cells with the C. jejuni cadF flpA double mutant, as this isolate does not activate the EGFR (Eucker et al., 2012). No difference was observed in the level of IL-8 secretion from INT 407 cells infected with the C. jejuni cadF flpA double mutant when compared to the wild-type strain (Fig. S3). This supports the finding that the EGFR is not contributing to IL-8 secretion.
To determine the role of p130Cas in IL-8 secretion, INT 407 cells were treated with siRNA to knockdown p130Cas expression and infected with C. jejuni. Uninfected cells served as a negative control. The level of IL-8 in supernatants was measured by ELISA. The level of IL-8 secreted from C. jejuni infected INT 407 cells treated with p130Cas siRNA was not different from C. jejuni infected cells treated with scrambled siRNA (Fig. 4C). This result suggests that p130Cas is not involved in triggering IL-8 secretion.
To determine the role of Src in IL-8 secretion, INT 407 cells were treated with Src kinase inhibitor PP2 and infected with C. jejuni. Uninfected cells served as a negative control. In contrast to the EGFR and p130Cas, treatment of INT 407 cells with PP2 significantly reduced the amount of secreted IL-8 following C. jejuni infection (Fig. 4A). We then probed lysates of C. jejuni infected INT 407 cells treated with PP2 for Erk 1/2 activation. We found that Src inhibition reduced the level of Erk 1/2 activation following C. jejuni infection (Fig. 4C). Taken together, these data suggest that Erk 1/2 activation is independent of the EGFR and p130Cas, but indicate that Src is playing a role in Erk 1/2 activation and IL-8 secretion.
Rac1 activation and host cell invasion are not required for IL-8 secretion
We have shown previously that the C. jejuni CiaC effector protein is delivered to host cells and is required for cell invasion. We further reported that a C. jejuni ciaC mutant is deficient in the activation of the Rho-GTPase Rac1. In agreement with the latter finding, knockdown of the Rac1 guanine exchange factor Dock180 reduced the internalization of the C. jejuni wild-type strain to that of the ciaC mutant (Eucker et al., 2012). Here we found that infection of INT 407 cells with the C. jejuni ciaC mutant, which is deficient in host cell invasion and Rac1 activation, triggered IL-8 secretion to the same extent as cells infected with a C. jejuni wild-type strain (Fig. S4A and B). We also found that a knockdown of Dock180 expression using siRNA did not result in a reduction in IL-8 secretion (Fig. S4C). These data indicate that host cell invasion and Rac1 activation are not required for an IL-8 response to C. jejuni.
Focal adhesion kinase (FAK) is critical for IL-8 secretion in response to C. jejuni
FAK is a non-receptor tyrosine kinase recruited to activated β1 integrin receptors (Mitra et al., 2006). Following association with the β1 integrin, FAK autophosphorylates Y-397, resulting in the recruitment and activation of Src (Mitra et al., 2005). Src activation leads to the subsequent phosphorylation of FAK at Y-925. To reduce FAK activation and subsequent phosphorylation events, INT 407 cells were treated with TAE 226, an inhibitor of FAK Y-397 and Y-925 phosphorylation. The cells were then infected with C. jejuni and Erk 1/2 phosphorylation (activation) was assessed by immunoblot analysis. Vehicle treated cells infected with C. jejuni and untreated cells served as positive and negative controls, respectively. Treatment of cells with the FAK inhibitor significantly reduced Erk 1/2 activation following C. jejuni infection, as compared to vehicle treated cells (Fig. 5A). IL-8 mRNA accumulation was then measured by RT-PCR following TAE 226 treatment of host cells. Treatment with TAE 226 significantly reduced IL-8 gene expression as compared to vehicle treated cells (Fig. 5B). We also measured the level of secreted IL-8 and found that TAE 226 treatment resulted in a dose-dependent decrease in secreted IL-8 as compared to vehicle treated cells infected with C. jejuni (Fig. 5C). This result is in agreement with the observation that inhibition of Src with PP2 reduces IL-8 secretion from C. jejuni infected cells as Src is required for full activation of FAK. To ensure that the effects observed in INT 407 cells were not cell-type specific, human colonic carcinoma cells (Caco-2 cells) were treated with TAE 226 and infected with C. jejuni. While Caco-2 cells secreted less IL-8 than the INT 407 cells in response to C. jejuni infection, the result obtained with TAE 226 treatment was consistent with the two cell lines (Fig. 5D). Collectively, these data suggest that FAK is an essential component of the IL-8 signaling cascade.
Fig. 5. FAK is critical to Erk 1/2 activation and IL-8 secretion in response to C. jejuni.

Panels: (A) INT 407 cells were treated with focal adhesion kinase (FAK) inhibitor TAE 226 and infected with C. jejuni. Immunoblots coupled with densitometric analysis were used to determine the level of Erk 1/2 activation following a 40 min incubation. (B) INT 407 cells were treated with TAE 226 and infected with C. jejuni. The level of IL-8 gene expression was measured by RT-PCR following a 6 hr infection. IL-8 expression was normalized to GAPDH transcript levels with uninfected cells serving as a negative control. (C) INT 407 cells were treated with TAE 226 and infected with C. jejuni. Supernatants were collected after 6 hr and IL-8 was quantified by ELISA. Multiple TAE 226 concentrations were tested to determine if there was a dose-dependent effect. Uninfected cells served as a negative control. (D) Caco-2 human intestinal epithelial cells were treated with TAE 226 and infected with C. jejuni. Supernatants were collected after 6 hr incubation, and IL-8 was quantified by ELISA. Uninfected cells served as negative control.
Focal adhesion kinase (FAK) activates PI3 kinase
FAK signaling leads to the recruitment and activation of PI3 kinase, which leads to the recruitment of molecules linked to numerous signaling cascades that could trigger IL-8 secretion from cells. To explore the importance of FAK, we further examined the PI3 kinase response of C. jejuni infected cells when FAK was inhibited with TAE 226 treatment. Because the activation state of PI3 kinase is not governed by posttranslational modification and is not directly measurable, we measured activation of the downstream signaling protein Akt as an indicator of PI3 kinase activity. A significant reduction in the level of Akt activation was observed following TAE 226 treatment (Fig. S5A). This finding supports the proposal that C. jejuni activation of FAK leads to PI3 kinase activation.
Activation of PI3 kinase and Akt can also lead to activation of NF-κB, another signaling protein required for transcriptional activation of IL-8 (Hoffmann et al., 2002). Because Akt can activate NF-κB, INT 407 cells were treated with TAE 226 or the PI3 kinase inhibitor LY294002, infected with C. jejuni, and NF-κB activation was measured by immunoblot analysis. Treatment of cells with TAE 226 or LY294002 did not alter NF-κB activation (Fig. S5B), indicating that the NF-κB pathway is not dependent on cross-talk with FAK or PI3 kinase. Noteworthy is that the inhibition of PI3 kinase by treatment of INT 407 cells with LY294002 resulted in a significant decrease in IL-8 secretion from C. jejuni infected INT 407 cells. This suggests that PI3 kinase is playing a role in modulating a host response (Fig S5C).
Paxillin is in a complex with the MAP kinase Erk 1/2 and is required for an IL-8 response
Paxillin, which is activated by the FAK-Src complex, leads to the recruitment of the inactive (non-phosphorylated) form of Erk 1/2 (Ishibe et al., 2003, Ishibe et al., 2004, Mitra et al., 2005, Sen et al., 2010). Because paxillin is activated in response to C. jejuni, we hypothesized that paxillin associates with Erk 1/2 and is necessary for secretion of IL-8 (Monteville et al., 2003). To determine if C. jejuni infection results in an increase in Erk 1/2 association, immunoprecipitation experiments were performed (Figure 6). We found that Erk 1/2 co-precipitated with paxillin from cells infected with C. jejuni, while uninfected cells did not yield a paxillin-Erk 1/2 co-immunoprecipitate (Fig. 6A). To determine if paxillin is involved in Erk 1/2 activation and IL-8 secretion, knockdown of paxillin expression was performed using siRNA. Cells transfected with scrambled siRNA served as a control. Cells treated with paxillin siRNA secreted significantly less IL-8 than cells treated with control siRNA (Fig. 6B). In addition, paxillin knockdown resulted in a significant decrease in IL-8 gene expression when compared to infected cells treated with control siRNA (Fig. 6C). The level of activated Erk 1/2 was also reduced in cells treated with paxillin siRNA when compared to infected cells treated with control siRNA, as judged by immunoblot analysis (Fig. 6D). These findings indicate that paxillin is in a complex with Erk 1/2 and that paxillin is required for IL-8 secretion.
Fig. 6. Erk 1/2 is recruited to and activated at paxillin in response to C. jejuni infection.

Panels: (A) INT 407 cells were infected with C. jejuni and incubated for 40 min. Uninfected cells served as a negative control. Paxillin was immunoprecipitated from cell lysates with anti-paxillin antibody. The cell lysates were analyzed by immunoblot and probed with antibody against total Erk 1/2. Immunoprecipitations using antibodies against IgG were used to examine nonspecific interactions. (B) INT 407 cells were treated with siRNA specific to paxillin and infected with C. jejuni. Lysates were collected and immunoblots probed with antibodies directed against phospho Erk 1/2 (pErk), with total Erk 1/2 (tErk) serving as a loading control. The efficiency of paxillin knockdown using siRNA treatment was tested by immunoblot analysis. (C) INT 407 cells were transfected with siRNA specific to paxillin or a scambled (Scrm) control and infected with C. jejuni. The level of IL-8 gene expression was measured by RT-PCR following a 6 hr infection. IL-8 expression was normalized to GAPDH transcript levels. Uninfected cells served as negative control. (D) ELISA quantification of IL-8 secretion by INT 407 cells treated with siRNA specific to paxillin or scrambled siRNA following incubation with C. jejuni. Uninfected cells served as a negative control.
Paxillin is required for IL-8 secretion from host epithelial cells in response to multiple bacterial pathogens
In this study, we determined that the β1 integrin, FAK, Src, and paxillin, which are all components of the focal complex, are required for maximal Erk 1/2 activation and IL-8 induction in response to infection with C. jejuni. Based on these findings, we hypothesized that Helicobacter pylori and Salmonella enterica Serovar Typhimurium also required paxillin for maximal activation of an IL-8 response in epithelial cells. To test this hypothesis, INT 407 cells were treated with siRNA to paxillin and the amount of IL-8 secreted from infected cells was measured (Figure 7A and B). Cells treated with scrambled siRNA served as a positive control and untreated cells served as a negative control. Knockdown of paxillin resulted in a significant reduction in the level of IL-8 secreted from cells infected with either H. pylori or S. Typhimurium. To determine if paxillin is a part of a conserved response of epithelial cells to other pathogenic bacteria, cells were also infected with Vibrio parahaemolyticus, Pseudomonas aeruginosa, and Staphylococcus aureus. We chose these pathogens because they are known to induce inflammation and have been shown to interact with the FC. INT 407 cells were also incubated with Citrobacter freundii, which is a commensal organism, as a negative control as it should not induce an inflammatory response. Paxillin siRNA treatment of INT 407 cells was also found to significantly reduce the amount of IL-8 secreted in response to V. parahaemolyticus, P. aeruginosa, and S. aureus (Fig. S6). Based on these findings, we concluded that the focal complex is a central player in epithelial cells detecting and responding to bacterial pathogens.
Fig. 7. Knockdown of paxillin results in a decrease in IL-8 secretion from epithelial cells in response to H. pylori and S. Typhimurium.
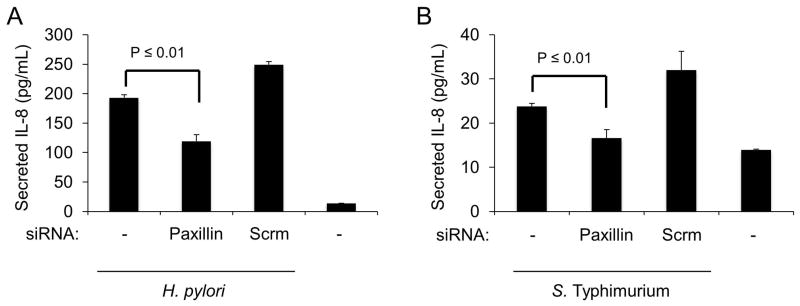
Panels: (A, B) INT 407 cells were treated with siRNA specific to paxillin or scrambled siRNA. The cells were infected with H. pylori or S. Typhimurium and supernatants were collected after a 5 hr incubation and tested for IL-8 by ELISA. Uninfected cells served as a negative control.
Discussion
The benefit of inflammation to invading bacterial pathogens is debatable. On the one side, inflammation and the resultant inundation by immune cells can be seen as detrimental to invasive pathogens. The recruited immune cells can kill the invading pathogenic bacteria and prevent dissemination into other tissues. However, some benefits to the pathogen can be derived from the effects of IL-8 induction. Macrophages recruited to infected tissues may disrupt the epithelium, allowing increased access to the basement membrane necessary for disease progression. Such a strategy of epithelial disruption has been reported for Shigella species (Sansonetti et al., 1999). Another benefit for pathogens is the increase in nutrients in the exudate as well as the clearance of competing organisms through host secreted antibacterial agents and increased peristaltic flushing (Winter et al., 2010). Bacteria may have evolved specific mechanisms for inducing inflammation due to the positive aspects of inflammation.
The purpose of this study was to identify the host epithelial cell signaling cascade that C. jejuni activate to cause IL-8 secretion. We utilized C. jejuni, which is known to trigger an intense inflammatory response in an infected host, to dissect the host cell components necessary for IL-8 secretion. We have shown that components of the focal complex, including β1 integrins, FAK, and Src, play a key role in the activation of Erk 1/2. We have also shown that Erk 1/2 activation is Src dependent, but independent of the EGFR and p130Cas. Noteworthy is that activated Erk 1/2 translocates to the nucleus where it activates transcription factors such as AP-1, resulting in IL-8 secretion (Romashkova et al., 1999, Tak et al., 2001, Hoffmann et al., 2002). We have demonstrated that Erk 1/2 is associated with paxillin in response to C. jejuni infection, as evidenced by paxillin immunoprecipation experiments. Knockdown of paxillin resulted in a decrease in Erk 1/2 activation and IL-8 secretion. Based on these data, we generated a model illustrating the focal complex components involved in triggering IL-8 secretion (Fig. 8). To our knowledge, this is the first report that identifies the host cell proteins required for Erk 1/2 activation in response to C. jejuni infection.
Fig. 8. The focal complex is required for IL-8 induction.

Shown is a model illustrating the signaling cascade leading to IL-8 secretion from C. jejuni infected cells. We propose that C. jejuni associates with fibronectin leading activation of β1 integrins. The β1 integrins serve as a scaffold for the association and activation of FAK. Src is then recruited to FAK and becomes activated. The FAK-Src complex phosphorylates paxillin, leading to the recruitment and activation of Erk 1/2, and IL-8 gene transcription. The bacterial proteins CadF and FlpA (yellow and blue dots on the bacteria respectively) and host proteins outlined with a dashed line (EGFR, p130Cas, Dock180, and Rac1) are required for C. jejuni host cell invasion, but do not play a significant role in IL-8 induction.
Many pathogens, including Salmonella Typhimurium and Helicobacter pylori, are known to exploit the focal complex to their advantage (Shi et al., 2006, Kwok et al., 2007). In the case of Salmonella, manipulation of the focal complex results in host cell invasion. One of the focal complex components that Salmonella requires for host cell invasion is FAK, but it does not require the kinase function of the protein, suggesting that FAK may be required solely as a scaffold (Shi et al., 2006). In contrast to Salmonella as well as C. jejuni, H. pylori does not invade host cells, but does interact with β1 integrins for the delivery of the effector protein CagA. Following delivery to the cytosol of the host cell, CagA promotes the activation of FAK (Kwok et al., 2007). In agreement with our findings, paxillin is known to be required for IL-8 secretion in response to H. pylori (Tabassam et al., 2011). Our results show that disruption of the focal complex results in a decrease in IL-8 secretion from cells infected with both S. Typhimurium and H. pylori. Collectively these findings show that IL-8 regulation in this system is independent of host cell invasion. Our findings further indicate that the role of the FC is not limited to one or two pathogenic organisms, but is a conserved epithelial response to pathogens.
The bacterial component(s) required for the activation of the IL-8 signaling cascade through the focal complex are not yet known. The C. jejuni CadF and FlpA proteins bind to fibronectin and promote integrin activation (Monteville et al., 2003). Thus, it was unexpected that the level of IL-8 in supernatants from INT 407 cells infected with the C. jejuni cadF flpA double mutant was not different (reduced) compared to cells infected with the C. jejuni wild-type strain (Fig. S3). While the result with the C. jejuni cadF flpA double mutant is confounding, especially given that the knockdown of the β1 integrin with siRNA resulted in a significant decrease in secreted IL-8 following C. jejuni infection (Fig. 1A), several explanations are possible for why the integrin β1 subunit is required for IL-8 production. The simplest explanation is that C. jejuni activates the β1 subunit via a third Fn-binding protein or a secreted effector protein. It is also possible that the cytoplasmic tail of integrin β1 subunit may be activated via inside-out signaling. Well documented is that the short cytoplasmic tail of the β subunit interacts with cytosolic scaffold and regulatory proteins, leading to the activation of signaling cascades (Hynes, 2002). Noteworthy is that C. jejuni delivers multiple effector proteins to the cytosol of epithelial cells that engage components of the focal complex that may promote inside-out signaling by engagement of the integrin β1 subunit. Studies are currently being performed in our laboratory to dissect the cellular mechanism of β1 subunit activation (outside-in versus inside-out signaling) in response to the C. jejuni cadF flpA double mutant.
We have shown that C. jejuni, S. Typhimurium, and H. pylori trigger IL-8 secretion from epithelial cells by activating the focal complex. Of additional interest is the fact that these pathogens bind to or have components that interact with fibronectin. C. jejuni has two fibronectin binding proteins, termed CadF and FlpA; H. pylori produces a fibronectin interacting toxin VacA; and S. Typhimurium interacts with fibronectin through ShdA and MisL (Konkel et al., 1997, Cover et al., 2005, Flanagan et al., 2009, Henderson et al., 2011). Although each of these pathogens interacts with fibronectin in a distinctive manner, we have shown that all of these pathogens require β1 integrin and paxillin to induce a maximal IL-8 response. Consistent with our model shown in Figure 8, others have shown that S. Typhimurium and H. pylori activate Erk 1/2 and that Erk 1/2 activation is required for IL-8 secretion (Mitsuno et al., 2001, Galdiero et al., 2002, Vitiello et al., 2004, Torok et al., 2005). Based on these findings, it is tempting to speculate that certain bacterial pathogens have evolved to activate the focal complex, resulting in an inflammatory response. Alternatively, host cells may have evolved mechanisms that allow them to detect pathogen association with fibronectin and/or integrin receptors.
Our understanding of the MAP kinase activation pathways has significantly increased over the past decade. Cellular biologists have shown that paxillin acts as a scaffold for Erk 1/2 activation following its phosphorylation by FAK/Src (Ishibe et al., 2003). Thus, paxillin has been found to respond to contact-dependent signaling, allowing for rapid activation of cell signaling cascades. We show that Erk 1/2 co-precipitates with paxillin in response to C. jejuni infection. Based on this finding, we propose that paxillin is acting as an Erk 1/2 scaffold upon C. jejuni infection. In the context of inflammation, the function of paxillin may not be limited to peripheral functions. Recent work has shown that paxillin translocates with Erk 1/2 to the nucleus where it interacts directly with DNA to regulate gene expression (Sen et al., 2010). Therefore, the knockdown of paxillin may alter the regulation of various genes. To address this possibility, we transfected INT 407 cells with a paxillin-GFP construct and infected them with C. jejuni. Paxillin was not observed in the nucleus of the C. jejuni-infected cells after 6 hr period, as judged by confocal microscopy (not shown). Based on this finding, we favor the hypothesis that components of the focal complex, particularly paxillin, are modulating a peripheral host cell response to pathogenic bacteria.
In summary, we have shown that several bacterial pathogens utilize components of the focal complex to induce IL-8 secretion. This result is significant because it provides deeper insight into the cellular mechanism of pathogen detection by an epithelial cell. Future work will focus on identifying the bacterial proteins (e.g., adhesins and effector proteins) that initiate host cell activation.
Experimental procedures
Cell culture and bacterial strains
Human INT 407 intestinal epithelial cells (ATCC CCL6; American Type Culture Collection, Manassas, VA) were grown in Minimal Essential Medium (MEM) supplemented with 10% fetal bovine serum (FBS). The C. jejuni F38011 clinical strain, ciaC mutant, and cadF flpA double mutant were grown on Mueller-Hinton blood (MHB) agar as described previously (Konkel et al., 1992). H. pylori strain 43504 was grown on MHB agar in 100% humidity. P. aeruginosa strain MEK1109, C. freundii ATCC 8090, S. aureus strain MEK1111, and V. parahaemolyticus strain MEK1110 were passaged on LB agar in microaerobic conditions (85% N2, 10% CO2, 5% O2, and 80% humidity). Prior to an assay, the bacteria were subcultured onto MHB plates for 18 to 24 hr.
Antibodies and Reagents
Paxillin antibody was purchased from BD Transduction Laboratories (San Diego, CA). Antibodies against β1 integrin (4706), pErk (4370), p c-Raf (9427), and pMEK1/2 (9154) were acquired from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Erk 1/2 (sc-94), Dock 180 (sc-13163), and actin (sc-1616) were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Antibody against p130Cas (610271) was purchased from BD Signal Transduction Laboratories (San Jose, CA, USA). Secondary goat anti-rabbit (A6154) and goat anti-mouse (A4416) HRP-conjugated antibodies were purchased from Sigma Aldrich (St Louis, MO, USA). All antibodies were used according to the manufacturer’s instructions.
The inhibitors were used at the following concentrations: Bacterial protein synthesis inhibitor: chloramphenicol at 1 mg/mL (Sigma), MEK 1/2: PD98059 at 50 μM (Selleck, Houston, TX, USA), FAK: TAE 226 at 5, 2.5, 1.25 and 0.625 μM (Novartis, Basel, Switzerland), Src: PP2 at 10 μg/mL (Sigma), Raf: Raf Inhibitor I at 9 nM (Merck, Darmstadt, Germany), PLX-4720 at 80 nM (Selleck), GDC-0879 at 25 nM (Selleck), and EGFR: erlotinib at 20 μM (LC Laboratories, Woburn, MA). Inhibitor experiments were performed with samples treated with each inhibitor or the appropriate vehicle. Using the trypan blue viability assay, we assessed host cell survival upon treatment with each inhibitor. All treatments reported herein resulted in no detectable change in host cell viability. The concentration of chloramphenicol has been previously demonstrated to completely prevent bacterial protein synthesis in this strain of C. jejuni (Konkel et al., 1992).
IL-8 secretion assay
INT 407 cells were seeded into 24-well tissue culture trays at a confluency of 1.5 × 105 cells per well and cultured overnight at 37°C. Inhibitors were added to cells 30 min prior to bacterial inoculation at the indicated concentrations. C. jejuni were re-suspended from MHB plates in MEM 1% FBS to an optical density of OD540 0.03 and then added to the INT 407 cells. The tissue culture plates were centrifuged for 1 min at 800 × g to promote host cell contact. After a 30 min incubation period at 37°C, supernatant was removed from each well and replaced with MEM 1% FBS containing the appropriate inhibitor. The trays were incubated overnight at 37°C and the supernatant was collected and analyzed by ELISA using the BD optEIA human IL-8 detection set. Samples for each condition were generated in quadruplicate and assayed by ELISA in duplicate. The results were confirmed by repeating the assays on multiple days.
Immunoblot analysis
INT 407 cells were seeded into 6-well tissue culture trays at a confluency of 2.5 × 105 cells per well and incubated at 37°C overnight. The cells were rinsed and serum starved in MEM without FBS for 3 hr. Prior to adding C. jejuni, the INT 407 cells were pretreated with each inhibitor for 30 min. Bacteria were added in serum free MEM at an optical density of OD540 0.03 and incubated for 40 min. Following incubation the cells were rinsed once with ice-cold PBS supplemented with 50 mM NaF and lysed in lysis buffer (10 mM HEPES, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 10% glycerol, 0.1% Triton X-100, pH 7.4) supplemented with protease inhibitor cocktail P2714 (Sigma). The protein in the cell lysates were separated by SDS-polyacrylamide gel electrophoresis (12% SDS-PAGE), transferred to Immobilon PSQ (PVDF) membranes (Millipore, Billerica, MA, USA), and probed with specific antibodies. Blots were developed with Perkin Elmer ECL Western Lightning and visualized with a GE ImageQuant LAS-4000 mini, (GE Healthcare Biosciences Pittsburgh, PA, USA).
siRNA treatment
The siRNA against β1 integrin (ITGB1HSS105559), paxillin (PXNHSS108927), and a High GC duplex (46-2000) negative control (Scrm) were obtained from Invitrogen (Grand Island, NY). Target gene knockdown was performed by reverse transfection according to the manufacturer’s instructions. Briefly, INT 407 cells were seeded into 24-well trays containing 1 pmol siRNA and Lipofectamine RNAi MAX (Invitrogen) at 1.5 × 106 cells per well. The cells were incubated 24 hr at 37°C prior to inoculation with C. jejuni according to the IL-8 secretion assay described above. After the supernatants were removed for IL-8 concentration determination, lysates of the INT 407 cells were prepared for immunoblot analysis to determine the effectiveness of protein knockdown.
Immunoprecipitation
INT 407 cells were grown in T75 flasks to 60% confluency, rinsed, and serum starved for 24 hr. Cells were infected with the C. jejuni wild-type strain and incubated for 40 min. The cells were rinsed with ice-cold PBS supplemented with 50 mM NaF prior to lysis. Lysates were generated using IP lysis buffer (25 mM Tris, 150 mM NaCl, 50 mM NaF, 1mM EDTA, 1 mM Na3VO4 5% glycerol, 0.1% Triton X-100 pH 7.4) and centrifuged at 12,000 × g at 4°C for 10 min to remove insoluble proteins. Following centrifugation, the protein concentration of each supernatant was determined by Precision Red protein quantification assay (Cytoskeleton, Denver, CO, USA) and normalized. The cell lysates were then incubated with mouse monoclonal anti-paxillin antibody (BD Transduction Labs) and protein A/G conjugated agarose beads (Santa Cruz) while rocking for 24 hr at 4°C. The beads were pelleted by centrifugation at 5000 × g for 3 min. The pellets were washed with IP wash buffer (20 mM HEPES, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 10% glycerol, 0.1% Triton X-100 pH 7.4) in three subsequent wash-centrifugation cycles. The final pellet was suspended in Laemmli sample buffer.
RT-PCR analysis
INT 407 cells were cultured in a 24-well tissue culture tray. For the use of chemical inhibitors the INT 407 cells were pretreated with vehicle or TAE 226 for 30 min and then infected with C. jejuni. INT 407 cells treated with siRNA were infected with C. jejuni 18 hr after transfection. After a 6 hr infection, the RNA was extracted using the Aurum Total RNA Fatty and Fibrous Tissue Kit, (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Following total RNA extraction cDNA was generated using SuperScript® III First-Strand Synthesis System (Life Technologies, Grand Island, NY, USA). The relative cDNA was quantified using Power Sybr Green (Life Technologies). Analysis of relative gene expression was performed using ΔΔCT, with GAPDH serving as the reference gene.
Statistical analysis
All results presented in this work represent at least three biological replicates from experiments repeated on multiple days. Averages and standard error mean (SEM) were generated from each sample set. F tests were performed to ensure normal deviation between sets. Those data that did not have significant differences in their deviations were tested with an unpaired, two-tailed Student’s t test. Data with significant differences between standard deviations were tested with an unpaired, two-tailed Welch’s t test. To make multiple comparisons within one data set the data was analyzed by one-way ANOVA and individual comparisons were then made by Tukey’s posthoc test.
Supplementary Material
Fig. S1. C. jejuni requires protein synthesis to induce an IL-8 response from epithelial cells.
C. jejuni was treated with the bacterial protein synthesis inhibitor chloramphenicol. INT 407 cells were infected with the antibiotic treated C. jejuni or vehicle treated bacteria. Uninfected cells served as a negative control. The level of IL-8 in supernatants collected after 24 hr incubation was measured by ELISA.
Fig. S2. Raf inhibition results in partial reduction of IL-8 secretion in response to C. jejuni.
INT 407 cells were treated with Raf inhibitors PLX-4720 or GDC-0879. The cells were infected with C. jejuni and incubated for 24 hr. Uninfected cells served as a negative control. Supernatants were collected and the amount of IL-8 secreted was quantified by ELISA.
Fig. S3. The CadF and FlpA fibronectin binding proteins are not required for IL-8 secretion from host cells.
INT 407 cells were infected with a C. jejuni wild-type strain and C. jejuni cadF flpA double mutant. Uninfected cells served as a negative control. IL-8 secretion was quantified by ELISA. There was no significant difference in the amount of IL-8 secreted in response to the C. jejuni mutant as compared to the wild-type strain.
Fig. S4. Rac1 activation is not required for a maximal IL-8 response.
Panels: (A) The invasiveness of the C. jejuni ciaC mutant was determined by the gentamicin protection assay. INT 407 cells were infected with the C. jejuni wild-type strain and C. jejuni ciaC mutant. Host cell association was assessed 30 min following infection. Gentamicin was added to infected cells 3 hr after infection and incubated for 3 additional hr. (B) INT 407 cells were infected with a C. jejuni wild-type strain and C. jejuni ciaC mutant. Uninfected cells served as a negative control. IL-8 secretion was quantified by ELISA. A significant difference was not detected in the amount of IL-8 secreted in response to the mutant as compared to the wild-type strain as judged by ELISA of IL-8 in supernatants. (C) The expression of the Rac1 activating guanine exchange factor Dock180 was reduced by siRNA treatment of INT 407 cells. Scrambled siRNA (Scrm) treated cells served as a transfection control and uninfected cells served as a negative control. The cells were infected with C. jejuni, supernatants collected after a 24 hr incubation, and IL-8 measured by ELISA. The efficiency of Dock180 knockdown was tested by immunoblot with a Dock180 antibody. The blot was re-probed with antibodies against actin as a loading control.
Fig. S5. FAK is required for PI3 kinase activation and IL-8 secretion but is not required for NF-κB activation in response to C. jejuni.
Panels: (A) INT 407 cells were treated with the FAK inhibitor TAE 226 and infected with C. jejuni. Vehicle treated cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control. Lysates were prepared after a 30 min infection and probed for activation of Akt by immunoblot with antibodies against phospho-Akt (pAkt). The blot was reprobed with antibodies against actin as a loading control. (B) INT 407 cells were treated with FAK inhibitor TAE 226 or with PI3 kinase inhibitor LY294002 and infected with C. jejuni. Lysates were prepared 30 min after infection and analyzed by immunoblot. The level of NF-κB activation in response to C. jejuni infection was measured with antibodies against phospho-NF-κB (pNF-κB). The blot was re-probed with antibodies against actin as a loading control. (C) INT 407 cells were treated with LY294002 and infected with C. jejuni. Vehicle treated cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control. IL-8 secretion was quantified by ELISA.
Fig. S6. Pseudomonas aeruginosa, Vibrio parahaemolyticus, and Staphylococcus aureus require paxillin to trigger maximal IL-8 secretion from epithelial cells.
INT 407 cells were treated with siRNA specific to paxillin or scrambled siRNA (Scrm). The amount of IL-8 in supernatants collected from INT 407 cells infected with Pseudomonas aeruginosa, Vibrio parahaemolyticus, and Staphylococcus aureus was quantified by ELISA. The IL-8 values were normalized to untreated cells infected with wild-type bacteria, and the value generated for the uninfected cells was subtracted from all values. Citrobacter freundii, a commensal organism, was included as a negative control.
Acknowledgments
We thank Dr. Levi O’Loughlin for critical review of this manuscript. This work was presented, in part, at the 112th General Meeting of the American Society for Microbiology (June 16–19, 2012). We thank Novartis for providing the TAE 226 compound.
Funding
This study was supported from funds awarded to MEK from the School of Molecular Biosciences at Washington State University and the National Institutes of Health (NIH, R56 AI088518-01A1). TPE was supported, in part, by Award Numbers T32GM008336 and T32GM083864 from the National Institute of General Medical Sciences (NIGMS). DRS was supported, in part, by Award Number T32GM083864 from the NIGMS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH and NIGMS. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- Cabodi S, Moro L, Bergatto E, Boeri Erba E, Di Stefano P, Turco E, et al. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF-dependent responses. Biochem Soc Trans. 2004;32:438–442. doi: 10.1042/BST0320438. [DOI] [PubMed] [Google Scholar]
- Chen HC, Appeddu PA, Isoda H, Guan JL. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol England. 2005:320–332. doi: 10.1038/nrmicro1095. [DOI] [PubMed] [Google Scholar]
- Eucker TP, Konkel ME. The cooperative action of bacterial fibronectin-binding proteins and secreted proteins promote maximal Campylobacter jejuni invasion of host cells by stimulating membrane ruffling. Cell Microbiol. 2012;14:226–238. doi: 10.1111/j.1462-5822.2011.01714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan RC, Neal-McKinney JM, Dhillon AS, Miller WG, Konkel ME. Examination of Campylobacter jejuni putative adhesins leads to the identification of a new protein, designated FlpA, required for chicken colonization. Infect Immun. 2009;77:2399–2407. doi: 10.1128/IAI.01266-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdiero M, Vitiello M, Sanzari E, D’Isanto M, Tortora A, Longanella A, Galdiero S. Porins from Salmonella enterica serovar Typhimurium activate the transcription factors activating protein 1 and NF-kappaB through the Raf-1-mitogen-activated protein kinase cascade. Infect Immun. 2002;70:558–568. doi: 10.1128/IAI.70.2.558-568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Hall JE, Fu W, Schaller MD. Focal adhesion kinase: exploring Fak structure to gain insight into function. Int Rev Cell Mol Biol. 2011;288:185–225. doi: 10.1016/B978-0-12-386041-5.00005-4. [DOI] [PubMed] [Google Scholar]
- Henderson B, Nair S, Pallas J, Williams MA. FEMS Microbiol Rev. England: Federation of European Microbiological Societies. Published by Blackwell Publishing Ltd; 2011. Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins; pp. 147–200. 2010. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Ishibe S, Joly D, Liu ZX, Cantley LG. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell. 2004;16:257–267. doi: 10.1016/j.molcel.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Ishibe S, Joly D, Zhu X, Cantley LG. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell. 2003;12:1275–1285. doi: 10.1016/s1097-2765(03)00406-4. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Cieplak W. Altered synthetic response of Campylobacter jejuni to cocultivation with human epithelial cells is associated with enhanced internalization. Infect Immun. 1992;60:4945–4949. doi: 10.1128/iai.60.11.4945-4949.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Garvis SG, Tipton SL, Anderson DE, Cieplak W. Identification and molecular cloning of a gene encoding a fibronectin-binding protein (CadF) from Campylobacter jejuni. Mol Microbiol. 1997;24:953–963. doi: 10.1046/j.1365-2958.1997.4031771.x. [DOI] [PubMed] [Google Scholar]
- Konkel ME, Larson CL, Flanagan RC. Campylobacter jejuni FlpA binds fibronectin and is required for maximal host cell adherence. J Bacteriol. 2010;192:68–76. doi: 10.1128/JB.00969-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel ME, Mead DJ, Cieplak W., Jr Kinetic and antigenic characterization of altered protein synthesis by Campylobacter jejuni during cultivation with human epithelial cells. J Infect Dis. 1993;168:948–954. doi: 10.1093/infdis/168.4.948. [DOI] [PubMed] [Google Scholar]
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature England. 2007:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- Matlawska-Wasowska K, Finn R, Mustel A, O’Byrne CP, Baird AW, Coffey ET, Boyd A. The Vibrio parahaemolyticus Type III Secretion Systems manipulate host cell MAPK for critical steps in pathogenesis. BMC Microbiol. 2010;10:329. doi: 10.1186/1471-2180-10-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Mitsuno Y, Yoshida H, Maeda S, Ogura K, Hirata Y, Kawabe T, et al. Helicobacter pylori induced transactivation of SRE and AP-1 through the ERK signalling pathway in gastric cancer cells. Gut. 2001;49:18–22. doi: 10.1136/gut.49.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteville MR, Yoon JE, Konkel ME. Maximal adherence and invasion of INT407 cells by Campylobacter jejuni requires the CadF outer-membrane protein and microfilament reorganization. Microbiology. 2003;149:153–165. doi: 10.1099/mic.0.25820-0. [DOI] [PubMed] [Google Scholar]
- Morrison DK. KSR: a MAPK scaffold of the Ras pathway? J Cell Sci. 2001;114:1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- Orr AW, Murphy-Ullrich JE. Regulation of endothelial cell function BY FAK and PYK2. Front Biosci. 2004;9:1254–1266. doi: 10.2741/1239. [DOI] [PubMed] [Google Scholar]
- Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes & Development. 2002;16:427–438. doi: 10.1101/gad.962902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelson DR, Eucker TP, Bell JA, Dybas L, Mansfield LS, Konkel ME. The Campylobacter jejuni CiaD effector protein activates MAP kinase signaling pathways and is required for the development of disease. Cell Commun Signal. 2013;11:79. doi: 10.1186/1478-811X-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansonetti PJ, Arondel J, Huerre M, Harada A, Matsushima K. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun. 1999;67:1471–1480. doi: 10.1128/iai.67.3.1471-1480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Ginsberg MH. Networks and crosstalk: integrin signalling spreads. Nature Cell Biology. 2002;4 doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- Sen A, O’Malley K, Wang Z, Raj GV, Defranco DB, Hammes SR. Paxillin regulates androgen- and epidermal growth factor-induced MAPK signaling and cell proliferation in prostate cancer cells. J Biol Chem. 2010;285:28787–28795. doi: 10.1074/jbc.M110.134064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Casanova JE. Invasion of host cells by Salmonella typhimurium requires focal adhesion kinase and p130Cas. Mol Biol Cell. 2006;17:4698–4708. doi: 10.1091/mbc.E06-06-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassam FH, Graham DY, Yamaoka Y. Paxillin is a novel cellular target for converging Helicobacter pylori-induced cellular signaling. Am J Physiol Gastrointest Liver Physiol. 2011;301:G601–611. doi: 10.1152/ajpgi.00375.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torok AM, Bouton AH, Goldberg JB. Helicobacter pylori induces interleukin-8 secretion by Toll-like receptor 2- and Toll-like receptor 5-dependent and -independent pathways. Infect Immun. 2005;73:1523–1531. doi: 10.1128/IAI.73.3.1523-1531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello M, D’Isanto M, Galdiero M, Raieta K, Tortora A, Rotondo P, Peluso L. Interleukin-8 production by THP-1 cells stimulated by Salmonella enterica serovar Typhimurium porins is mediated by AP-1, NF-kappaB and MAPK pathways. Cytokine. 2004;27:15–24. doi: 10.1016/j.cyto.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Watson RO, Galán JE. Signal transduction in Campylobacter jejuni-induced cytokine production. Cell Microbiol. 2005;7:655–665. doi: 10.1111/j.1462-5822.2004.00498.x. [DOI] [PubMed] [Google Scholar]
- Winter SE, Keestra AM, Tsolis RM, Baumler AJ. The blessings and curses of intestinal inflammation. Cell Host Microbe. 2010;8:36–43. doi: 10.1016/j.chom.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–119. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Zouq NK, Keeble JA, Lindsay J, Valentijn AJ, Zhang L, Mills D, et al. FAK engages multiple pathways to maintain survival of fibroblasts and epithelia: differential roles for paxillin and p130Cas. J Cell Sci. 2009;122:357–367. doi: 10.1242/jcs.030478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. C. jejuni requires protein synthesis to induce an IL-8 response from epithelial cells.
C. jejuni was treated with the bacterial protein synthesis inhibitor chloramphenicol. INT 407 cells were infected with the antibiotic treated C. jejuni or vehicle treated bacteria. Uninfected cells served as a negative control. The level of IL-8 in supernatants collected after 24 hr incubation was measured by ELISA.
Fig. S2. Raf inhibition results in partial reduction of IL-8 secretion in response to C. jejuni.
INT 407 cells were treated with Raf inhibitors PLX-4720 or GDC-0879. The cells were infected with C. jejuni and incubated for 24 hr. Uninfected cells served as a negative control. Supernatants were collected and the amount of IL-8 secreted was quantified by ELISA.
Fig. S3. The CadF and FlpA fibronectin binding proteins are not required for IL-8 secretion from host cells.
INT 407 cells were infected with a C. jejuni wild-type strain and C. jejuni cadF flpA double mutant. Uninfected cells served as a negative control. IL-8 secretion was quantified by ELISA. There was no significant difference in the amount of IL-8 secreted in response to the C. jejuni mutant as compared to the wild-type strain.
Fig. S4. Rac1 activation is not required for a maximal IL-8 response.
Panels: (A) The invasiveness of the C. jejuni ciaC mutant was determined by the gentamicin protection assay. INT 407 cells were infected with the C. jejuni wild-type strain and C. jejuni ciaC mutant. Host cell association was assessed 30 min following infection. Gentamicin was added to infected cells 3 hr after infection and incubated for 3 additional hr. (B) INT 407 cells were infected with a C. jejuni wild-type strain and C. jejuni ciaC mutant. Uninfected cells served as a negative control. IL-8 secretion was quantified by ELISA. A significant difference was not detected in the amount of IL-8 secreted in response to the mutant as compared to the wild-type strain as judged by ELISA of IL-8 in supernatants. (C) The expression of the Rac1 activating guanine exchange factor Dock180 was reduced by siRNA treatment of INT 407 cells. Scrambled siRNA (Scrm) treated cells served as a transfection control and uninfected cells served as a negative control. The cells were infected with C. jejuni, supernatants collected after a 24 hr incubation, and IL-8 measured by ELISA. The efficiency of Dock180 knockdown was tested by immunoblot with a Dock180 antibody. The blot was re-probed with antibodies against actin as a loading control.
Fig. S5. FAK is required for PI3 kinase activation and IL-8 secretion but is not required for NF-κB activation in response to C. jejuni.
Panels: (A) INT 407 cells were treated with the FAK inhibitor TAE 226 and infected with C. jejuni. Vehicle treated cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control. Lysates were prepared after a 30 min infection and probed for activation of Akt by immunoblot with antibodies against phospho-Akt (pAkt). The blot was reprobed with antibodies against actin as a loading control. (B) INT 407 cells were treated with FAK inhibitor TAE 226 or with PI3 kinase inhibitor LY294002 and infected with C. jejuni. Lysates were prepared 30 min after infection and analyzed by immunoblot. The level of NF-κB activation in response to C. jejuni infection was measured with antibodies against phospho-NF-κB (pNF-κB). The blot was re-probed with antibodies against actin as a loading control. (C) INT 407 cells were treated with LY294002 and infected with C. jejuni. Vehicle treated cells infected with C. jejuni served as a positive control and uninfected cells served as a negative control. IL-8 secretion was quantified by ELISA.
Fig. S6. Pseudomonas aeruginosa, Vibrio parahaemolyticus, and Staphylococcus aureus require paxillin to trigger maximal IL-8 secretion from epithelial cells.
INT 407 cells were treated with siRNA specific to paxillin or scrambled siRNA (Scrm). The amount of IL-8 in supernatants collected from INT 407 cells infected with Pseudomonas aeruginosa, Vibrio parahaemolyticus, and Staphylococcus aureus was quantified by ELISA. The IL-8 values were normalized to untreated cells infected with wild-type bacteria, and the value generated for the uninfected cells was subtracted from all values. Citrobacter freundii, a commensal organism, was included as a negative control.