

Abstract
We conducted a meta analysis of Parkinson’s disease genome-wide association studies using a common set of 7,893,274 variants across 13,708 cases and 95,282 controls. Twenty-six loci were identified as genome-wide significant; these and six additional previously reported loci were then tested in an independent set of 5,353 cases and 5,551 controls. Of the 32 tested SNPs, 24 replicated, including 6 novel loci. Conditional analyses within loci show four loci including GBA, GAK/DGKQ, SNCA, and HLA contain a secondary independent risk variant. In total we identified and replicated 28 independent risk variants for Parkinson disease across 24 loci. While the effect of each individual locus is small, a risk profile analysis revealed a substantial cummulative risk in a comparison highest versus lowest quintiles of genetic risk (OR=3.31, 95% CI: 2.55, 4.30; p-value = 2×10−16). We also show 6 risk loci associated with proximal gene expression or DNA methylation.
Increasing evidence supports the extensive and complex genetic contribution to Parkinson’s disease (PD). Genome-wide association (GWA) studies have shed light on the genetic basis of this disease, with the identification and replication of risk loci that fit the common disease common variant hypothesis.1–17 The loci identified have both affirmed the central role of genes previously linked to PD and implicated new proteins in the pathogenic cascade.18 These data have also shown that thus far only a small portion of the heritable component of PD has been identified.19 Experience in other complex diseases and traits demonstrates that ever greater resolution of genetic risk can be achieved through larger sample sizes and that common genetic variability may play a more substantial role in complex traits than previously anticipated.20–22 With each of these factors in mind, we performed a meta-analysis of all existing European ancestry PD GWA study data and a replication study in an independent data set.
We performed a meta-analysis of genome-wide SNP data from 13,708 PD patients and 95,282 controls. This approach required imputation using the August 2010 release of the 1000 Genomes Project European ancestry haplotype reference set to standardize data to over 11 million variants.23 Only markers that were successfully imputed in at least three datasets and that had a meta-analysis wide sample size weighted minor allele frequency of 0.1% or more were included (n=7,893,274). The genomic inflation factor for each of the datasets ranged from 0.889 to 1.056 (based on lambda values standardized to a scale of 1000 cases and 1000 controls, see Supplementary Table 1 for study specific details). Fixed-effect meta-analysis of the summary statistics from each set revealed 26 loci associated with risk for disease in the discovery phase, based on a widely-accepted genome-wide p-value threshold of 5×10−8 (Table 1 and Figure 1 with additional details in Supplementary Table 2 and Supplementary Figure 1).24
Table 1.
Results of discovery and replication association analyses.
| SNP Information | Discovery phase (13,728 cases and 95,282 controls) | Replication phase (5,353 cases and 5,551 controls) | Joint phase (19,081 cases and 100,833 controls) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| SNP | C | Position (bp) | Nearest gene(s) | Effect allele | Alternate allele | Effect allele frequency | Odds ratio | P | Odds ratio | P | Odds ratio | P |
| Genome Wide Significant, Discovery Phase | ||||||||||||
|
| ||||||||||||
| rs35749011* | 1 | 155,135,036 | GBA/SYT11 | a | g | 0.017 | 1.762 | 6.09×10-23 | 2.307 | 7.48×10-09 | 1.824 | 1.37×-29 |
| rs823118 | 1 | 205,723,572 | RAB7L1/NUCKS1 | t | c | 0.559 | 1.126 | 1.36×10-13 | 1.109 | 1.43×10-04 | 1.122 | 1.66×-16 |
| rs10797576 | 1 | 232,664,611 | SIPA1L2 | t | c | 0.14 | 1.139 | 1.19×10-08 | 1.11 | 3.38×10-03 | 1.131 | 4.87×-10 |
| rs6430538 | 2 | 135,539,967 | ACMSD/TMEM163 | t | c | 0.43 | 0.873 | 5.56×10-15 | 0.882 | 9.42×10-06 | 0.875 | 9.13×-20 |
| rs1474055* | 2 | 169,110,394 | STK39 | t | c | 0.128 | 1.213 | 7.12×10-16 | 1.218 | 1.07×10-06 | 1.214 | 1.15×-20 |
| rs115185635* | 3 | 87,520,857 | KRT8P25/APOOP2 | c | g | 0.035 | 1.789 | 2.18×10-08 | 0.931 | 0.846 | 1.142 | 0.022 |
| rs12637471 | 3 | 182,762,437 | MCCC1 | a | g | 0.193 | 0.844 | 3.32×10-16 | 0.836 | 3.72×10-07 | 0.842 | 2.14×-21 |
| rs34311866 | 4 | 951,947 | TMEM175/GAK/DGKQ | t | c | 0.809 | 0.784 | 3.58×10-33 | 0.791 | 6.29×10-12 | 0.786 | 1.02×-43 |
| rs11724635 | 4 | 15,737,101 | BST1 | a | c | 0.553 | 1.122 | 8.07×10-13 | 1.138 | 2.73×10-06 | 1.126 | 9.44×-18 |
| rs6812193 | 4 | 77,198,986 | FAM47E/SCARB2 | t | c | 0.364 | 0.897 | 7.17×10-11 | 0.935 | 0.011 | 0.907 | 2.95×-11 |
| rs356182 | 4 | 90,626,111 | SNCA | a | g | 0.633 | 0.737 | 3.23×10-67 | 0.822 | 1.75×10-12 | 0.760 | 4.16×-73 |
| rs9275326* | 6 | 32,666,660 | HLA-DQB1 | t | c | 0.094 | 0.797 | 5.82×10-13 | 0.9 | 0.018 | 0.826 | 1.19×-12 |
| rs199347 | 7 | 23,293,746 | GPNMB | a | g | 0.59 | 1.123 | 2.37×10-12 | 1.072 | 7.66×10-03 | 1.110 | 1.18×-12 |
| rs117896735* | 10 | 121,536,327 | INPP5F | a | g | 0.014 | 1.767 | 1.21×10-11 | 1.404 | 1.10×10-03 | 1.624 | 4.34×-13 |
| rs3793947* | 11 | 83,544,472 | DLG2 | a | g | 0.443 | 0.912 | 2.59×10-08 | 0.976 | 0.201 | 0.929 | 3.96×-07 |
| rs329648 | 11 | 133,765,367 | MIR4697 | t | c | 0.354 | 1.1 | 1.65×10-08 | 1.121 | 4.38×10-05 | 1.105 | 9.83×-12 |
| rs76904798 | 12 | 40,614,434 | LRRK2 | t | c | 0.143 | 1.17 | 1.33×10-12 | 1.11 | 3.69×10-03 | 1.155 | 5.24×-14 |
| rs11060180 | 12 | 123,303,586 | CCDC62 | a | g | 0.558 | 1.101 | 2.14×10-08 | 1.114 | 7.26×10-05 | 1.105 | 6.02×-12 |
| rs11158026 | 14 | 55,348,869 | GCH1 | t | c | 0.335 | 0.889 | 7.13×10-11 | 0.948 | 0.039 | 0.904 | 5.85×-11 |
| rs1555399* | 14 | 67,984,370 | TMEM229B | a | t | 0.468 | 0.872 | 5.53×10-16 | 0.971 | 0.144 | 0.897 | 6.63×-14 |
| rs2414739 | 15 | 61,994,134 | VPS13C | a | g | 0.734 | 1.114 | 4.13×10-09 | 1.109 | 7.96×10-04 | 1.113 | 1.23×-11 |
| rs14235 | 16 | 31,121,793 | BCKDK/STX1B | a | g | 0.381 | 1.094 | 3.89×10-08 | 1.133 | 7.72×10-06 | 1.103 | 2.43×-12 |
| rs17649553 | 17 | 43,994,648 | MAPT | t | c | 0.226 | 0.771 | 4.86×10-37 | 0.764 | 7.03×10-15 | 0.769 | 2.37×-48 |
| rs12456492 | 18 | 40,673,380 | RIT2 | a | g | 0.693 | 0.905 | 5.12×10-09 | 0.9 | 2.16×10-04 | 0.904 | 7.74×-12 |
| rs62120679* | 19 | 2,363,319 | SPPL2B | t | c | 0.314 | 1.141 | 2.53×10-09 | 0.999 | 0.518 | 1.097 | 5.57×-07 |
| rs8118008* | 20 | 3,168,166 | DDRGK1 | a | g | 0.657 | 1.111 | 2.32×10-08 | 1.113 | 1.18×10-04 | 1.111 | 3.04×-11 |
|
| ||||||||||||
| Previously Reported as Significant in Genome Wide Studies | ||||||||||||
|
| ||||||||||||
| rs34016896 | 3 | 160,992,864 | NMD3 | t | c | 0.319 | 1.08 | 7.68×10-06 | 1.028 | 0.174 | 1.067 | 1.08×-05 |
| rs591323 | 8 | 16,697,091 | FGF20 | a | g | 0.275 | 0.921 | 1.30×10-05 | 0.902 | 6.16×10-04 | 0.916 | 6.68×-08 |
| rs60298754 | 8 | 89,373,041 | MMP16 | t | c | 0.024 | 1.078 | 0.181 | - | - | 1.078 | 0.181 |
| rs7077361 | 10 | 15,561,543 | ITGA8 | t | c | 0.874 | 1.11 | 3.24×10-05 | 1.044 | 0.154 | 1.092 | 4.16×-05 |
| rs11868035 | 17 | 17,715,101 | SREBF/RAI1 | a | g | 0.298 | 0.937 | 2.17×10-04 | 0.947 | 0.036 | 0.939 | 5.98×-05 |
| rs2823357 | 21 | 16,914,905 | USP25 | a | g | 0.37 | 1.036 | 0.032 | 1.018 | 0.267 | 1.031 | 0.027 |
C - Chromosome; OR - odds ratio
replication genotyping for these SNPs failed assay design or quality control and a suitable proxy variant was selected (rs35749011, proxy rs71628662; rs1474055, proxy rs1955337; rs115185635, proxy rs62267708; rs117896735, proxy rs118117788; rs3793947, proxy rs12283611; rs1555399, proxy rs1077989; rs62120679, proxy rs10402629; rs8118008, proxy rs55785911). Note, only replication phase p-values are one-sided. Nearest gene or previously published proximal gene names included in table.
Figure 1.

Manhattan plot of discovery phase meta-analyses.
To identify which of the putatively associated loci were truly disease-related, we attempted to replicate each locus in an independent sample series using a semi-custom genotyping array called NeuroX. This array typed >240,000 exonic variants available on Illumina’s Infinium HumanExome BeadChip and an additional ~24,000 variants proven or hypothesized to be relevant in neurodegenerative disease [Nalls et al., in review]. Within the custom content we included the 26 genome-wide significant candidate loci implicated in PD from the primary meta-analysis. For each independent locus the most significantly associated SNP and a series of proxy variants were included in the array design. Following stringent quality control, high quality genotype data were available for a sample set of 5,353 cases and 5,551 controls (see Online Methods for complete details). Association analysis revealed replication of 22 of 26 loci tested based on a nominal 1-sided p-value of <0.05 and consistent direction of association which incorporates the premise of prior knowledge for most loci based on previous meta-GWAS (Table 1); of these loci, six were novel (SIPA1L2, INPP5F, MIR4697, GCH1, VPS13C, DDRGK1). In addition we examined association at six loci previously reported to be associated with risk for PD but that did not show association at p<5×10−8 in the discovery phase.1,2,4,25 While these loci have been reported in samples derived from some of the cohorts included in the discovery phase of this meta-analysis, individuals in the replication samples were distinct from those used to nominate these loci. We found evidence for association based on a nominal 1-sided p-value of <0.05 in the replication data at two of these loci in our replication phase analyses (FGF20 and SREBF/RAI1; Table 1). We do note that some loci did not replicate, these loci included regions of high effect heterogeneity and low effect sizes (~1.1 odds ratio), for which our replication series may have been slightly underpowered. For example, at an odds ratio of 1.1 and an allele frequency of 5%, our replication series were only at a power of ~35% to reach our designated alpha; while under the assumption of no effect heterogeneity across the replication samples and no winner’s curse phenomenon, we were at ~80% power to reach our target alpha for replication if the allele frequency is increased to 40% and the odds ratio remains 1.1. We recognize that study heterogeneity contributed to some of this non-replication (as evidenced by the I2 metric in Supplementary Table 2), particularly in the discovery phase of analyses. In the discovery phase, the associations at rs115185635 and rs1555399 were driven almost completely by the IPDGC-UK cohort and highly heterogenous across cohorts (I2 estimates at 91.0 and 97.2 respectively). In addition, rs115185635 was a very difficult variant to impute likely due to its frequency, as it passed quality control in only 8 of our participating studies. We do not believe there is any major issue with the UK data based on both previously published studies, as well as consistent effects at more established loci evidenced by I2 metrics in Supplementary Tables 2 and 3, as well as in Supplementary Table 1 and Supplementary Figures 2 – 4 describing study specific effect estimates in addition to genomic inflation factors. Our strategy of a distinct replication phase was instituted primarily to confirm suspect novel loci and to exclude any types of systematic issues that may lead to false positives at individual loci.
We tested whether multiple independent risk alleles existed at any of the 26 genome-wide significant loci identified in the discovery phase. For each locus we tested all variants within 1 million base pairs of the index SNP with the most extreme p-value. To identify risk alleles independent of the primary effect, the index SNP was included as a covariate in the model (0, 1 or 2 copies of the minor allele). Additional independent risk variants were identified at 8 of the loci (9.31×10−6 to 7.09×10−19) and were also included on the replication array. Four of these variants revealed significant association upon conditional analysis of the replication phase data (Table 2 and Supplementary Table 3 for additional details).
Table 2.
Results of conditional association analyses.
| Significant conditional SNP, signifiying secondary locus | rs114138760 | rs79217002 | rs34884217 | rs1596117* | rs7681154* | rs13201101* | rs10886515 | rs117022814 | |
| Most significant SNP from discovery phase, used as covariate | rs35749011 | rs12637471 | rs34311866 | rs6812193 | rs356182 | rs9275326 | rs117896735 | rs62120679 | |
| Nearest gene(s) | GBA/SYT11 | MCCC1 | TMEM175/GAK/DGKQ | FAM47E/SCARB2 | SNCA | HLA-DQB1 | INPP5F | SPPL2B | |
| R2 between SNPs based on 1000 Genomes Project European ancestry samples | 0.000 | 0.003 | 0.012 | 0.028 | 0.209 | 0.002 | 0.000 | 0.006 | |
| Conditional SNP information | C | 1 | 3 | 4 | 4 | 4 | 6 | 10 | 19 |
| Position (bp) | 154898185 | 183011072 | 944210 | 77151490 | 90763703 | 32343604 | 121343589 | 2209647 | |
| Effect allele | c | a | a | t | a | t | t | t | |
| Alternate allele | g | g | c | c | c | c | c | c | |
| Effect allele frequency | 0.012 | 0.9907 | 0.9126 | 0.2005 | 0.5021 | 0.0529 | 0.7145 | 0.0262 | |
| Summary statistics from conditional analyses | Odds ratio | 1.574 | 0.669 | 1.247 | 1.115 | 0.841 | 1.192 | 1.100 | 1.341 |
| P | 3.80×10-07 | 9.31×10-06 | 1.10×10-06 | 2.80×10-07 | 7.09×10-19 | 3.84×10-06 | 9.19×10-07 | 1.95×10-06 | |
| Summary statistics from discovery phase | Odds ratio | 1.497 | 0.688 | 1.344 | 1.094 | 0.997 | 1.179 | 1.105 | 1.319 |
| P | 2.18×10-06 | 1.69×10-05 | 1.56×10-12 | 6.05×10-06 | 0.854 | 4.95×10-06 | 2.59×10-08 | 2.00×10-06 | |
| Summary statistics from replicaiton phase | Odds ratio | 1.586 | 1.076 | 1.105 | 1.036 | 0.934 | 1.217 | 1.023 | 1.094 |
| P | 5.72×10-04 | 0.714 | 0.017 | 0.189 | 8.02×10-03 | 8.33×10-03 | 0.234 | 0.174 | |
| Summary statistics from combined discovery and replication phases | Odds ratio | 1.519 | 0.789 | 1.232 | 1.083 | 0.981 | 1.185 | 1.084 | 1.255 |
| P | 9.73×-09 | 1.08×-03 | 2.51×-11 | 9.45×-06 | 0.171 | 2.50×-07 | 2.26×-07 | 4.82×-06 |
Replication genotyping for these SNPs failed assay design or quality control and a suitable proxy variant was selected (rs1596117, proxy rs4859430; rs7681154, proxy rs3910105; rs13201101, proxy rs8192591; based on discovery series comparison, the minor allele for rs3910105 tags the major allele of rs7681154 therefore risk is consistent across proxy and discovery SNP). Note, only replication phase p-values are one-sided. Nearest gene or previously published proximal gene names included in table.
Risk profiles were generated for each subset of replication samples separately using all SNPs with replication phase p-values less than the marginal one-sided p-value of 0.05 (discovery, candidate and conditional phases, Tables 1 and 2). These 28 SNPs were used to compute genetic risk profile scores (for additional risk profiling methods, please see Online Methods). Similar to previous studies we showed marginal predictive power for genetic risk profile scores, with areas under the receiver operator curves of 0.616 without age and sex included as covariates and 0.633 with age and sex (Figure 2, Supplementary Table 4, Supplementary Figure 5).3 As expected, those individuals with a genetic risk profile score above one standard deviation from the population mean, indicative of a roughly 34% increase in genetic risk scores above the control mean, had a significantly higher risk of PD (from meta-analysis odds ratio = 1.51, 95% CI = 1.38–1.66, p=2×10−16). In an analysis of outliers, we compared the 5th quintile of genetic risk scores to the 1st quintile of genetic risk as a reference, the odds ratio was 3.31 (95% CI = 2.55–4.30, p=2×10−16). These odds ratios are larger compared to earlier publications and may be due to finer scale imputation used in the discovery phase of this project, as well as the inclusion of additional loci and to some degree differing distributions of cumulative genetic risk scores across populations in the analysis.3,4 Cohort level summary statistics were significantly heterogeneous for both trend based analyses (I2 = 0.74, heterogeneity p-value = 0.003) and the comparisons of the highest versus lowest risk quintiles (I2 = 0.70, heterogeneity p-value = 0.01). Therefore, a random effects model was used to account for the heterogeneity of effect.
Figure 2.
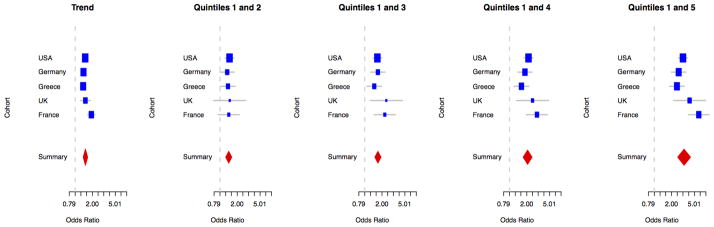
Forest plots describing cohort level and summary effects of risk profile analyses.
For each of the 28 SNPs included in the risk profile analyses, we attempted to infer functional consequences in frontal cortex and cerebellar tissue samples from neurologically normal individuals that were assayed for both genome-wide methylation and expression levels.26 These analyses may shed light on potential disease mechanisms for follow-up in future studies. We tested cis associations (any methylation or expression probes +/− 1Mb from each SNP) in each of the datasets. After quality control, 25 SNPs of interest from our meta-analysis passed quality control in the mRNA expression datasets and all 28 SNPs of interest passed quality control the CpG methylation datasets. We tested multiple probes per SNP in each set of analyses. A total of 336 unique SNP-probe pairs were tested in the frontal cortex mRNA expression dataset, 865 pairs in the frontal cortex CpG methylation dataset, 333 pairs in the cerebellar mRNA expression dataset and 1,097 pairs in the cerebellar CpG methylation dataset. Associations were tested using linear regression adjusting for appropriate covariates and resulting p-values adjusted based on the false-discovery rate correction (see Online Methods for details).
After correcting for multiple tests, we found 30 significant associations between SNPs of interest and either CpG methylation or mRNA expression (Supplementary Table 5) across six loci. Of particular interest were associations at rs199347 on chromosome 7 and rs823118 on chromosome 1, as both SNPs are significantly associated with both methylation and expression changes in each brain region. The risk allele (A) at rs199347 on chromosome 7 was associated with increased expression of two probes tagging NUPL2 as well as with decreased methylation of GPNMB in both brain regions. These data suggest that risk at the locus containing rs199347 might be due to increased transcription of NUPL2 further bolstered by decreased methylation. On chromosome 1, the risk allele (T) at rs823118 was associated with decreased expression of NUCKS1 and increased expression of RAB7L1, as well as increased DNA methylation detected by two probes close to FLJ32569 in both brain tissues. These data suggest a complicated risk locus at the NUCKS1/RAB7L1/FLJ32569 region, where the same risk allele is associated with both increased expression of RAB7L1 as well as increased regulation of nearby genes NUCKS1 and FLJ32569. The possibility of multiple functionally active risk variants at this locus seems likely and is evident in the results of our conditional phase of analyses (Table 2). The complicated nature of this locus may be suggestive of some type of interaction or epistatic effect as well and it is likely that future functional and deep sequencing studies will be required to understand the basis of association at this region.
In total here we have identified 28 independent risk loci for PD; 22 found in the discovery phase and confirmed by replication, two previously reported loci confirmed in the replication phase, and four loci identified by a second risk allele exerting an effect independent of the primary risk allele.
URL’s
MACH2QTLv1.11 (http://www.sph.umich.edu/csg/abecasis/MaCH/download/) MiniMac (http://genome.sph.umich.edu/wiki/Minimac) 1000 Genomes haplotypes (http://www.sph.umich.edu/csg/abecasis/MaCH/download/) Summary statistics of this study have been made available at http://www.pdgene.org (Note to the editor/reviewers: Summary statistics of the discovery, conditional and replication phase of this study will be made available to the community at the listed URL upon publication of this study)
ONLINE METHODS
Discovery Methods
All studies willing to participate with genome-wide genotyping data on Parkinson’s diseases cases and controls were included in this effort. For specific details on these studies, please refer to individual publications [IPDGC, PD GWAS Consortium, 23&Me, CHARGE, PDGENE, and Ashkenazi studies].2–6,10,17,27–30 For this analysis, the 23andMe cohort was split into two subsets “v2” and “v3”. The “v2” designation refers to a subset of samples genotyped on the Illumina HumanHap550+ BeadChip, the “v3” designation refers to a subset of samples genotyped on the Illumina Human OmniExpress+ BeadChip. Besides different genotyping arrays, sample handling, ascertainment, quality control and analytic methods were identical across the 23andMe subsets. For 3 out of the 15 studies contributing to the discovery phase of analyses, population controls were utilized to some degree, totaling 8,156 samples (Supplementary Table 1). Based on a prevalence of 2 in 1,000, we could estimate a misclassification of approximately 16 samples across datasets. This would likely have a small impact based on our large sample size, meta-analytic design and interest in relatively common allele frequencies.
In general standard quality control was applied by each study for sample inclusion, including case age at onset over 18 years, no known mutations in genes associated with Mendelian forms of PD (SNCA, PARK2, DJ-1, PINK1, LRRK2), minimum sample call rate > 95%, European ancestry confirmed through principal components or multidimensional scaling analyses, no relation to other samples in the meta-analysis (checked when possible by use of dbGAP available data) at the cousin level or closer (except in the case of Framingham Heart Study). Studies deriving samples from the same geographic region (or globally in the case of the 23AndMe dataset) cross-checked for relatedness when data access was permitted by using identity by descent filtering to remove related samples both within and across datasets contributing to the meta-analysis. If samples overlapped studies involved in the meta-analysis, the sample was excluded from the series with less dense genotyping. All participants donated DNA samples and provided informed consent for participation in genetics studies. Prior to imputation, SNPs were filtered using study specific criteria including a minimum call rate of > 95%, a minor allele frequency > 1%, Hardy-Weinberg equilibrium p-values > 1×10−4 in controls and non-random missingness by phenotype or haplotype at p-values > 1×10−4. SNPs ambiguous to strand (A/T and G/C) were also removed. Imputation to a standard reference panel from the 1000 Genomes Project (August 2010 release, European ancestry only) was then carried out using miniMac on default settings.31
Imputed dosages were then analyzed using logistic regression for case-control studies or cox regression for cohort studies (Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium cohorts with incident cases) adjusting for the first 2 eigenvectors from principal components analysis, age at onset (cases) or exam (controls), and sex. Eigenvectors from principal components for use as covariates were generated on a study specific level, with each dataset applying its own adjustment separately. This was also repeated in the replication by generating unique eigenvectors for each ancestry stratified dataset for use as covariates. The Framingham Heart Study utilized generalized estimating equations clustered on pedigrees to account for family relationships.
Meta-analysis was conducted based on the fixed-effect model as implemented in METAL by combining summary statistics across datasets.32 At the meta-analytic level, summary statistics were filtered for inclusion after meeting a minimum imputation quality (RSQ from miniMac) of 0.30, minor allele frequency greater than 0.1% across studies, realistic beta coefficients where the absolute value of beta was less than 5, and passing initial quality control in at least three of the contributing studies. In addition, we tested novel random-effects approaches from Han and Eskin, 2011; however, this method did not identify any novel loci and since the results across both methodologies were near identical, thus only fixed-effect results are reported here.33
Conditional Methods
Conditional analyses were undertaken using identical statistical models as in the discovery phase except for the inclusion of allele dosages from the most significant SNP per locus as an additional covariate. For each locus identified as genome-wide significant in the discovery phase, we reran cohort level analyses in a subset of 7 datasets with the largest counts of cases (due to primary data availability at the participant level, only IPDGC-NIA, IPDGC-NL, IPDGC-GE, 23&Me, PROGENI-GenePD, NGRC and HIHG were included) testing all SNPs within +/− 1MB from the 26 genome-wide significant SNPs in Table 1, while adjusting for the SNP with the most extreme p-value per locus. These summary statistics were then meta-analyzed in the same manner as the discovery phase analyses. The threshold for multiple test correction across secondary loci for conditional analyses was set to 1×10−5 based on Bonferroni correction for the number of SNPs tested across all regions. These methods were also applied to look for tertiary signals at all loci; three tertiary loci were identified, but these signals were not included in the replication array and are therefore not shown (data available upon request to corresponding author).
Replication Methods
Replication genotyping was carried out using the Illumina NeuroX genotyping array, with all samples genotyped at the National Institute on Aging’s Laboratory of Neurogenetics (LNG). In brief, the NeuroX array includes over 24,000 neurodegenerative-focused-variants added to the existing >240,000 exonic variants already available on Illumina’s Infinium HumanExome BeadChip. Of these neurodegenerative-focused variants, over 9,000 variants are dedicated to Parkinson’s disease and include tagging SNPs, proxies and technical replicates for loci of interest related to the discovery phase of this study. For each genome-wide significant locus identified in the discovery phase of this study and in the conditional analyses, loci were covered on the array by either 5 additional proxy SNPs or 5 technical replicates if no proxy SNPs were available. Loci were defined as any SNP reaching a genome-wide significant p-value and correlated at r2 < 0.50 with any other significant SNPs within 250 kilobases within each genomic region of interest. Proxies were selected based on this 250kb threshold and LD defined using 1000 genomes European ancestry samples from the same panel in which the discovery series was imputed. Proxies were ranked by discovery phase p-value after meeting the LD threshold minimum of r2 > 0.50 with the most significant SNP in the locus. Nominated proxies with the smallest discovery phase p-value were given precedence in replication analyses when the most significant SNP from the discovery phase was not available or not successfully assayed on the NeuroX array. For replication, 39 SNPs or their highest ranked proxy in terms of discovery phase or conditional p-value were utilized in the replication analyses. All summary statistics for these replication SNPs will be made available on the PDgene website (PDGene database (http://www.pdgene.org). Genotypes were called using Illumina GenomeStudio, and all PD related SNPs analyzed for this study were manually clustered and visually inspected. Standard exome content variants included on the NeuroX array were called using a cluster file from the CHARGE Consortium based on over 60,000 samples and these variants were used for sample quality control.34 Over 14,000 samples genotyped on the array at LNG were used in the variant calling process.
From called genotypes, PD cases and neurologically normal controls were extracted and underwent quality control as per standard GWAS, with slight deviations from normal practices to account for the bias in NeuroX array content. Variants with GenTrain scores > 0.70 (indicative of quality genotype clusters) from the standard content of the NeuroX array were extracted first to calculate call rates. Samples with call rates < 95% were excluded, as well as samples whose genetically determined sex did not match that from clinical data and samples exhibiting excess heterozygosity. After these initial exclusions, SNPs overlapping with HapMap Phase 3 samples were extracted from the previous subset and pruned for LD (SNPs excluded if r2 > 0.50 within a 50 SNP sliding window), as well as concurrently excluding SNPs with MAF < 5%, HWE p-values < 1×10−5 in controls, and per SNP missingness rates > 5%. At this stage, pairwise identity by descent filtering was then used to remove samples that were cryptically related and principal components analysis was used to identify samples that were to be excluded due to genetic ancestry not consistent with primarily European descent based on comparisons with HapMap Phase 3 reference populations. After these exclusions, the samples passing quality control were separated into distinct datasets based on the country and center of origin. All samples in the replication series were ascertained as follows: case ascertainment was based on UK brain bank criteria from clinic visit, or use of PD medication or medical records of PD diagnosis by clinician; recruited controls include individuals free of known neurological disease either by clinical assessment and/or self-report. The final replication set consisted of 5,353 cases and 5,551 controls with all relevant phenotypic data for this analysis stratified across American (2,407 cases and 2,782 controls), French (553 cases, 474 controls), German (1,044 cases and 871 controls), Greek (944 cases and 877 controls) and British (405 cases and 547 controls) participants.
Within each subset of samples passing quality control, principal components analysis was used to generate eigenvectors for use as covariates to account for population substructure within each cohort based on common high quality SNPs that have also been LD pruned as described above. Within each subset of samples, logistic regression adjusting for the first 2 eigenvectors from principal components analysis, age at onset (cases) or exam (controls), and sex was used to examine associations of each nominated SNP with PD. After subset summary statistics were generated, fixed-effect meta-analyses were used to generate aggregate summary statistics and quantify heterogeneity across subsets for all 39 replication target SNPs placed on the array. As a note, regions around these SNPs +/− 500kb were not included in the sample QC procedure, and these 39 SNPs were manually clustered to evaluate quality genotyping.
Risk Profiling Methods
Risk profiles were generated incorporating three groups of SNPs. The first group included genome-wide significant index SNPs (or their proxies) from the discovery phase that replicated in the independent replication phase (n=22). The second group comprised conditional SNPs that validated in the replication phase (n=4). The third group consisted of previously reported SNPs that did not quite meet genome-wide significance in the discovery phase but provided evidence of association in the replication phase (n=2). These risk profiles were generated using weights based on effect estimates from the discovery phase of this study using methodologies described in detail elsewhere.3,4,11,35 In brief, genetic risk scores were scaled on a per SNP basis using effect estimates from the discovery phase then applied to the genotype data generated for the samples in the replication phase to create the dataset for analyses of the risk profiles. Within each subset of the replication sample series, overall trend tests for PD risk were evaluated using logistic regression with the risk profile score predicting affection status adjusted for the first 2 eigenvectors from PCA as well as age and sex. Each subset was also divided into quintiles based on their risk profile scores, and similar logistic regression models were used to estimate risk associated in each of the 4 higher risk quintiles compared to the lowest risk quintile. All risk profile summary statistics were meta-analyzed across subsets using random-effects to account for effect heterogeneity. To evaluate clinical predictability of PD, all risk profiles were combined into one model across replication subsets, adjusting for age, sex and cohort/subset membership in receiver-operator curve analyses.
Expression and Methylation Quantitative Trait Locus Methods
Overlapping SNPs identified in the discovery phase (Tables 1 and 2) that were successfully genotyped or imputed in the combined North American Brain Expression Consortium (NABEC) and United Kingdom Brain Expression Consortium (UKBEC) datasets were tested for association with proximal expression and methylation levels (GSE36192). Allelic dosages of SNPs of interest were tested for association with all methylation and expression probes within +/− 1MB from each SNP using linear regression adjusting for covariates of gender, age at death, the first two component vectors from multi-dimensional scaling, post mortem interval (PMI), brain bank and batch in which preparation or hybridization were performed, using MACH2QTLv1.11 (http://www.sph.umich.edu/csg/abecasis/MaCH/download/). These analyses were run separately for frontal cortex and cerebellar regions, each with analyses focusing on methylation (292 samples) or expression (399 samples). For these four analyses, significance was based on standard FDR adjustments for multiple testing. Further details on consortia membership, acknowledgments and full methods for the expression and methylation portions of this study, please see Supplementary Note.
EXPRESSION AND METHYLATION METHODS
Frozen frontal cortex and cerebellar samples were obtained from > 399 self-reported European ancestry samples without determinable neuropathological evidence of disease.26,36 Genomic DNA was extracted with phenol-chloroform. Bisulfite converted DNA and assayed at > 27,000 sites on the Illumina Infinium HumanMethylation27 BeadChips. MRNA expression levels were assayed using Illumina HumanHT-12 v3 Expression Beadchips. In brief, Individual probes were excluded from analyses if the p-value for detection was > 0.01 or there was less than 95% completeness of data per probe, and samples were excluded if <95% of probes were detected. Probes were also removed if an analyzed SNP was within the probe or the probe mapped ambiguously to multiple locations in the genome. Expression data were cubic spline normalized and log2-transformed prior to analyses.
Each tissue sample was genotyped using the Illumina HumanHap550 v3, Human610-Quad v1 or Human660W-Quad v1 Infinium Beadchips, shared SNPs were extracted prior to QC and imputation. Standard GWAS quality control was undertaken with inclusion criteria such as: minimum call rate 95% for both participants and SNPs, MAF > 0.01, HWE > 1×10−7, no first-degree relatives in the sample collection (identity by descent score < 0.125 in PLINK), and European ancestry confirmed by multidimensional scaling analyses.
Data were imputed using MiniMac (http://genome.sph.umich.edu/wiki/Minimac) to the most recent data freeze of 1000 Genomes haplotypes (http://www.sph.umich.edu/csg/abecasis/MaCH/download/1000G.2012-03-14.html) using default settings. All imputed SNPs were filtered for a minimum imputation quality of 0.30. After quality control, data were available for > 10 million SNPs, with expression data on 399 samples (9814 probes from the frontal cortex and 9587 probes in cerebellum) and methylation data on 292 samples (27465 CpG sites in the frontal cortex tissue samples and 27419 CpG sites in the cerebellum).
Linear regression models were utilized to estimate associations between allele dosages of per SNP and gene expression or methylation levels adjusted for covariates of gender, age at death, the first two component vectors from multi-dimensional scaling, post mortem interval (PMI), brain bank and batch in which preparation or hybridization were performed, using MACH2QTLv1.11 (http://www.sph.umich.edu/csg/abecasis/MaCH/download/). Analyses were carried out separately for each brain region and each array type. Only probes within +/− 1MB were analyzed to test only cis associations. From these analysis results, data were mined for 28 replciated SNPs of interest included in Table 1 and Table 2.
Supplementary Material
Acknowledgments
We would like to thank all of the subjects who donated their time and biological samples to be a part of this study. For funding and additional acknowledgements please see Supplementary Note.
Footnotes
AUTHOR CONTRIBUTIONS
Overall study design: MAN, NP, JB, ADS, BF, MS, JAH, NWW, TG, WKS, LB, NE, TF, AS
Design and/or management of the individual studies: MAN, CBD, JB, CS, XL, JL, RC, GMH, JSP, AG, KM, ADS, RHM, LNC, JAH, PH, HC, MS, MAI, JCB, NWW, HH, HP, AB, WKS, TG, NE, TF, ABS
Genotyping: DGH, EK, SA, CL, CE, HP
Phenotyping: TF, GMH, JSP, KM, GX, HC, NWW, HH, AB, TF, WKS
Statistical Methods and Data Analysis: MAN, NP, CL, DGH, EK, MS, CS, JPAI, MFK, MM, ADS, WKS, LB, NE, TF, ABS
Writing Group: MAN, NP, CL, TF, ABS
Critical Review of the Manuscript: MAN, NP, CL, CBD, DGH, EK, JB, CS, MFK, GMH, MM, AG, BF, MS, GX, RHM, LNC, JAH, PH, HC, NWW, HH, HP, AB, WKS, TG, LB, NE, TF, ABS, JSP
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Lill CM, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Do CB, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011;7:e1002141. doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Parkinson Disease Genomics Consortium et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–9. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Parkinson’s Disease Genomics Consortium (IPDGC) & Wellcome Trust Case Control Consortium 2 (WTCCC2) A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet. 2011;7:e1002142. doi: 10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards TL, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pankratz N, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pankratz N, et al. Meta-analysis of Parkinson’s Disease: Identification of a novel locus, RIT2. Ann Neurol. 2012;71:370–384. doi: 10.1002/ana.22687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simón-Sánchez J, et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet EJHG. 2011;19:655–661. doi: 10.1038/ejhg.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamza TH, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, et al. Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med Genet. 2011;12:104. doi: 10.1186/1471-2350-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez DG, et al. Genome wide assessment of young onset Parkinson’s disease from Finland. PloS One. 2012;7:e41859. doi: 10.1371/journal.pone.0041859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pihlstrøm L, et al. Supportive evidence for 11 loci from genome-wide association studies in Parkinson’s disease. Neurobiol Aging. 2013;34:1708. e7–13. doi: 10.1016/j.neurobiolaging.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 13.Sharma M, et al. Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology. 2012;79:659–667. doi: 10.1212/WNL.0b013e318264e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saad M, et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum Mol Genet. 2011;20:615–627. doi: 10.1093/hmg/ddq497. [DOI] [PubMed] [Google Scholar]
- 15.Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 16.Elbaz A, et al. Independent and joint effects of the MAPT and SNCA genes in Parkinson disease. Ann Neurol. 2011;69:778–792. doi: 10.1002/ana.22321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Psaty BM, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macleod DA, et al. RAB7L1 Interacts with LRRK2 to Modify Intraneuronal Protein Sorting and Parkinson’s Disease Risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller MF, et al. Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum Mol Genet. 2012;21:4996–5009. doi: 10.1093/hmg/dds335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei Z, et al. Large Sample Size, Wide Variant Spectrum, and Advanced Machine-Learning Technique Boost Risk Prediction for Inflammatory Bowel Disease. Am J Hum Genet. 2013;92:1008–1012. doi: 10.1016/j.ajhg.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willems SM, Mihaescu R, Sijbrands EJG, van Duijn CM, Janssens ACJW. A Methodological Perspective on Genetic Risk Prediction Studies in Type 2 Diabetes: Recommendations for Future Research. Curr Diab Rep. 2011;11:511–518. doi: 10.1007/s11892-011-0235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Cohorts for Heart and Aging Research in Genetic Epidemiology (CHARGE) Consortium et al. Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat Genet. 2013 doi: 10.1038/ng.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Bakker PIW, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van der Walt JM, et al. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am J Hum Genet. 2004;74:1121–1127. doi: 10.1086/421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbs JR, et al. Abundant Quantitative Trait Loci Exist for DNA Methylation and Gene Expression in Human Brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofman A, et al. The Rotterdam Study: 2012 objectives and design update. Eur J Epidemiol. 2011;26:657–686. doi: 10.1007/s10654-011-9610-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ton TG, et al. Post hoc Parkinson’s disease: identifying an uncommon disease in the Cardiovascular Health Study. Neuroepidemiology. 2010;35:241–249. doi: 10.1159/000319895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikram MA, et al. Genomewide association studies of stroke. N Engl J Med. 2009;360:1718–1728. doi: 10.1056/NEJMoa0900094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eriksson N, et al. Genetic variants associated with breast size also influence breast cancer risk. BMC Med Genet. 2012;13:53. doi: 10.1186/1471-2350-13-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta- analysis of genomewide association scans. Bioinforma Oxf Engl. 2010;26:2190– 2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grove ML, et al. Best Practices and Joint Calling of the HumanExome BeadChip: The CHARGE Consortium. PloS One. 2013;8:e68095. doi: 10.1371/journal.pone.0068095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ripatti S, et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376:1393–1400. doi: 10.1016/S0140-6736(10)61267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hernandez DG, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.