

Abstract
Feeding can be inhibited by multiple cues, including those associated with satiety, sickness or unpalatable food. How such anorexigenic signals inhibit feeding at the neural circuit level is incompletely understood. While some inhibitory circuits have been identified, it is not yet clear whether distinct anorexigenic influences are processed in a convergent or parallel manner. The amygdala central nucleus (CEA) has been implicated in feeding control, but its role is controversial. The lateral subdivision of CEA (CEl) contains a subpopulation of GABAergic neurons, marked by protein kinase C-δ. Here we show that CEl PKC-δ+ neurons in mice are activated by diverse anorexigenic signals in vivo, required for the inhibition of feeding by such signals, and strongly suppress food intake when activated. They receive pre-synaptic inputs from anatomically distributed neurons activated by different anorexigenic agents. These data suggest that CEl PKC-δ+ neurons constitute an important node that mediates the influence of multiple anorexigenic signals.
Introduction
Feeding is a goal-directed behavior that is essential for the survival of all metazoan organisms1. Elucidating the control of feeding behavior requires an understanding of both the positive and negative regulation of this activity. Although the positive regulation of feeding in mammals is increasingly well-understood1-4, the negative regulation of food intake is comparatively under-studied5,6. Food intake can be attenuated by multiple signals, including those associated with satiety, sickness and unpalatable tastants. How these diverse anorexigenic signals inhibit feeding at the neural circuit level is not clear. In particular, it is not known whether different anorexigenic signals act through parallel pathways, or whether they are integrated in a convergent manner by a central node at a relatively early stage in processing, and if so where that node is located.
The central nucleus of the amygdala (CEA)has long been suggested to play a role in regulating feeding3,7-9, but the nature of this role is uncertain. A number of neuromodulators, including those regulating the melanocortin system or opioid signaling, can affect food intake when infused into the CEA10-14. However lesion studies have generated conflicting or negative results, reporting small increases15,16, decreases17, or no change18,19 in food intake. The CEA is a heterogeneous nucleus that comprises several different subdivisions, each containing multiple neuronal subclasses20-22. Thus one potential explanation for the conflicting data on the role of CEA in feeding is that different manipulations may affect different cell types, in an undefined manner.
Here we have investigated the role in feeding control of a genetically identified subpopulation of CEA neurons, which express protein kinase C-delta (PKC-δ)20 and are located in the lateral subdivision of CEA (CEl). We find that this subpopulation is activated by multiple anorexigenic signals in vivo. Functional manipulation of these neurons indicates that they are required for the inhibition of feeding by several anorexigenic signals, and that their optogenetic activation robustly and acutely inhibits food intake. Monosynaptic retrograde tracing experiments indicate that upstream neurons activated by different anorexigenic signals and located in multiple brain regions converge onto CEl PKC-δ+ neurons, rather than onto a common input to these neurons. These data suggest that CEl PKC-δ+ neurons mediate the inhibitory influence on feeding of multiple anorexigenic signals, implying that they constitute an important node in the integration and processing of such signals.
Results
CEl PKC-δ+ neurons are activated by diverse anorexigenic signals
PKC-δ labels ∼50% of CEl GABAergic neurons20. To investigate the involvement of CEl PKC-δ+ neurons in the regulation of feeding, we first monitored c-Fos expression after intraperitoneal injection of several known anorexigenic signals: cholecystokinin (CCK) which mimics satiety23, lithium chloride (LiCl) which induces nausea and visceral malaise24, and lipopolysaccharide (LPS) which triggers a wide range of inflammatory and sickness responses25,26. Double immunostaining for c-Fos and PKC-δ indicated that these anorexigenic agents induced significant c-Fos expression in CEl PKC-δ+ neurons, and more importantly that PKC-δ marks a sizeable fraction of c-Fos+ neurons activated by CCK or LiCl (but not LPS; Fig. 1a, d-e, and Supplementary Fig. 1a). Notably, almost 80% of Fos+ neurons activated by CCK in CEl were PKC-δ+ (Supplementary Fig. 1a).
Figure 1. CEl PKC-δ+ neurons are activated by diverse anorexigenic signals.

a, d-e. Representative histology (a) and quantification (d-e) of c-Fos like immunoreactivity in CEl after intraperitoneal injection of saline, CCK, LiCl, or LPS. b, f-g. Histology (b) and quantification (f-g) of CEl c-Fos expression in 24 h fasted mice with or without 3 hr refeeding. c, h-i. Histology (c) and quantification (h-i) of CEl c-Fos expression in mice with oral infusion of water, quinine, or sucrose solution. Box plots (d, f, h) show mean (+), median, quartiles (boxes), and range (whiskers) in each CEl brain section; Values in (e, g, i) are means ± s.e.m. n = 6 – 17 brain sections from 4 – 5 animals in each condition. One-way ANOVA (F(3, 24) = 45.5, p < 0.0001 (d); F(2, 36) = 73.2, p < 0.0001 (h)) with post-hoc Bonferroni t-test indicated a significant difference between the saline-injected and drug-injected groups; Unpaired t-tests are used in (e), t(12) = 6.85, p < 0.0001 (CCK), t(12) = 7.03, p < 0.0001 (LiCl), t(14) = 1.00, p = 0.33 (LPS); t(30) = 8.43, p < 0.0001 (f); t(32) = 8.09, p < 0.0001 (re-fed) (g); t(28) = 3.42, p = 0.0019 (quinine) (i). n.s. not significant, ** p < 0.01, *** p < 0.001.
Because CCK has been suggested to be a mediator of satiety23, we investigated whether CEl PKC-δ+ neurons become activated during satiation. Indeed, we observed a significant c-Fos induction in CEl PKC-δ+ neurons 3 hr after ad libitum feeding by mice that had been food-deprived for24 h (Fig. 1b, f-g, and Supplementary Fig. 1b). Finally, we examined activation by bitter tastants, which also reduce food intake. We observed c-Fos induction in CEl PKC-δ+ neurons after oral infusion of quinine solution but not water or sucrose (Fig. 1c, h-i, and Supplementary Fig. 1c). These data suggest that CEl PKC-δ+ neurons are activated by diverse anorexigenic signals including those associated with satiety, nausea and unpalatability.
PKC-δ+ neuronal activity is required for anorexigenic influences
We next investigated whether the activity of CEl PKC-δ+ neurons is required for feeding inhibition by the anorexigenic agents that activated these cells. To do this, we pharmacogenetically inhibited CEl PKC-δ+ neurons using an inhibitory DREADD GPCR (hM4Di) that is activated by the pharmacologically inert ligand CNO27. Selective expression of hM4Di in these neurons was achieved using a transgenic mouse line in which Cre recombinase is specifically expressed in PKC-δ+ neurons20. Because PKC-δ is expressed in multiple brain regions20, we restricted expression of hM4Di to CEl via intracranial stereotaxic injection of a Cre-dependent adeno-associated virus (AAV) encoding hM4Di (AAV8-hSyn-DIO-hM4Di-mCherry)28. Electrophysiological analysis in acute amygdala slices20 confirmed that CNO inhibited spiking in hM4Di-mCherry-expressing PKC-δ+ neurons (Fig. 2a).
Figure 2. Activity of CEl PKC-δ+ neurons is required for the influence of anorexigenic agents.
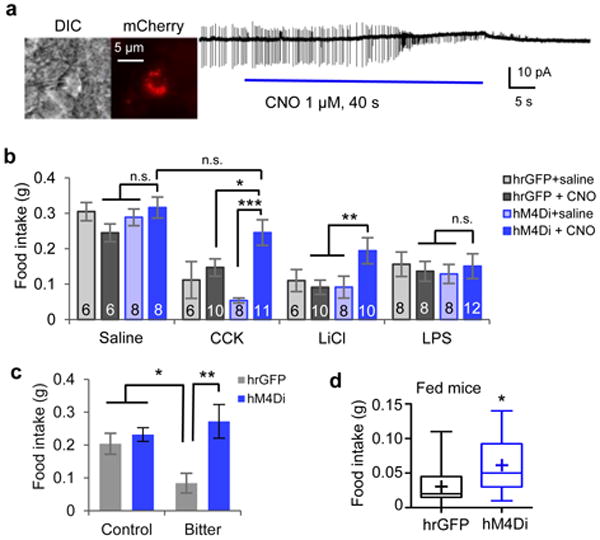
a. Cell-attached slice recording from a CEl PKC-δ+ neuron expressing hM4Di-mCherry that is silenced by bath application of CNO. b. Food intake in 24 h fasted animals after administration of different anorexigenic agents. The number of animals in each condition is indicated in the bars. Values are means ± s.e.m.. Two-way ANOVA with post-hoc Bonferroni t-tests showed a significant effect of CNO silencing, F(1, 24) = 0.363, p = 0.55 (saline); F(1, 31) = 11.7, p = 0.0018 (CCK); F(1, 30) = 4.50, p = 0.042 (LiCl); F(1, 32) = 0.001, p = 0.98 (LPS). c. Intake of control vs. quinine-laced food in 24 h fasted animals expressing hrGFP (n = 5 animals) or hM4Di (n = 5 animals), after intraperitoneal injection of CNO. Note that addition of quinine (bitter) inhibited food intake in controls. Values are means ± s.e.m.. Two-way ANOVA (F(1, 16) = 9.91, p = 0.0062) with post-hoc Bonferroni t-test showed a significant effect of silencing. d. Food intake in CNO-treated fed mice expressing hrGFP (n = 13 animals) or hM4Di (n = 14 animals) in CEl PKC-δ+ neurons. Box plots show mean (+), median, quartiles (boxes), and range (whiskers). Unpaired t-test, t(25) = 2.2, p = 0.045. n.s., not significant; * p < 0.05, ** p < 0.01.
Anorexigenic agents such as CCK, LiCl and LPS strongly inhibit feeding in food-deprived mice. Because these agents activate PCKC-δ+ neurons, we tested whether pharmacogenetic inhibition of PKC-δ+ neuronal activity could overcome the effect of these anorexigenic agents. c-Fos induction by CCK, LiCl and LPS in PKC-δ+ neurons expressing hM4Di was significantly reduced by CNO co-administration (Supplementary Fig. 2a, b), indicating that pharmacogenetic inhibition of these neurons was effective in vivo. Importantly, inhibition of these neurons rescued the effect of CCK to inhibit feeding in food-deprived animals (Fig. 2b, CCK, light-blue vs. dark-blue bars). Food intake (measured 20 min after drug administration) was restored to a level statistically indistinguishable from control mice that did not receive CCK (Fig. 2b, saline vs. CCK, dark-blue bars). Administration of saline rather than CNO to CCK-treated hM4Di-expressing mice failed to rescue feeding (Fig. 2b, CCK, light-blue bar). Similarly, CNO failed to rescue feeding in CCK-treated mice expressing humanized Renilla GFP (hrGFP)29, rather than hM4Di, in CEl PKC-δ+ neurons (Fig. 2b, CCK, black bar). Pharmacogenetic inhibition of CEl PKC-δ+ neurons also overcame the anorexigenic effect of LiCl (Fig. 2b, LiCl), while feeding inhibition induced by LPS was unaffected (Fig. 2b, LPS). Strikingly, this “rank-order potency” of rescue from anorexia (CCK>LiCl>LPS) mirrored the rank order potency of c-Fos induction in CEl PKC-δ+ neurons (Fig. 1d-e). Pharmacogenetic silencing of CEl PKC-δ+ neurons did not increase food intake in 24 h food-deprived animals in the absence of anorexigenic drugs (Fig. 2b, saline, light-blue vs. dark-blue bars), indicating a true epistatic interaction with the drugs rather than an independent compensating effect to promote feeding.
To extend these observations, we asked whether hM4Di/CNO-mediated silencing of CEl PKC-δ+ neurons could overcome the suppression of feeding by bitter tastants in food-deprived mice. Indeed, addition of quinine to food pellets significantly reduced food intake, and this reduction was reversed by CNO administration to mice expressing hM4Di in PKC-δ+ neurons (Fig. 2c). Surprisingly, however, pharmacogenetic silencing of PKC-δ+ neurons did not reduce sensitivity to bitter tastants as determined using quantitative lickometer assays (Supplementary Fig. 3a-b). This result suggests that these neurons gate the intake of potentially toxic food resources, rather than controlling gustatory sensitivity or discrimination. Taken together, these data suggest that CEl PKC-δ+ neurons mediate feeding inhibition by many (although not all) anorexigenic agents.
Silencing CEl PKC-δ+ neurons increases feeding in satiated mice
Because the satiety signal CCK, as well as re-feeding of food-deprived mice to satiety, induced c-Fos in CEl PKC-δ+ neurons (Fig. 1a-b, d-g), we asked whether silencing CEl PKC-δ+ neurons would increase food intake in fed mice. Indeed, CNO injection caused a robust (∼2-fold) and significant increase in food intake in fed mice expressing hM4Di in CEl PKC-δ+ neurons (Fig. 2d). To confirm this effect using an independent method, we silenced CEl PKC-δ+ neurons using eNpHR3.030. Brain slice recordings confirmed that CEl PKC-δ+ neurons expressing eNpHR3.0 can be strongly silenced by 593 nm laser light (Fig. 3a-b, and Methods). For in vivo behavioral testing, light was delivered through optic ferrule fibers implanted just above the injection sites (Fig. 3c). Consistent with our pharmacogenetic results, bilateral optogenetic silencing of PKC-δ+ neurons also significantly increased food intake in fed mice (Fig. 3d). These data suggest that the activity of CEl PKC-δ+ neurons is required to limit food intake in sated mice.
Figure 3. Optogenetic silencing of CEl PKC-δ+ neurons increases food intake in fed mice.

a-b. Sample slice recordings from PKC-δ+ neurons expressing eNpHR3.0; orange lines, 593 nm light. See Methods for further details. c. Diagram illustrating bilateral optogenetic silencing of CEl PKC-δ+ neurons (red). d. Food intake in fed animals. e-i. Raster plot of bouts (e), latency to the first feeding bout (f), number of bouts (g), bout duration (h), and cumulative bouts (i) in fed animals expressing hrGFP or eNpHR3.0 in CEl PKC-δ+ neurons. Solid lines in (i) are fits using the equation Bt=Bmax * (1-1/[1+(t-Ti)/T0.5]), where Bt is the cumulative number of bouts at time t; T0.5 is the time to reach the half maximal value of B, (Bmax, plateau phase), T0.5(eNpHR) = 8.2 min, T0.5(hrGFP) = 4.2 min; Ti is the time at which feeding is initiated. n = 7 animals in each group. Box plots show mean (+), median, quartiles (boxes), and range (whiskers). Values in (i) are means ± s.e.m.. Unpaired t-test, t(12) = 2.5, p = 0.026 (d); t(12) = 1.4, p = 0.18 (e); t(12) = 3.5, p = 0.0048 (f); t(12) = 0.24, p = 0.82 (g). * p< 0.05, ** p < 0.01.
The time-resolved manipulations afforded by optogenetic silencing of PKC-δ+ neurons allowed us to examine the role of these neurons during re-feeding of food-deprived mice. eNpHR3.0-mediated silencing significantly increased the number of feeding bouts, but not the average bout duration, and caused a trend to a decreased latency to feed (Fig. 3e-h). Cumulative bout analysis (Fig. 3i) indicated that mice with silenced PKC-δ+ neurons took longer to reach plateau than controls (T0.5(eNpHR) = 8.2 min, T0.5(hrGFP) = 4.2 min), suggesting that they achieved satiety more slowly. Together, these data indicate that the normal kinetics of feeding reduction as animals reach satiety requires PKC-δ+ neuronal activity.
Activation of CEl PKC-δ+ neurons inhibits feeding
The foregoing loss-of-function data raised the question of whether artificial activation of CEl PKC-δ+ neurons would suffice to inhibit food intake. To test this, we expressed ChR2 in CEl PKC-δ+ neurons using a Cre-dependent AAV (AAV2-EF1α-DIO-ChR2-EYFP)31. Whole-cell patch clamp recordings in CEA brain slices from virus-injected PKC-δ-Cre mice confirmed that 473 nm light pulses triggered robust spiking in PKC-δ+ neurons (Fig. 4a).
Figure 4. Activation of CEl PKC-δ+ neurons inhibits feeding.
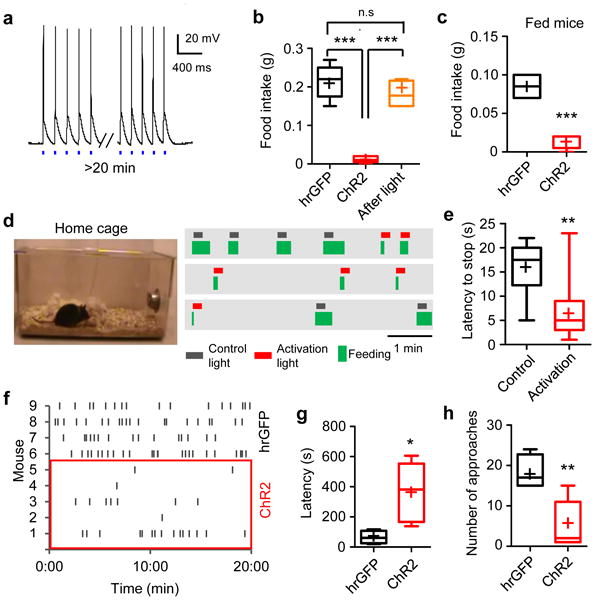
a. Brain slice whole-cell patch clamp recording traces showing action potentials triggered in PKC-δ+ neurons expressing ChR2 by 5 Hz, 10 ms 473 nm laser pulses. b-c. Food intake by 24 h fasted (b) and fed (c) animals expressing control protein hrGFP (n = 4) or ChR2 (n = 5). Mice expressing ChR2 were allowed to continue feeding for an additional 20 min after photostimulation offset (b, orange bar, n = 4). One-way ANOVA adjusted with Bonferroni's correction, F(2, 10) = 45.1, p < 0.0001 (b); Unpaired t-test, t(7) = 8.02, p < 0.0001 (c). d. Raster plots showing feeding episodes by one animal in its home cage. Activation light (473 nm) or control light (571 nm) was triggered 1-2 seconds after feeding began. 5 Hz, 10 ms light pulses were delivered for 10 seconds. e. Latency to stop feeding in response to photostimulation. n = 8 (control), and 11 (activation) trials from 4 animals. Unpaired t-test, t(17) = 3.15, p = 0.0058. f. Raster plot of food approaches in 24 h fasted animals. g. Latency of the first approach to food. Unpaired t-test, t(7) = 2.9, p = 0.022. h. Number of food approaches during a 20 min test. Unpaired t-test, t(7) = 3.6, p = 0.0083. Box plots show mean (+), median, quartiles (boxes), and range (whiskers). n.s., not significant; * p < 0.05, ** p < 0.01, *** p < 0.001.
Bilateral optogenetic activation of CEl PKC-δ+ neurons in vivo at 5 Hz (see Methods), a spiking rate comparable to that measured for these neurons in vivo20,32, strongly inhibited food intake during a 20 min test in 24 h food-deprived mice (Fig. 4b). Normal feeding resumed after the offset of photostimulation (Fig. 4b, orange bar). A similar inhibition was observed in fed mice (Fig. 4c), while controls expressing hrGFP in PKC-δ+ neurons showed no effect (Fig. 4b, c). Photoactivation of PKC-δ+ neurons also interrupted ongoing feeding in the home cage within a few seconds following the onset of photostimulation (Fig. 4d, e, and Movie S1). Activation also transiently inhibited drinking in water-deprived mice, although this effect was modest, and, in contrast to the effect on food intake, was only observed during the first few minutes of the 20 min test (Supplementary Fig. 3c-g). Importantly, feeding inhibition by PKC-δ+ neuron activation was not rescued when drinking water was provided (Supplementary Fig. 3h), suggesting that the suppression of food intake is not due to increased thirst.
To gain more insight into the inhibitory effect of CEl PKC-δ+ neurons on feeding, we performed a time-resolved analysis of feeding in 24 h food-deprived mice. This analysis revealed that activation of CEl PKC-δ+ neurons increased the latency to approach the food (Fig. 4f, g; “approach” defined as when the animal's nose touched the food pellet), and decreased the number of approaches (Fig. 4f, h). Thus, activation of CEl PKC-δ+ neurons inhibited both the appetitive and consummatory phases of feeding behavior. Importantly, mating behavior towards a female intruder mouse was not affected (Supplementary Fig. 3i), suggesting that PKC-δ+ neurons do not inhibit approach to any appetitive stimulus, but specifically inhibit approach to food resources.
Activation of CEl PKC-δ+ neurons does not increase anxiety
The CEA is well known to promote anxiety and fear33-35. This raised the possibility that activation of CEl PKC-δ+ neurons could inhibit feeding indirectly, by promoting anxiety or fear. Several lines of evidence suggest that this is not the case. First, in three different assays of anxiety, including the elevated plus maze, open field test and light-dark box test, optogenetic activation of CEl PKC-δ+ neurons was anxiolytic rather than anxiogenic (Fig. 5a-c). Second, the inhibition of feeding caused by optogenetic activation of CEl PKC-δ+ neurons was not reversed by the anxiolytic drug diazepam (DZ; Fig. 5d), although DZ did increase food intake in control mice (Fig. 5e). Third, activation of CEl PKC-δ+ neurons did not promote freezing or reduced locomotion (Supplementary Fig. 4a-b). Finally, activation did not produce conditioned place aversion, a measure of discomfort or unpleasantness36 (Supplementary Fig. 4c-e). These data suggest that the anorexic effect of CEl PKC-δ+ neuronal activation is unlikely to be due to increased fear or anxiety
Figure 5. Activation of CEl PKC-δ+ neurons does not increase anxiety.
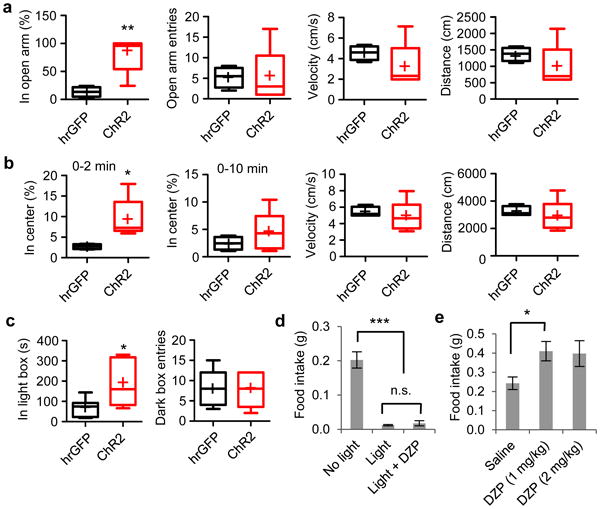
a-c. Optogenetic activation of PKC-δ+ neurons during elevated plus maze (a), open field test (b), and light-dark box test (c). n = 5 mice expressing ChR2 and n = 4 - 7 mice expressing control protein hrGFP. Box plots show mean (+), median, quartiles (boxes), and range (whiskers). Unpaired t-test, t(7) = 4.01, p = 0.0051 (a, in open arm); t(7) = 0.014, p = 0.99 (a, open arm entries); t(7) = 1.12, p = 0.30 (a, velocity); t(7) = 1.12, p = 0.30 (a, distance moved); t(7) = 2.74, p = 0.029 (b, in center, 0 – 2 min); t(7) = 1.04, p = 0.33 (b, in center, 0 – 10 min); t(7) = 0.60, p = 0.57 (b, velocity, 0 – 10 min); t(7) = 0.60, p = 0.57 (b, distance moved, 0 – 10 min); t(7) = 2.50, p = 0.031 (c, in light box), t(7) = 0.14, p = 0.90 (c, dark box entries). d. Food intake after injection of the anxiolytic drug diazepam (DZP). n = 4 (no light), 5 (light), 5 (light+ DZP) mice expressing ChR2 in each group. One-way ANOVA (F(2, 13) = 44.5, p < 0.0001) with post-hoc Bonferroni t-test indicated. e. Diazepam injected at a dose of 1 mg/kg increases food intake in wild type animals. n = 4 animals in each group. Unpaired t-test, t(6) = 2.78, p = 0.032. Values are means ± s.e.m.; n.s., not significant; * p < 0.05, ** p < 0.01, *** p < 0.001.
To investigate whether CEl PKC-δ+ neuron activation might inhibit feeding by promoting visceral malaise, we asked whether it could serve as an unconditional stimulus (US) in conditioned taste aversion (CTA) assays. Stimuli such as LiCl that cause visceral malaise are able to act as a US in such assays24. However, activation of PKC-δ+ neurons did not promote CTA, although in parallel controls LiCl administration to mice expressing hrGFP in CEl PKC-δ+ neurons did serve as an effective US for CTA (Supplementary Fig. 4f). This positive control indicates that expression of virally encoded proteins in these neurons does not interfere with CTA per se. Together, these data suggest that activation of PKC-δ+ neurons does not inhibit feeding by promoting visceral malaise.
Monosynaptic anorexigenically activated inputs to PKC-δ+ neurons
The observation that CEl PKC-δ+ neurons are activated by, and required for the effect of, several types of anorexigenic agents raised the question of the circuits through which these agents act on these neurons. To address this question, we performed Cre-dependent, rabies virus-based monosynaptic retrograde tracing from CEl PKC-δ+ neurons37,38, in combination with c-Fos labeling following treatment with anorexigenic agents (Fig. 6, Supplementary Fig. 5 and 6). On the one hand, these agents might all act through a final common pathway pre-synaptic to CEl PKC-δ+ neurons, in which case c-Fos+ neurons in a single location would be retrogradely labeled (Supplementary Fig. 6a, left); on the other hand, if c-Fos+ neurons in multiple areas were retrogradely labeled, it would suggest convergence from several pathways onto CEl PKC-δ+ neurons (Supplementary Fig. 6a, right).
Figure 6. Identification of monosynaptic inputs to CEl PKC-δ+ neurons that are activated by anorexigenic drugs.
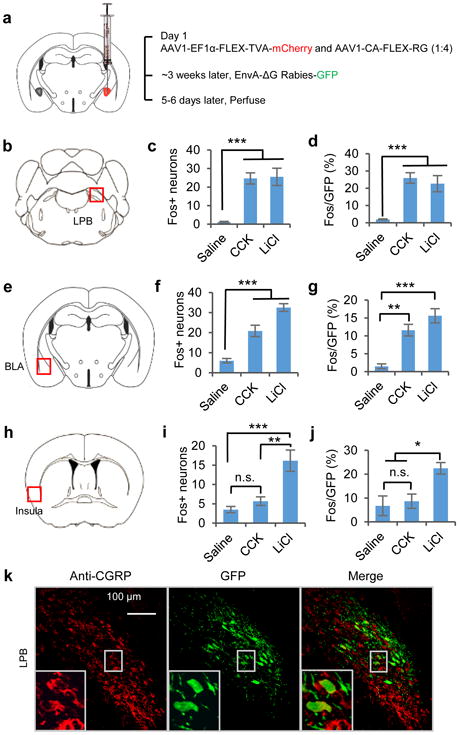
a. Virus injection procedure for rabies retrograde tracing. b-j. Quantification of c-Fos expression and GFP double-labelling in LPB (b-d), BLA (e-g), and insula (h-j). Representative histology is shown in Supplementary Fig. 6. n = 6 sections from two injection experiments in each group. One-way ANOVA (F(2, 15) = 23.1, p < 0.0001 (c); F(2, 15) = 12.0, p = 0.0008 (d); F(2, 15) = 42.4, p < 0.0001 (f); F(2, 15) = 25.1, p < 0.0001 (g); F(2, 15) = 14.6, p = 0.0003 (i); F(2, 15) = 18.8, p < 0.0001 (j)) with post-hoc Bonferroni t-test shows significant differences between groups (asterisks). k. Histology of rabies-GFP+ (monosynaptic retrograde labeling from CEl PKC-δ+ neurons) and CGRP+(antibody staining) neurons in LPB. GFP/CGRP (%) = 14.5 ± 1.5; CGRP/GFP (%) = 63.2 ± 4.5; n = 7 sections from two injection experiments. Values are means ± s.e.m.. n.s., not significant; * p < 0.05, ** p < 0.01, *** p < 0.001.
These experiments yielded retrogradely labeled (GFP+) cells within CEA itself, as well as in the basolateral amygdala (BLA), insular cortex, lateral parabrachial nucleus (LPB), and several other brain regions (Supplementary Fig. 5). Moreover, different anorexigenic agents activated c-Fos expression (relative to saline-injected controls) in these retrogradely labeled cells, in a combinatorial manner: LiCl and quinine induced c-Fos expression in Rabies-GFP+ neurons in all of these areas (LPB, BLA, and insula), while CCK induced c-Fos expression in retrogradely labeled neurons within the LPB and BLA, but not in the insula (Fig. 6a-j, Supplementary Fig. 6 and 7). Slice recordings using ChR2 assisted circuit mapping39 confirmed that CEl PKC-δ+ neurons receive monosynaptic excitatory inputs from the insula and LPB (Supplementary Fig. 8a-g). Taken together, these data argue against the idea that the upstream neurons activated by these anorexigenic agents converge on a common target pre-synaptic to CEl PKC-δ+ neurons, but rather that they converge on PKC-δ+ neurons themselves (Supplementary Fig. 6a, right).
Recently, Carter et al. showed that activation of CGRP+ neurons in the LPB inhibits feeding, and that this inhibition is mediated by projections to CEl5. Cre-dependent monosynaptic retrograde tracing from CEl PKC-δ+ neurons combined with anti-CGRP immunostaining revealed that over 60% of retrogradely labeled LPB neurons were CGRP+ (Fig. 6k). These data suggest that PKC-δ+ neurons are synaptic targets of CGRP+ LPB neurons that project to CEl, but are also targets of CGRP-LPB neurons. Consistent with these retrograde tracing experiments, slice recordings revealed that CEl PKC-δ+ neurons receive excitatory inputs from the LPB (Supplementary Fig. 8e-g).
Local CEA inhibitory circuits mediate feeding inhibition
As a first step towards investigating the circuit-level mechanisms through which CEl PKC-δ+ neurons might exert their inhibitory influence on feeding, we mapped their downstream projections using Cre-dependent fluorescent tracers40. These experiments revealed local projections of PKC-δ+ neurons in CEA (lateral and medial subdivisions), as well as longer-range projections to the bed nucleus of the stria terminalis (BNST) and the LPB (Supplementary Fig. 8h). Using ChR2-assisted circuit mapping39, we confirmed that CEl-PKC-δ+ neurons mono-synaptically inhibit PKC-δ− CEA neurons20,32 (20 out of 20 neurons tested evoked IPSCs, inhibitory postsynaptic currents), as well as BNST neurons (IPSCs evoked in 9 out of 10 neurons); however inhibition of LPB neurons was weak (only 1 out of 6 neurons showed a small IPSC) (Supplementary Fig. 8i-n).
To determine which of these outputs might contribute to the influence of CEl PKC-δ+ neurons on feeding, we first optogenetically activated the projections of these neurons to the LPB and BNST35. No inhibition of feeding was observed (n = 5 animals tested for each projection; data not shown). To test whether intra-amygdalar inhibition is involved, we bilaterally infused the GABAA receptor antagonist bicuculline into CEA while optogenetically activating PKC-δ+ neurons (Fig. 7a). Amygdala acute slice recordings indicated that bicuculline blocked IPSCs in PKC-δ− neurons evoked by optogenetic activation of PKC-δ+ neurons (Fig. 7b). Photostimulation-induced inhibition of food intake was blocked by bicuculline infusion (Fig. 7c), suggesting that local GABAergic signaling in CEA is required for the inhibitory influence of GABAergic PKC-δ+ neurons on feeding.
Figure 7. Local CEA inhibitory circuits mediate feeding inhibition by PKC-δ+ neurons.

a. Schematic illustrating combined in vivo light and drug delivery to CEA. b, IPSC of PKC-δ− neurons by optogenetic activation of PKC-δ+ neurons is blocked by bicuculline. c, Food intake in 24 h fasted animals photostimulated and infused with ACSF or 0.2 mM bicuculline. n = 7 mice in each group. Two-way ANOVA (F(1, 12) = 0.869, p = 0.37) with post-hoc Bonferroni t-test indicated that ChR2 induced feeding inhibition is blocked by bicuculline. d. CEl PKC-δ−, but not PKC-δ+, neurons are silenced following CEl injection of Cre-out rAAV-eNpHR3.0 (schematic). e. Silencing of CEl PKC-δ− neurons reduces food intake in 24 h fasted animals. n = 9 mice in each group. Paired t-test, t(16) = 3.46, p = 0.0030. See Methods and Supplementary Fig. 9 for additional details. f-h. Food intake in 24 h fasted (g) or fed (h) animals after activation of PKC-δ− neurons (f). n = 10 (control) and 9 (activation) animals. Unpaired t-test, t(17) = 0.458, p = 0.65 (g); t(17) = 0.39, p = 0.70 (h). Box plots show mean (+), median, quartiles (boxes), and range (whiskers). i. Food intake in 24 h fasted animals treated with CCK while activating CEl PKC-δ− neurons using Cre-out ChR2. Unpaired t-test, t(17) = 2.64, p = 0.0093 (saline + activation vs. CCK + activation); t(15) = 2.78, p = 0.014 (CCK + control vs. CCK + activation). Values are means ± s.e.m.. n.s., not significant; *p < 0.05; ** p < 0.01; *** p < 0.001.
The preceding results prompted us to investigate the intra-amygdalar targets through which PKC-δ+ neurons might inhibit feeding. As described earlier, inhibition of these neurons using hM4Di not only reduced c-Fos induction in PKC-δ+ cells by anorexigenic signals, but concurrently increased c-Fos expression in CEl PKC-δ− neurons (Supplementary Fig. 2). These data, and the fact that CEl PKC-δ+ neurons monosynaptically inhibit CEl PKC-δ− neurons20,32 (Fig. 7b), raised the possibility that the latter neurons exert an influence on feeding opposite to that of PKC-δ+ neurons. If so, then inhibition of these PKC-δ− neurons might inhibit feeding, similar to the effect of activating CEl PKC-δ+ neurons.
To test this hypothesis, we used a “Cre-out” strategy, in which PKC-δ-Cre mice were injected in CEA with an rAAV designed so that Cre recombinase excises, rather than activates, the eNpHR3.0 coding sequence41 (Fig. 7d). In this way, PKC-δ− neurons in CEA, rather than PKC-δ+ neurons, should express eNpHR3.0 (Fig. 7d). Slice recordings confirmed photostimulation-dependent silencing of PKC-δ− neurons, while PKC-δ+ neurons were not obviously affected (Fig. 7d, traces, and Methods). Food intake in 24 h food-deprived mice was partially but significantly inhibited upon silencing PKC-δ− neurons in vivo (Fig. 7e), in a manner that scaled with the level of eNpHR3.0 expression in CEl (Supplementary Fig. 9). These data support the idea that CEl PKC-δ− neurons exert a net positive influence on feeding, in contradistinction to PKC-δ+ neurons.
To extend this result, we asked whether activation of PKC-δ− neurons might, conversely, increase food intake. To do this, we first activated genetically defined subsets of CEl PKC-δ− neurons using two different Cre lines: Tac2-Cre (obtained from the Allen Institute for Brain Science; http://connectivity.brain-map.org/transgenic/experiment/131034318) and CRF-Cre42 (of which 50% and 70% are PKC-δ−, respectively; Supplementary Fig. 10a-c). However optogenetic activation of these neurons yielded neither increased nor decreased feeding (Supplementary Figure 10d-g). To activate other populations of CEl PKC-δ− neurons that might not be covered by these Cre lines, we expressed ChR2 in them using the same “Cre-out” approach41 described earlier for eNpHR3.0 (Fig. 7f). Activation of these PKC-δ− neurons neither increased food intake in 24 h fasted nor in fed mice (Fig. 7g, h). This may reflect a ceiling effect on feeding, or incomplete elimination of ChR2 expression from PKC-δ+ neurons that exert a counteracting influence.
Since inhibition of PKC-δ+ neurons likewise failed to increase food intake in 24 h fasted mice, but reversed the inhibitory effect of some anorexigenic agents (Fig. 2b), we asked whether activation of PKC-δ− neurons would similarly attenuate the effects of anorexigenic agents. Indeed, optogenetic activation of CEA PKC-δ− neurons significantly rescued the inhibition of feeding by CCK, although complete recovery of food intake to control levels was not achieved (Fig. 7h). Inhibition of feeding by LiCl was not significantly rescued (not shown). These data suggest that the influence of at least one anorexigenic signal (CCK) may be exerted, at least in part, through inhibition of CEA PKC-δ− neurons.
Discussion
CEl PKC-δ+ and PKC-δ− neurons exert opposing influences on feeding
Although the CEA has been implicated in the control of feeding, prior results have been contradictory, likely reflecting the cellular heterogeneity of this structure20-22 and the inadequate specificity of the perturbational methods used. Here we show that PKC-δ+ neurons in CEl play a central role in mediating the inhibitory influence on feeding of diverse anorexigenic signals, as well as in satiety, and can exert a profound and rapid inhibitory influence on feeding when activated. We also show that some CEA PKC-δ− neurons may exert an opposite-direction influence on feeding (although the resolution of our method does not allow us to distinguish whether these neurons are in CEl, CEm or both). Furthermore, both populations mediate the influence of some anorexigenic agents to suppress food intake, but in opposite directions: PKC-δ+ neurons must be active, while PKC-δ− neurons must be inhibited, to achieve complete suppression of feeding. These functional data are consistent with our observation that anorexigenic agents increase c-Fos expression in CEl PKC-δ+ neurons and reduce its expression in PKC-δ− neurons (Supplementary Fig. 2). The presence of intermingled populations of neurons in CEA with opposing influences on feeding may explain previous inconsistencies in the literature.
CEl PKC-δ+ neurons as a locus for multiple anorexigenic influences
CEl PKC-δ+ neurons are activated by multiple anorexigenic signals, as well as by bitter tastants and satiety. Inhibition of these neurons impairs the influence of some but not all anorexigenic agents tested (CCK and LiCl, but not LPS which induces a wide range of inflammatory and sickness response25,26). It also blocks the influence of quinine, a bitter tastant, to inhibit feeding and increases the time to satiation in food-deprived animals. Taken together, these data suggest that PKC-δ+ neurons mediate the effects of multiple (but clearly not all) inhibitory influences on feeding. This finding raises the question of whether these influences converge on a final common input to PKC-δ+ neurons, or rather act through multiple, parallel circuits that converge at the level of these neurons themselves. Our combined c-Fos and monosynaptic retrograde tracing data argue against the former, and in favor of the latter possibility. However further studies will be required to determine whether each of these upstream inputs acts through the same or through different PKC-δ+ neurons.
A recent study has shown that LPB CGRP+ neurons inhibit feeding via projections to CEl5. Our data indicate that ∼60% of LPB inputs to PKC-δ+ neurons derive from CGRP+ neurons. Whether the CEl PKC-δ+ neurons that receive input from CGRP+ neurons are activated by anorexigenic agents and mediate feeding inhibition, and whether inputs from CGRP- LPB neurons also inhibit feeding via inputs to these neurons, is unclear and remains to be investigated. We note, moreover, that LPB CGRP+ neurons mediate the anorexigenic influences of LiCl and LPS but not CCK5, while CEl PKC-δ+ neurons mediate the inhibitory influences of CCK and LiCl, but not LPS. This suggests that other inputs to PKC-δ+ neurons may mediate the inhibitory influence of CCK, and that the LPB represents but one of multiple anorexigenic pathways that converge on these CEl neurons. Interestingly, our ChR2-assisted circuit-mapping studies indicated that most if not all of the upstream inputs to PKC-δ+ neurons that we have tested are excitatory. The fact that these CEl neurons are GABAergic suggests, therefore, that they may represent the first inhibitory relay in the central processing of inhibitory influences on food intake.
CEl PKC-δ+ neurons, feeding and fear
The CEA is thought to be involved in promoting a variety of aversive states, including fear, anxiety, and visceral malaise33-35,43. Because fear and anxiety reduce appetitive behaviors, including feeding, it was possible that activation of CEl PKC-δ+ neurons might inhibit feeding indirectly, by promoting such aversive states. However, using multiple behavioral assays of fear and anxiety, we found no evidence that activation of CEl PKC-δ+ neurons, under conditions that inhibit feeding, makes mice more fearful or anxious; to the contrary the influence of their activation is anxiolytic. These data are consistent with previous studies indicating that CEl PKC-δ+ neurons exert an inhibitory influence on conditioned fear20. Moreover, innate fear, as expressed by unconditional freezing, was not evoked by optogenetic activation of PKC-δ+ neurons at frequencies that inhibit feeding. Finally, while fear and anxiety not only inhibit feeding, but also other appetitive behaviors such as mating, activation of CEl PKC-δ+ neurons, in contrast, had no inhibitory effect on mating.
These data not with standing, we cannot exclude the possibility that the optogenetic activation of PKC-δ+ neurons inhibits feeding indirectly, via an influence on some other behavioral or motivational state that in turn reduces food intake. For example, the CEA also plays a role in autonomic processes such as the control of heart rate and blood pressure33, and we have not ruled out that the effect of CEl PKC-δ+ neuron activation to inhibit feeding may involve some of these physiological influences. Nevertheless, the fact that PKC-δ+ neurons are activated by anorexigenic drugs in vivo, and, most importantly, that inhibiting these neurons reduces the anorexigenic influence of some of these drugs, makes it more likely, in our opinion, that these neurons play a role in mediating the influence of certain anorexigenic signals.
The relationship between the CEl PKC-δ+ neurons involved in the control of feeding, and those involved in the regulation of conditioned fear20, remains to be established. Our c-Fos data indicate that ∼20-25% of PKC-δ+ neurons are activated by anorexigenic drugs (Supplementary Fig. 1). This 20-25% may represent a deterministic subset of the PKC-δ+ population. In other words, there may be different subsets of CEl PKC-δ+ neurons that regulate the expression of conditioned fear, and the influence of anorexigenic agents, respectively. Our previous study provided evidence of molecular heterogeneity within the CEl PKC-δ+ population: for example, 40% of these neurons express the neuropeptide enkephalin, while 65% express the oxytocin receptor20. Whether such heterogeneity is relevant to the control of fear vs. feeding is not clear. Alternatively, a common or overlapping set of PKC-δ+ neurons in CEl may play a role in both fear and in feeding, according to their state of activity and/or the functional networks in which they participate. Further studies will clearly be required to resolve these alternatives.
How do CEl PKC-δ+ neurons inhibit feeding?
The neural circuits through which CEl PKC-δ+ neurons exert their inhibitory influence on feeding remains to be established. Because feeding is promoted by hypothalamic structures such as the arcuate nucleus (Arc) and lateral hypothalamus (LH)1-3, it seems likely that direct or indirect projections from CEA to these structures are involved in the inhibition of feeding. Whether these projections derive from CEl PKC-δ+ neurons, or from other CEA populations that are targets of PKC-δ+ neurons, is not yet clear. It has recently been shown that GABAergic neurons in BNST promote feeding via projections to the LH46. Because CEl PKC-δ+ neurons project to, and make inhibitory synapses in, the BNST it is attractive to think that these GABAergic neurons might suppress feeding via inhibition of LH-projecting BNST neurons. However, our attempts to inhibit feeding by stimulating CEl PKC-δ+ neuron projections to the BNST were unsuccessful, although we cannot rule out technical reasons for this negative result (e.g., insufficient levels of ChR2 expression in projections).
While other extra-amygdalar projections of CEl PKC-δ+ neurons (http://connectivity.brain-map.org) may be involved in feeding suppression, these neurons also make extensive projections within the amygdala. We found that local infusion of bicuculline in CEA can block feeding inhibition caused by optogenetic activation of PKC-δ+ neurons. This epistasis suggests that intra-amygdalar GABAergic synapses made by PKC-δ+ neurons are required for their anorexigenic function, although we cannot rule out an indirect effect due to dis-inhibition of other neighboring amygdala populations. Whether these synapses inhibit amygdalar subpopulations that make long-range (direct or indirect) projections to feeding centers, or rather minimize local feedback inhibition onto PKC-δ+ neurons, e.g., from CEl PKC-δ− neurons20,32 is not clear and will require further study.
The studies presented here have identified a population of central amygdale GABAergic neurons that plays an important role in mediating the influence of signals that inhibit feeding behavior. The fact that optogenetic activation of these neurons strongly reduces food intake further suggests that these cells could provide a target for novel therapeutic interventions to treat obesity, anorexia or other eating disorders.
Online Methods
All experimental protocols were conducted according to US National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee (IACUC) at the California Institute of Technology.
Mice
Male mice were housed on a 12 hour light (7 am)/dark (7 pm) cycle with ad libitum access to water and rodent chow (Lab Diet, 5001) unless otherwise noted. Male mice were used for all the behavioral experiments, male and a few female mice were used in brain slice electrophysiology. Ai14 Cre-reporter mice47 have been described previously.
Virus
All the AAV viruses were produced at the Gene Therapy Center Vector Core in the University of North Carolina at Chapel Hill. Rabies virus were purchased from Salk Gene Transfer, Targeting and Therapeutics Core. Behavioral experiments were usually performed 4 weeks after virus injection. For rabies virus tracing, AAV1-EF1α-FLEX-TVAmCherry and AAV1-CA-FLEX-RG were mixed at a ratio of 1:4 and then injected in CEA; around 3 weeks later, EnvA G-deleted Rabies-GFP were injected in the same place; 5 - 6 days later, mice were perfused and their brains were sectioned to check the monosynaptic upstream neurons. Each virus and its serotype were indicated in the main text.
Animal surgery
Mice survival surgeries were performed as described previously48. Briefly, mice 2-4 months old were deeply anaesthetized with 5% isoflurane in oxygen and kept at 1.5% isoflurane during surgery. Surgery was performed with a stereotaxic frame (Kopf). Viral injection coordinates (in mm, midline, Bregma, dorsal surface): CEA (±2.85, -1.40, -4.72), LPB (±1.50, -5.30, -3.40), LH (±1.20, -1.55, -5.20), ventral BNST (±1.00, +0.05, -4.70), insula (±3.90, +0.05, -4.00). Ferrule fibers were implanted ∼0.5 mm above the injection sites. After ferrule fiber implanted, dental cement (Metabond) was used to anchor the fiber to the skull. For behavioral experiments that require drug infusion, guide cannulas (26 G, PlasticsOne) were implanted ∼0.8 mm above the injection sites. Mice were single housed after surgery. At least 3-4 weeks were allowed for mice recovery and viral expression after surgery.
In vivo photostimulation
Several lasers (Shanghai DreamLaser: 473 nm, 50 mW; 593 nm, 50 mW; Crystal Laser: 445 nm, 50 mW; 473 nm, 50 mW; 561 nm, 50 mW) were used to deliver light. An Accupulser Signal Generator (World Precision Instruments, SYS-A310) were used to control the frequency and pulse width of the laser light. Light was delivered to the brain through an optic fiber (200 μm diameter core, NA 0.22, Doric Lenses) connected with the implanted ferrule fiber by a zirconium sleeve. The light power in the brain regions 0.5 mm below the fiber tip was calibrated according to the description49. The calibrated light power density (0.5 mm below the fiber tip) used in light activation experiment was 5-6 mW/mm2. A 5 Hz, 10 ms pulse width light were used in all the optogenetic activation experiments.5 Hz light stimulation was used because (1) PKC-δ+ neurons show an average firing rate of 3-5 Hz in in vivo recordings50, (2) CEA neurons fires at a rate of less than 5 Hz at innocuous stimulus while fires more than 10 Hz in response to noxious stimuli51, (3) PKC-δ+ neurons can be triggered robustly to fire action potentials by ≤ 10 Hz light pulses as shown by whole-cell patch clamp slice recordings48.
Continuous light illumination in the brain over a long time generates heat52, so pulsed light was used for optogenetic silencing during the 20 min feeding test. And because PKC-δ+ neurons do not have rebound firing after the light is off as tested in slice recording (Fig. 3a), a protocol of alternating 20 seconds light on and 10 seconds light off was used to silence CEl PKC-δ+ neurons (Fig. 3b). Because some type of PKC-δ− neurons have rebound firing after hyperpolarization48, a different protocol of 20 Hz, 20 ms light pulse (calibrated light power in CEA, ∼ 2 mW/mm2) was used to silence PKC-δ− neurons (Fig. 7d). As tested in brain slices, neurons expressing eNpHR3.0 can be hyperpolarized by different wavelengths (we tested 445 nm, 473 nm, 561 nm, 593 nm, and 633 nm); and because mice eat normally with 20 Hz, 20 ms 593 nm light pulses at a power less than 10 mW/mm2 (food intake in mice with light: 0.29 ± 0.03 g; food intake in mice without light: 0.30 ± 0.02 g; t test, p = 0.78.); we use the food intake when mice were implanted with optic fiber but received no light as control (Fig. 6e).
Pharmacology
CNO (Enzo life science-Biomol, BML-NS105-0005) were freshly dissolved in injection saline (0.9% NaCl) and intraperitoneally injected at 5 mg/kg for hM4Di silencing, or 2 mg/kg for hM3Dq activation. Other compounds used for intraperitoneal injection: CCK (5 μg/kg, Tocris), Diazepam (1 mg/kg or 2 mg/kg, Hospira), LiCl (150 mg/kg, Sigma, prepared in 150 mM), LPS (0.1 mg/kg, Sigma, L4516-1MG). Behavioral test were usually performed 20-30 min after drug injection.
Behavioral tests
Feeding behavior
For 24 h fasted feeding test, mice were food-deprived the day before test. Mice were briefly anaesthetized with isoflurane and coupled with optic fibers. 15 min after recovery, mice were introduced into a novel empty cage with a regular food pellet (Lab Diet, 5001), and allowed for feeding for 20 min. The weight of the food pellet, including the food debris left in the cage floor after test, was measured to calculate the food intake. For fed feeding test, mice were not food deprived before feeding test. For optogenetic experiments, the light was started just before the mice were introduced into the testing cage. The feeding behavior was videotaped and manually analyzed. For pharmacogenetic experiments, CNO was injected 20 min before the feeding test. All the feeding test were performed between 2 pm and 7 pm. Bitter food was prepared by immersing regular food pellets in 10 mM quinine (Sigma) solution for 10 min and dried overnight, and the control taste food was prepared by immersing the food pellets in dH2O for 10 min and dried overnight. In the conditioned taste aversion experiment (Supplementary Fig. 4f), bacon taste food pellets (Newco Distributors, 10 mg Tablet) were used as a novel taste food.
Intraoral infusion
A standard ball-tipped gavage needle was put just in the mouth and ∼0.2 ml water was slowly infused in ∼15 seconds. To induce c-Fos expression by bitter tastant, mice were oral-infused with ∼0.2 ml 10 mM quinine solution for 15 seconds, animals infused with water or 0.5 M sucrose solution were used as controls. Mice were perfused 1-1.5 hour after infusion.
Anxiety test
Standard elevated plus maze, open field, and light/dark box were used. A platform (74 cm above the floor) with two opposing open arms (30 × 5 cm) and two opposing closed arms (30 × 5 × 14 cm) was used as elevated plus maze. Mice were placed into the center of the plus and their behavior was tracked for 5 min. A square plastic box (50 × 50 × 30 cm, a 25 × 25 cm square center was defined as “center” in analysis) was used as open box. Mice were placed individually in the center of the box, and their behavior was tracked for 10 min. A plastic box consisting of a dark (black with cover, 16 × 50 cm) and a bright (white no cover, 34 × 50 cm) compartment, which are connected by a central opening (7 × 14 cm) at the floor level, was used as a light/dark box. Mice were placed individually in the center of the light area. The behavior was tracked for 10 min. All the behaviors were videotaped and analyzed offline with Ethovision.
Place preference test
Place preference test were performed with a three-chamber system described previously53, in which Chamber A and Chamber B have different visual and tactile cues by having distinct walls and floors, while the center chamber is a neutral plastic enclosure. Mice were allowed to explore all three chambers for 15 min on day 1, and restricted in either chamber A or B for 20 min with light delivered on day 2. Mice were then allowed to explore all three chambers for 15 min on day 4. The behavior were videotaped and analyzed offline with Ethovision.
Behavioral tests were performed by an investigator with knowledge of the identity of the experimental groups versus control groups.
Immunohistochemistry and histology
All mice after behavioral test were perfused and checked for virus expression and implanted fiber or cannula location. For immunofluorescent staining, mice were transcardially perfused with 20 ml PBS followed by 20 ml 4% paraformaldehyde in PBS. For cryosection staining, brains were dissected out and immersed in 15% sucrose overnight. Cryosections of 30 μm thickness were processed. For the vibratome sectioning, brains were removed and postfixed in 4% paraformaldehyde overnight, then the section were cut with a vibratome (Leica, VT1000S) at 100 μm thickness. Sections were stained with primary antibody at 4 °C overnight, in a blocking solution contains 1% BSA or 5% donkey serum and 0.5% Triton X-100. After 3 × 10 min wash in PBS, standard Alexa Fluor secondary antibodies (Invitrogen, 1:250) were used at room temperature for 1 hour. Sections were then washed 3 × 10 min in PBS and mounted in Fluo Gel (17985-10; Electron Microscopy Sciences, with DAPI) and viewed under an Olympus confocal microscope. Primary antibodies used: mouse anti-PKC-δ (BD Biosciences, 610398, 1:500), rabbit anti-CGRP (Bachem, T4032, 1:500), goat anti-c-Fos (Santa Cruz Biotech, sc-52-G, 1:250).
Electrophysiological slice recordings
Mouse brain slice was prepared as described48. In brief, mouse coronal sections of 250 μm thickness were cut with a vibratome (Leica, VT1000S), using ice-cold glycerol-based ACSF containing (in mM): 252 glycerol, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, 11 Glucose, oxygenated in carbogen (95% O2 balanced with CO2) for at least 15 min before use. Slice were recovered for at least 1 hour at 32°C and then kept at room temperature in regular ACSF containing (in mM): 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, 11 Glucose, oxygenated with carbogen. The fluorescence of the cells were detected by a fluorescence video microscopy (Olympus BX51). Whole-cell voltage, current clamp, or cell-attached recordings were performed with a MultiClamp 700B amplifier and Digidata 1440A (Molecular Devices). The patch pipette with a resistance of 5-8 MΩ was filled with an intracellular solution containing (in mM): 135 potassium gluconate, 5 EGTA, 0.5 CaCl2, 2 MgCl2, 10 HEPES, 2 MgATP and 0.1 GTP, pH 7.2, 290–300 mOsm. Data were sampled at 10 kHz, filtered at 3 kHz and analyzed with pCLAMP10 software.
For photostimulation in brain slice, several lasers (Shanghai DreamLaser, 473 nm, 50 mW; 593 nm, 50 mW; Crystal Laser, 561 nm, 50 mW) were used to deliver pulsed light or continuous light, the typical power at the specimen ranges from 0.1 to 10 mW/mm2, as measured with a photodiode. In ChR2 assisted circuits mapping experiments, 2 ms light pulses were used to trigger IPSC or EPSC, and a 0.1 μs 0.1 mV was applied at the same time of light trigger to help identify the start of light pulse.
Statistics
Data are represented as means ± s.e.m. or box-and-whister plots. P value were calculated by paired (within subjects comparisons) or un-paired (between subjects comparisons) Student's t-test, and a value of smaller than 0.05 was consider significant. For comparisons across more than two groups, data were analyzed using one-way ANOVA and adjusted with the Bonferroni's correction. For data with more than one independent variable, two-way ANOVA was used. Each test and significance were indicated in figure legends. Data were analyzed with GraphPad Prism Software.
Supplementary Material
Acknowledgments
We thank B. Lowell for providing Cre-dependent DREADDs constructs and viruses for pilot experiments; C. Saper for providing the original hrGFP construct; Z. Josh Huang for CRF-Cre mice; Allen Institute for Brain Science for Tac2-Cre mice; C. Xiao and H. Lester for electrophysiology training and advice and K. Deisseroth for providing viruses for optogenetics. We thank J.S. Chang, C. Chiu, and A. Chang for technical assistance on histology and behavior scoring, M. Martinez and M. McCardle for tail genotyping, G. Mosconi and C. Chiu for lab management and G. Mancuso for administrative support. This work was supported by NIH grant R01MH085082 and a grant from the Klarman Foundation to D.J.A. H.C. is supported by a postdoctoral fellowship of the Hilda and Preston Davis Foundation and a NARSAD Young Investigator Award, D.J.A. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions: H.C. designed and performed the experiments, and co-wrote the manuscript; W.H. generated BAC constructs for PKC-δ+ transgenic mice; T.A. made the virus constructs for Cre-out ChR2 and Cre-out eNpHR. D.J.A. contributed to experimental design and co-wrote the manuscript.
Competing Financial Interests: The authors declare no competing financial interests.
A Supplementary Methods Checklist is available.
References
- 1.Sternson SM. Hypothalamic survival circuits: blueprints for purposive behaviors. Neuron. 2013;77:810–824. doi: 10.1016/j.neuron.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. nature05026 [pii] 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 3.Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 4.Woods SC, Seeley RJ, Porte D, Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science. 1998;280:1378–1383. doi: 10.1126/science.280.5368.1378. [DOI] [PubMed] [Google Scholar]
- 5.Carter ME, Soden ME, Zweifel LS, Palmiter RD. Genetic identification of a neural circuit that suppresses appetite. Nature. 2013;503:111–114. doi: 10.1038/nature12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Q, Clark MS, Palmiter RD. Deciphering a neuronal circuit that mediates appetite. Nature. 2012;483:594–597. doi: 10.1038/nature10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishi T, Elmquist JK. Body weight is regulated by the brain: a link between feeding and emotion. Mol Psychiatry. 2005;10:132–146. doi: 10.1038/sj.mp.4001638. 4001638 [pii] 10.1038/sj.mp.4001638. [DOI] [PubMed] [Google Scholar]
- 8.Betley JN, Cao ZF, Ritola KD, Sternson SM. Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell. 2013;155:1337–1350. doi: 10.1016/j.cell.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King BM. Amygdaloid lesion-induced obesity: relation to sexual behavior, olfaction, and the ventromedial hypothalamus. American journal of physiology Regulatory, integrative and comparative physiology. 2006;291:R1201–1214. doi: 10.1152/ajpregu.00199.2006. [DOI] [PubMed] [Google Scholar]
- 10.Kask A, Schioth HB. Tonic inhibition of food intake during inactive phase is reversed by the injection of the melanocortin receptor antagonist into the paraventricular nucleus of the hypothalamus and central amygdala of the rat. Brain Res. 2000;887:460–464. doi: 10.1016/s0006-8993(00)03034-1. S0006-8993(00)03034-1 [pii] [DOI] [PubMed] [Google Scholar]
- 11.Beckman TR, Shi Q, Levine AS, Billington CJ. Amygdalar opioids modulate hypothalamic melanocortin-induced anorexia. Physiol Behav. 2009;96:568–573. doi: 10.1016/j.physbeh.2008.12.007. S0031-9384(08)00386-7 [pii] 10.1016/j.physbeh.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fekete E, Vigh J, Bagi EE, Lenard L. Gastrin-releasing peptide microinjected into the amygdala inhibits feeding. Brain Res. 2002;955:55–63. doi: 10.1016/s0006-8993(02)03362-0. [DOI] [PubMed] [Google Scholar]
- 13.Fekete EM, Bagi EE, Toth K, Lenard L. Neuromedin C microinjected into the amygdala inhibits feeding. Brain Res Bull. 2007;71:386–392. doi: 10.1016/j.brainresbull.2006.10.007. S0361-9230(06)00308-X [pii] 10.1016/j.brainresbull.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Kovacs A, et al. Microinjection of RFRP-1 in the central nucleus of amygdala decreases food intake in the rat. Brain Res Bull. 2012;88:589–595. doi: 10.1016/j.brainresbull.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Bovetto S, Richard D. Lesion of central nucleus of amygdala promotes fat gain without preventing effect of exercise on energy balance. Am J Physiol. 1995;269:R781–786. doi: 10.1152/ajpregu.1995.269.4.R781. [DOI] [PubMed] [Google Scholar]
- 16.Lenard L, Hahn Z. Amygdalar noradrenergic and dopaminergic mechanisms in the regulation of hunger and thirst-motivated behavior. Brain Res. 1982;233:115–132. doi: 10.1016/0006-8993(82)90934-9. 0006-8993(82)90934-9 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Box BM, Mogenson GJ. Alterations in ingestive behaviors after bilateral lesions of the amygdala in the rat. Physiol Behav. 1975;15:679–688. doi: 10.1016/0031-9384(75)90119-5. 0031-9384(75)90119-5 [pii] [DOI] [PubMed] [Google Scholar]
- 18.Kemble ED, Studelska DR, Schmidt MK. Effects of central amygdaloid nucleus lesions on ingestion, taste reactivity, exploration and taste aversion. Physiol Behav. 1979;22:789–793. doi: 10.1016/0031-9384(79)90250-6. 0031-9384(79)90250-6 [pii] [DOI] [PubMed] [Google Scholar]
- 19.Petrovich GD, Ross CA, Mody P, Holland PC, Gallagher M. Central, but not basolateral, amygdala is critical for control of feeding by aversive learned cues. J Neurosci. 2009;29:15205–15212. doi: 10.1523/JNEUROSCI.3656-09.2009. 29/48/15205 [pii] 10.1523/JNEUROSCI.3656-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haubensak W, et al. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day HE, Curran EJ, Watson SJ, Jr, Akil H. Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria terminalis: evidence for their selective activation by interleukin-1beta. J Comp Neurol. 1999;413:113–128. [PubMed] [Google Scholar]
- 22.Marchant NJ, Densmore VS, Osborne PB. Coexpression of prodynorphin and corticotrophin-releasing hormone in the rat central amygdala: evidence of two distinct endogenous opioid systems in the lateral division. J Comp Neurol. 2007;504:702–715. doi: 10.1002/cne.21464. [DOI] [PubMed] [Google Scholar]
- 23.Moran TH. Cholecystokinin and satiety: current perspectives. Nutrition. 2000;16:858–865. doi: 10.1016/s0899-9007(00)00419-6. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto T, Ueji K. Brain mechanisms of flavor learning. Frontiers in systems neuroscience. 2011;5:76. doi: 10.3389/fnsys.2011.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dantzer R. Cytokine-induced sickness behavior: mechanisms and implications. Annals of the New York Academy of Sciences. 2001;933:222–234. doi: 10.1111/j.1749-6632.2001.tb05827.x. [DOI] [PubMed] [Google Scholar]
- 26.Haba R, et al. Lipopolysaccharide affects exploratory behaviors toward novel objects by impairing cognition and/or motivation in mice: Possible role of activation of the central amygdala. Behavioural brain research. 2012;228:423–431. doi: 10.1016/j.bbr.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 27.Lee HM, Giguere PM, Roth BL. DREADDs: novel tools for drug discovery and development. Drug discovery today. 2013 doi: 10.1016/j.drudis.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krashes MJ, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011 doi: 10.1172/JCI46229. 46229 [pii] 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gautron L, Lazarus M, Scott MM, Saper CB, Elmquist JK. Identifying the efferent projections of leptin-responsive neurons in the dorsomedial hypothalamus using a novel conditional tracing approach. J Comp Neurol. 2010;518:2090–2108. doi: 10.1002/cne.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gradinaru V, et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141:154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 32.Ciocchi S, et al. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. nature09559 [pii] 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- 33.Maren S, Fanselow MS. The amygdala and fear conditioning: has the nut been cracked? Neuron. 1996;16:237–240. doi: 10.1016/s0896-6273(00)80041-0. [DOI] [PubMed] [Google Scholar]
- 34.Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends in neurosciences. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 35.Tye KM, et al. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. nature09820 [pii] 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panksepp J. Cross-species affective neuroscience decoding of the primal affective experiences of humans and related animals. PloS one. 2011;6:e21236. doi: 10.1371/journal.pone.0021236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wall NR, Wickersham IR, Cetin A, De La Parra M, Callaway EM. Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21848–21853. doi: 10.1073/pnas.1011756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron. 2012;74:858–873. doi: 10.1016/j.neuron.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 39.Petreanu L, Huber D, Sobczyk A, Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nature neuroscience. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- 40.Oh SW, et al. A mesoscale connectome of the mouse brain. Nature. 2014;508:207–214. doi: 10.1038/nature13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee H, et al. Scalable control of mounting and attack by Esr1 neurons in the ventromedial hypothalamus. Nature. 2014 doi: 10.1038/nature13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taniguchi H, et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- 44.Lin D, et al. Functional identification of an aggression locus in the mouse hypothalamus. Nature. 2011;470:221–226. doi: 10.1038/nature09736. nature09736 [pii] 10.1038/nature09736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salzman CD, Fusi S. Emotion, cognition, and mental state representation in amygdala and prefrontal cortex. Annual review of neuroscience. 2010;33:173–202. doi: 10.1146/annurev.neuro.051508.135256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD. The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science. 2013;341:1517–1521. doi: 10.1126/science.1241812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haubensak W, et al. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010;468:270–276. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aravanis AM, et al. An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. Journal of neural engineering. 2007;4:S143–156. doi: 10.1088/1741-2560/4/3/S02. [DOI] [PubMed] [Google Scholar]
- 50.Ciocchi S, et al. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010;468:277–282. doi: 10.1038/nature09559. nature09559 [pii] 10.1038/nature09559. [DOI] [PubMed] [Google Scholar]
- 51.Neugebauer V, Li W. Processing of nociceptive mechanical and thermal information in central amygdala neurons with knee-joint input. J Neurophysiol. 2002;87:103–112. doi: 10.1152/jn.00264.2001. [DOI] [PubMed] [Google Scholar]
- 52.Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 53.Vrontou S, Wong AM, Rau KK, Koerber HR, Anderson DJ. Genetic identification of C fibres that detect massage-like stroking of hairy skin in vivo. Nature. 2013;493:669–673. doi: 10.1038/nature11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.